Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(5):1512-1522. doi:10.7150/jca.66241 This issue Cite
Research Paper
Multi-omics analysis identifies distinct subtypes with clinical relevance in lung adenocarcinoma harboring KEAP1/NFE2L2
Xiaodong Yang1#, Ming Li2#, Zhencong Chen2#, Xiaobin Fan3, Liang Guo1, Bo Jin4, Yiwei Huang2, Qun Wang2, Liang Wu5 ![]() , Cheng Zhan2
, Cheng Zhan2 ![]()
1. Department of Thoracic Surgery, Shanghai Pulmonary Hospital, Tongji University, Shanghai, China.
2. Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai, China.
3. Department of General Surgery, Xingtai Third Hospital, Hebei Province, China.
4. Department of Cardiothoracic Surgery, Zhangqiu District People's Hospital, Shandong Province, China.
5. Department of Thoracic Surgery, Shanghai General Hospital, Shanghai, China.
#These authors contributed equally to this work.
Received 2021-8-18; Accepted 2022-1-26; Published 2022-2-28
Abstract

Backgrounds: Lung adenocarcinoma is one of the most common malignant tumors, in which KEAP1-NFE2L2 pathway is altered frequently. The biological features and intrinsic heterogeneities of KEAP1/NFE2L2-mutant lung adenocarcinoma remain unclear.
Methods: Multiplatform data from The Cancer Genome Atlas (TCGA) were acquired to identify two subtypes of lung adenocarcinoma harboring KEAP1/NFE2L2 mutations.
Bioinformatic analyses, including immune microenvironment, methylation level and mutational signature, were performed to characterize the intrinsic heterogeneities. Meanwhile, initial results were validated by using in silico assessment of common lung adenocarcinoma cell lines, which revealed consistent features of mutant subtypes. Furthermore, drug sensitivity screening was conducted based on public datasets.
Results: Two mutant subtypes (P1 and P2) of 89 patients were identified in TCGA. P2 patients had significantly higher levels of smoking and worse survival compared with P1 patients. The P2 subset was characterized by active immune microenvironment and more smoking-induced genomic alterations with respect to methylation and somatic mutations. Validations of the corresponding features in 20 mutant cell lines were achieved. Several compounds which were sensitive to mutant subtypes of lung adenocarcinoma were identified, such as inhibitors of PI3K/Akt and IGF1R signaling pathways.
Conclusions: KEAP1/NFE2L2-mutant lung adenocarcinoma showed potential heterogeneities. The intrinsic heterogeneities of KEAP1/NFE2L2 were associated with immune microenvironment and smoking-related genomic aberrations.
Keywords: lung adenocarcinoma, KEAP1, mutation, NFE2L2
Introduction
Lung cancer is the leading cause of cancer-associated morbidity and mortality worldwide, among which lung adenocarcinoma accounts for the highest proportion with increasing incidence rate [1-7]. Previous studies promoted a paradigm shift regarding classifying lung tumors based on the significant genomic alterations for therapeutic targets, such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) [8-10]. The Kelch-like ECH-associated protein 1 (KEAP1) and the nuclear factor erythroid-2-related factor 2 (NFE2L2) mutations were found in more than 20% patients with non-small cell lung cancer, which represented one of the most important genomic subtypes [11,12]. Moreover, the genomic alterations of KEAP1 and NFE2L2 were reported to play crucial roles in lung adenocarcinoma [13-15].
Abnormal regulations of reactive oxygen species contribute to the occurrence and development of malignant tumors [16]. The KEAP1 and NFE2L2 are the two main components in the stress response pathways. KEAP1 mediates the degradation of NFE2L2 to act as an adaptor protein of the Cullin 3 (CUL3) E3 ubiquitin ligase so as to maintain the redox homeostasis. In the presence of oxidative stress, the inactivation of KEAP1 results in the release, accumulation, and nucleus translocation of NFE2L2 to counteract the damage [17,18]. The KEAP1/NFE2L2 mutations, representing the dysfunctional activations of the stress response pathway, have been found in many malignant tumors, including lung adenocarcinoma [19-21]. The KEAP1-NFE2L2 can be hijacked by cancer cells, and the activation of the pathway leads to increased tumor growth and progression [22-24]. Nevertheless, the biological features and clinical implications of KEAP1/NFE2L2 mutations remain elusive and contradictory [25]. In a retrospective study of 9243 patients (4647 with lung cancer), KEAP1/NFE2L2 mutations were associated with higher tumor mutational burden and higher programmed death-ligand 1 expression. Improved survival was observed in the subset of patients treated with immune checkpoint inhibitors [26]. On the contrary, patients with KEAP1/NFE2L2 mutations had inferior survival compared with wild-type patients in subgroup analyses from several trials regarding immunotherapy [27-29]. Concurrent mutations with KEAP1/NFE2L2 may also affect patients' benefits from immunotherapy [30,31]. Moreover, Hellyer et al suggested that KEAP1/NFE2L2 mutations might represent a mechanism of intrinsic resistance to EGFR-tyrosine kinase inhibitor therapy [32]. Chemoresistance was also reported to be associated with KEAP1/NFE2L2 mutations [33,34].
In our study, multiplatform data from The Cancer Genome Atlas (TCGA) were acquired to identify two subtypes of lung adenocarcinoma harboring KEAP1/NFE2L2 mutations. Bioinformatic analyses, including immune microenvironment and methylation level, were performed to characterize potential mutant subgroups. The initial results were validated by using in silico assessment of common lung adenocarcinoma cell lines, which revealed consistent features of KEAP1/NFE2L2-mutant subtypes. Furthermore, cell line samples were adopted for drug sensitivity screening based on public datasets. Potential drugs which were sensitive to each mutant subtype of lung adenocarcinoma were explored.
Methods
Patient cohort and cell lines data
First, we selected all patients (565 patients) with primary lung adenocarcinoma in TCGA database. Level 3 RNA sequencing data, DNA methylation data (Illumina Infinium HumanMethylation 450K BeadChip), miRNA expression data and clinical information of patients with lung adenocarcinoma were downloaded from TCGA (https://protal.gdc.cancer.gov/). Somatic mutation data were selected based on previous studies by comprehensive analyses accounting for variance and batch effects [35]. Copy number variations (CNV) were estimated using the GISTIC2 method from the University of California Santa Cruz Xena website (https://xena.ucsc.edu). Patients with missing data types in the above were excluded (67 of 565 patients). According to the mutation data, patients with KEAP1/NFE2L2 mutations were selected as the main study cohort (89 patients). The remaining 409 patients without KEAP1/NFE2L2 mutations were regarded as the wild-type group in the subsequent analyses.
RNA sequencing data, miRNA expression levels, copy number values and gene mutation status of common lung adenocarcinoma cell lines were downloaded from the Cancer Cell Line Encyclopedia (CCLE, https://portals.broadinstitute.org/ccle). Also, DNA methylation levels (Illumina Infinium HumanMethylation 450K BeadChip) of selected cancer cell lines were acquired from the Gene Expression Omnibus (GEO, (https://www.ncbi.nlm.nih.gov/geo) (GSE68379). The drug sensitivity data of selected cancer cell lines were obtained from the Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene.org/). Histological information of each cell line was confirmed based on GDSC, CCLE and Cellosaurus database [36,37]. Cell lines with unknown data types were removed. In total, 20 lung adenocarcinoma cell lines with KEAP1/NFE2L2 mutations were identified.
Data processing and clustering
For the DNA methylation data, probes in sex chromosomes or overlapping single nucleotide polymorphisms were removed. Cross-reactive probes were also excluded according to Chen et al [38]. The frequencies of six base substitutions (C > A, C > G, C > T, T > A, T > C, and T > G) were calculated. For some datasets, features or probes with more than 20% missing values were deleted. The k-nearest neighbor algorithm was adopted to impute the remaining missing data.
All five data types (RNA sequencing, DNA methylation, miRN, copy number and base substitution) were integrated using the similar network fusion (SNF) method for both lung adenocarcinoma patients and cell lines. The SNF method constructs networks of samples for each available genome-wide data and efficiently fuses them into one network, which represents the full spectrum of underlying features and provides a comprehensive view under a given condition [39]. The SNF method has been used and validated in different types of diseases based on multi-omics data [40-43]. In this study, the SNF method fused all five datasets into one by creating a similarity matrix for each data type. A non-linear method based on the theory of message-passing was adopted to iteratively update and converge datasets. Afterwards, consensus clustering was performed to identify distinct KEAP1/NFE2L2 mutated subgroups of lung adenocarcinoma patients and cell lines [44].
Bioinformatic analyses to characterize KEAP1/NFE2L2-mutant subgroups
Mutant subgroups were preliminarily characterized by subjecting clusters for both patients and cell lines to Gene Set Enrichment Analysis (GSEA) using Hallmark, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) (MSigDB v7.0) gene sets [45]. Normalized enrichment score >1, nominal P-value <0.05, and false discovery rate Q-value < 0.25 were used as screening thresholds for GSEA. Moreover, we studied potential concurrent mutations in KEAP1/NFE2L2-mutant subsets of lung adenocarcinoma patients.
The features of tumor immune microenvironment in KEAP1/NFE2L2-mutant lung adenocarcinoma were evaluated according to several previous studies. Saltz et al proposed a leukocyte fraction by estimating tumor-infiltrating leukocytes on hematoxylin and eosin stained slides using deep learning techniques [46]. We also used the “Estimation of STromal and Immune cells in MAlignant Tumours using Expression data (ESTIMATE)” method for the assessment of tumor immune microenvironment [47]. Li et al developed a public resource (Tumor IMmune Estimation Resource, TIMER) to study tumor-infiltrating immune cells by computational approaches based on RNA sequencing [48]. The levels of specific immune cell infiltration, like CD8+ T cell and macrophage, between mutant subgroups were compared. Furthermore, we compared the number of immunogenic mutations per sample stratified by the KEAP1/NFE2L2 mutant status.
The global methylation levels (β value) between KEAP1/NFE2L2-mutant patient subgroups and cell line subsets were compared to investigate epigenomic alterations and potential clinical associations. Next, a list of smoking-related DNA methylation probes was obtained from a previous study conducted by Vaz et al. Vaz et al performed two repeated experiments with respect to chronic-cigarette-smoking-induced hypermethylated probes [49].The union of all reported probes was extracted and their levels stratified by the mutant subsets were compared. Somatic mutation status of KEAP1/NFE2L2-mutant patients was analyzed to extract mutational signatures using the SignatureAnalyzer [50]. Similarities were studied based on previously reported thirty mutational signatures in the Catalogue Of Somatic Mutations In Cancer (COSMIC, https://cancer.sanger.ac.uk/cosmic) to identify the potential clinical associations and etiologies.
Cancer-associated drug sensitivity data of lung adenocarcinoma cell lines were also downloaded from two sub-datasets of GDSC. Drug samples that were tested in < 50% cell lines were excluded. The natural log value of the fitted half-maximal inhibitory concentration [LN(IC50)] of each drug was adopted to select caner-associated drugs which were specifically sensitive to mutant subtypes (C1 and C2). Many attempts have been made to perform in vitro pharmacogenomic response analyses based on the publicly available GDSC datasets [51-53]. The parameter IC50 was also adopted in previous studies [54-56]. The criteria for KEAP1/NFE2L2-mutant specific drugs were as follows: LN(IC50)C1 or C2 < LN(IC50)C2 or C1, P < 0.05; LN(IC50)C1 or C2 < LN(IC50)WT, P < 0.05; and LN(IC50)C2 or C1 ≈ LN(IC50)WT, P > 0.05. However, only one C2-specific drug could be identified using the revised criteria: LN(IC50)C2 < LN(IC50)C1, P < 0.1; LN(IC50)C2 < LN(IC50)WT, P < 0.1; and LN(IC50)C1 ≈ LN(IC50)WT, P > 0.1.
Statistical analysis
All statistical analyses in this study were conducted using R version 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria) and IBM SPSS Statistics 22.0 (IBM, Inc., NY, USA). Comparisons of immunological features and drug sensitivities were performed using the Kruskal-Wallis H test and Mann-Whitney U test. Baseline characteristics and co-mutations were studied by the chi-square test. Survival curves were estimated and compared following the Kaplan-Meier method and the log-rank test. A two-tailed P-value less than 0.05 was considered statistically significant.
Results
Identification of subtypes of KEAP1/NFE2L2-mutant lung adenocarcinoma
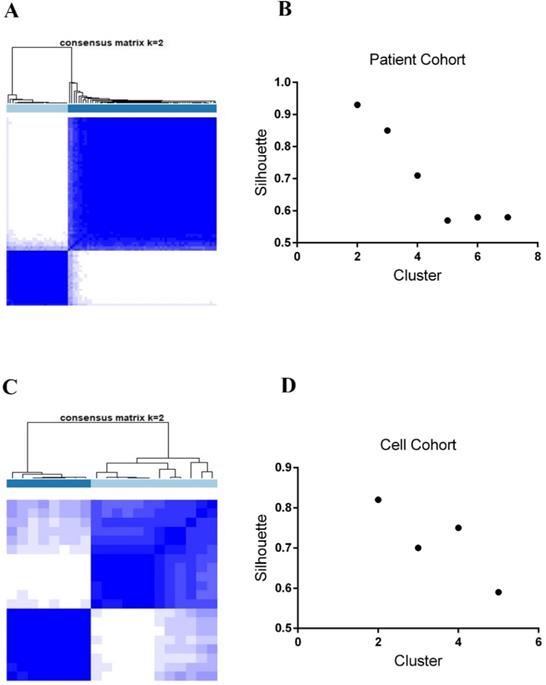
As previously stated in the Methods section, we integrated five data subtypes and clustered 89 KEAP1/NFE2L2-mutant lung adenocarcinoma patients into two subgroups (P1 and P2 groups, Figure 1A). Similarly, two subtypes were identified in 20 lung adenocarcinoma cell lines harboring KEAP1/NFE2L2-mutations (C1 and C2 groups, Figure 1C). Clustering with two classes in both patients and cell line samples showed the highest silhouette values (silhouette = 0.93 and 0.83, Figure 1B and 1D).
The SNF fused five types of datasets and consensus clustering identifies subsets of KEAP1/NFE2L2-mutant lung adenocarcinoma in patients and cell lines. A. Two subsets of KEAP1/NFE2L2-mutant patients were identified. B. Silhouette values of patient clustering with the k = 2 to 7. C. Two subsets of KEAP1/NFE2L2-mutant cell lines were identified. D. Silhouette values of cell line clustering with the k = 2 to 5.
Clinicopathological differences of the KEAP1/NFE2L2-mutant subtypes
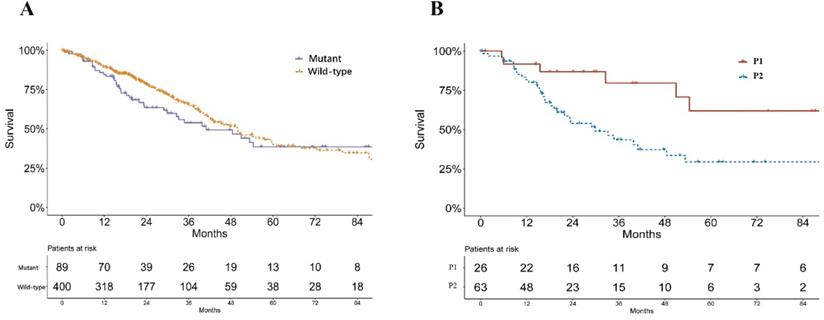
A significant difference was found in the smoking status of patients among P1, P2 and wild-type groups (P = 0.033, Table 1). The P2 group consisted of the highest proportions of current smokers and reformed smokers for ≤15 years, while P1 groups consisted of more reformed smokers ≥ 15 years (Table 1). No significant difference of pathological stage was found among patients of P1, P2 and KEAP1/NFE2L2 wild-type lung adenocarcinoma (P = 0.233, Table 1). Mutant samples contained a significantly higher proportion of female patients (P = 0.003, Table 1). Survival analysis showed no significant difference in overall survival between subgroups of KEAP/NFE2L2-mutant and wild-type lung adenocarcinoma (P = 0.212, Figure 2A). However, the P2-mutant subgroup was associated with a significantly worse survival than the P1 subgroup (P = 0.020, Figure 2B).
Baseline characteristics of wild type and KEAP1/NFE2L2-mutant subgroups of patients with lung adenocarcinoma in TCGA
| Wild type | Mutant P1 group | Mutant P2 group | P-value | |
|---|---|---|---|---|
| Age | 65.3 ± 9.9 | 67.6 ± 7.1 | 64.3 ± 11.2 | |
| Gender | 0.003 | |||
| Female | 234 (57.2) | 12 (46.2) | 22 (34.9) | |
| Male | 175 (42.8) | 14 (53.8) | 41 (65.1) | |
| Pathological Stage | 0.233* | |||
| Stage I | 226 (55.3) | 14 (53.8) | 30 (47.6) | |
| Stage II | 99 (24.2) | 5 (19.2) | 17 (27.0) | |
| Stage III | 67 (16.4) | 5 (19.2) | 9 (14.3) | |
| Stage IV | 15 (3.7) | 2 (7.7) | 7 (11.1) | |
| Unknown | 2 (0.5) | 0 (0) | 0 (0) | |
| Smoking Status | 0.033* | |||
| Non-smoker | 66 (16.1) | 1 (3.8) | 5 (7.9) | |
| Current smoker | 96 (23.5) | 5 (19.2) | 16 (25.4) | |
| Reformed smoker (> 15 years) | 105 (25.7) | 12 (46.2) | 11 (17.5) | |
| Reformed smoker (≤ 15 years) | 127 (31.1) | 8 (30.8) | 28 (44.4) | |
| Unknown | 15 (3.7) | 0 (0) | 3 (4.8) |
* Samples with unknown information were removed when comparisons were conducted among groups.
Survival curves of lung adenocarcinoma patients in TCGA. A. Survival curves of KEAP1/NFE2L2-mutant and wild-type patients (P = 0.212). B. Survival curves of KEAP1/NFE2L2-mutant patient subgroups (P1 and P2) (P = 0.020).
Basic biological features of KEAP1/NFE2L2-mutant subtypes
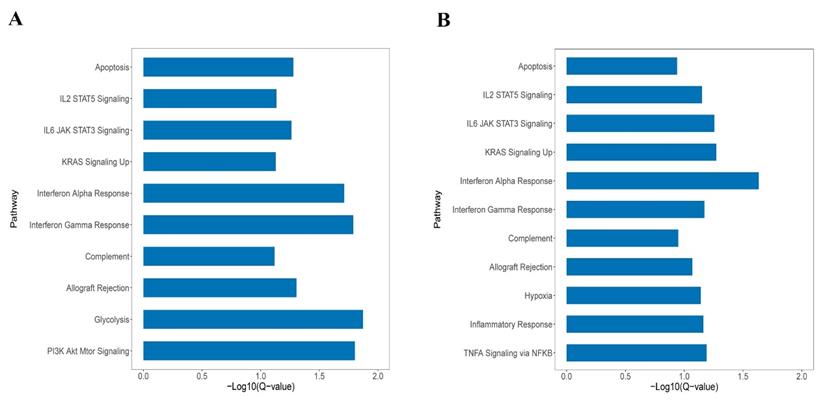
GSEA was performed in KEAP1/NFE2L2-mutant subtypes in both patients and cell line cohorts. As shown in Figure 3A and 3B, the P2 and C2 subtypes were both enriched in the same pathways, such as KRAS signaling, IL2/STAT5 signaling, apoptosis, and interferon alpha and gamma response. GSEA revealed similarities between the P2 and C2 subtypes, validating the integration and clustering process to some degree.
A. The enriched pathways in Hallmark of KEAP1/NFE2L2-mutant P2 patient subgroup. B. The enriched pathways in Hallmark of KEAP1/NFE2L2-mutant C2 cell line subgroup.
Moreover, both P2 and C2 subtypes were associated with regulations of immune-related pathways, such as activations of T cells and macrophages (Supplement Figure 1A and 1B). The results revealed that the P2 and C2 subgroups displayed active immune pathways compared with P1 and C1 subgroups, respectively.
The P2 subgroup was found associated with higher proportions of TP53 (P < 0.001), PCLO (P = 0.011), NF1 (P = 0.029) and PTPRT (P = 0.040) mutations, while the P1 subgroup may have more patients with STK11 (P = 0.008) mutation (Supplement Table 1). However, we did not validate the mutational associations in lung adenocarcinoma cell lines due to the small sample size.
Immunological features of the KEAP1/NFE2L2-mutant subtypes
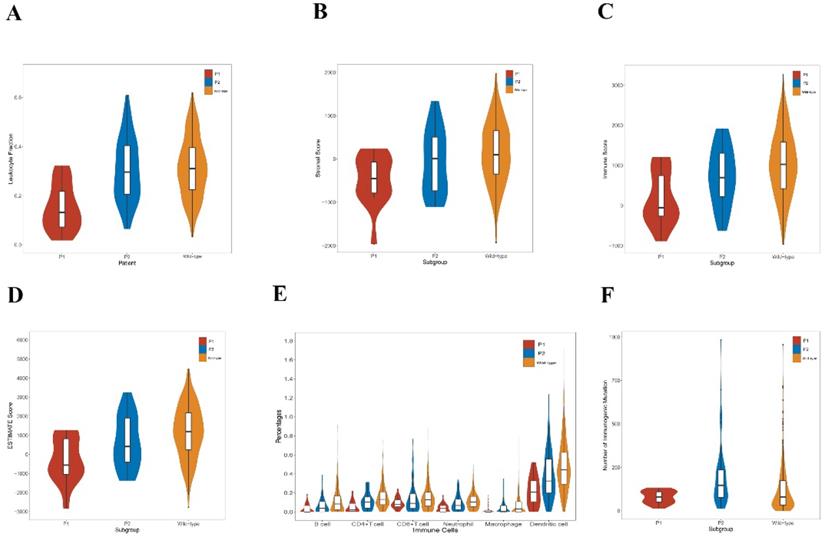
The tumor-infiltrating lymphocyte fractions were compared according to Saltz et al stratified by the mutation status [46]. Compared with the wild-type samples, lung adenocarcinoma harboring KEAP1/NFE2L2 had a significantly lower lymphocyte fractions (P = 0.001, Figure 4A). Subgroup analyses revealed that the P2 group exhibited significantly higher lymphocyte fractions compared with the P1 group (P < 0.001, Figure 4A). We also observed that significant differences of ESTIMATE scores exist among three groups, in which P1 was related to the lowest score (Figure 4B, 4C and 4D). Based on TIMER, a significant decrease was found in the infiltrating levels of CD4+ T cells (P < 0.001), CD8+ T cells (P = 0.011), B cells (P < 0.001), neutrophils (P < 0.001), dendritic cells (P < 0.001), and macrophages (P = 0.008) in the mutant subgroup (Figure 3B). Moreover, the P1 subgroup was associated with reduced infiltrations of B cells (P = 0.017), CD4+ T cells (P = 0.001), neutrophils (P = 0.002) and dendritic cells (P = 0.006) (Figure 4E). Furthermore, the P2 subtype was associated with higher number of immunogenic mutations than the P1 group (Figure 4F).
Immunological features of lung adenocarcinoma patients in TCGA. A. Comparison of leukocyte fraction stratified by KEAP1/NFE2L2-mutant (P1 and P2) and wild-type patient subgroups (mutant group vs. wild-type group, P = 0.001; P1 group vs. P2 group, P < 0.001). B. Comparison of stromal score calculated by ESTIMATE algorithm stratified by KEAP1/NFE2L2-mutant (P1 and P2) and wild-type patient subgroups (P1 vs. P2 vs. wild-type group, P = 0.005). C. Comparison of immune score calculated by ESTIMATE algorithm stratified by KEAP1/NFE2L2-mutant (P1 and P2) and wild-type patient subgroups (P1 vs. P2 vs. mutant group, P = 0.001). D. Comparison of ESTIMATE score calculated by ESTIMATE algorithm stratified by KEAP1/NFE2L2-mutant. (P1 and P2) and wild-type patient subgroups (P1 vs. P2 vs. wild-type group, P = 0.001). E. Comparison of tumor-infiltrating immune cells stratified by KEAP1/NFE2L2-mutant (P1 and P2) and wild-type patient subgroups based on TIMER database. [mutant group vs. wild-type group: CD4+ T cells (P < 0.001), CD8+ T cells (P = 0.011), B cells (P < 0.001), neutrophils (P < 0.001), dendritic cells (P < 0.001), and macrophages (P = 0.008); P1 group vs. P2 group: B cells (P = 0.017), CD4+ T cells (P = 0.001), CD8+ T cells (P = 0.375), neutrophils (P = 0.002), macrophages (P = 0.113), and dendritic cells (P = 0.006)]. F. Comparison of the number of immunogenic mutations per sample stratified by KEAP1/NFE2L2-mutant (P1 and P2) and wild-type patient subgroups (P1 vs. P2 vs wild-type group, P < 0.001).
Smoking-related genomic features of the KEAP1/NFE2L2-mutant subtypes of lung adenocarcinoma
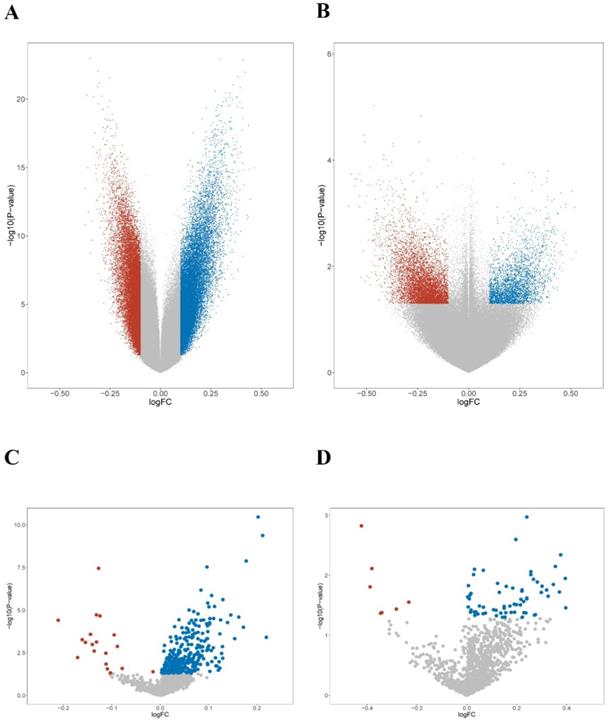
First, the methylation levels were compared across mutant subgroups. 84,700 and 64,204 differentially hypermethylated probes were found in the P1 and P2 groups, respectively (Figure 5A). Meanwhile, 8,981 hypermethylated probes were found in the C1 group, while 5,933 hypermethylated probes were found in the C2 group (Figure 5B). Next, unique smoking-related probes were extracted according to Vaz et al [49]. Both P2 and C2 groups displayed a similar trend of hypermethylation compared with the P1 and C1 groups (Figure 5C-D). The results suggested that smoking-related epigenomic alterations might play essential roles in KEAP1/NFE2L2-mutant subgroups. The epigenomic similarities confirmed a potential resemblance between patient and cell line mutant subsets.
Epigenomic features of KEAP1/NFE2L2-mutant subgroups of lung adenocarcinoma patients and cell lines. A. Volcano plot of the global DNA methylation difference between patient mutant subgroups (P1 and P2). B. Volcano plot of the global DNA methylation difference between cell line mutant subgroups (C1 and C2). C. Volcano plot of the smoking-related methylation signatures between patient mutant subgroups (P1 and P2). D. Volcano plot of the smoking-related methylation signatures between cell line mutant subgroups (C1 and C2).
Second, we assessed the somatic mutational patterns of all lung adenocarcinoma patients and obtained four distinctive signatures (Supplement Figure 2A). Among them, signature 2 subgroup (W2) was like Signature 4 and 29 of the thirty known somatic mutational signatures in the COSMIC database, which were closely associated with smoking and tobacco chewing (coefficient of cosine similarity = 0.805 and 0.740). Then, we compared the normalized activities of the identified W2 mutational signature between KEAP1/NFE2L2-mutant subgroups. We found that the P2 subset had significantly higher activities of W2 signature than the P1 subset (Supplement Figure 2B, P = 0.004), which further indicated possible different roles of smoking in the mutant subgroups.
Screening for compounds with potential sensitivity to the KEAP1/NFE2L2-mutant subtypes
After characterizing the clinical and biological features of the mutant subtypes, possible cancer-associated drugs which were sensitive to each subtype were explored. More than 400 drugs and compounds were tested on KEAP1/NFE2L2-mutant and wild-type lung adenocarcinoma cell lines in GDSC. This part aimed to target cancer-associated drugs and compounds with potential specific sensitivity to the C1 or C2 subset. 38 drugs, which were potentially sensitive to the C2 mutant subtype, were discovered (Supplement Table 2). Although the criteria were adjusted, only one C1-specific compound was identified (Supplement Table 2).
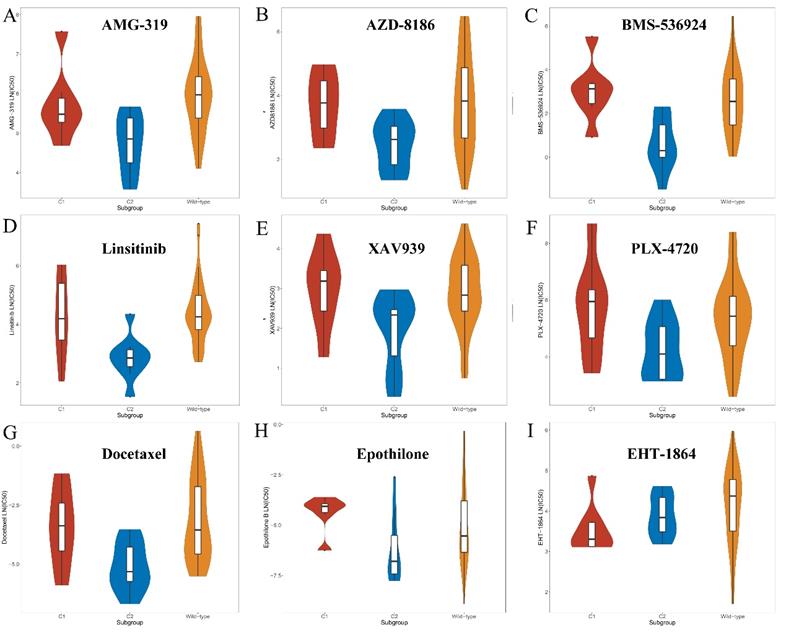
C2-specific drugs were found to be mainly composed of the following types. First, inhibitors of the PI3K/Akt signaling pathways, such as afuresertib, AZD8186 and AMG-319 might be sensitive to the C2 subgroup compared with the C1 and wild-type groups (Figure 6 and Supplement Table 2). Second, inhibitors of IGF1R signaling, such as BMS-536924, linsitinib and NVP-ADW742, showed better efficacy in the C2 subset (Figure 6 and Supplement Table 2). Moreover, drugs that target Wnt and MAPK/Erk signaling pathways were more toxic to the C2 subgroup (Figure 6 and Supplement Table 2). In addition, chemotherapy drugs, such as docetaxel, epothilone B and vinorelbine were found to preferentially kill tumor cells of the C2 subgroup (Figure 6 and Supplement Table 2). Nevertheless, only one compound (EHT-1864) was found that might be sensitive to the C1 subset (Figure 6 and Supplement Table 2). The selected compound, EHT-1864, is an inhibitor of Rac1, Rac2 and Rac3 and mediated the reorganization of actin cytoskeleton.
Screened drugs with selective sensitivity toward the KEAP1/NFE2L2-mutant subtypes. A-H. Drugs that selectively killed tumor cells of the C2 subset. I. Drug that selectively killed tumor cells of the C1 subset.
Discussion
The KEAP1/NFE2L2 mutations were observed in many common malignant tumors, including lung adenocarcinoma [11,12,19,21], which might define a molecular subset of rapidly progressing tumor [57]. In this study, the multiplatform data from TCGA were adopted to identify subsets of lung adenocarcinoma with KEAP1/NFE2L2 mutations. Clinicopathological and bioinformatics analyses, such as immune microenvironment and methylation level, were performed to further explore the intrinsic heterogeneities of KEAP1/NEFE2L2-mutant disease. Moreover, cell line samples were used for drug sensitivity screening based on public datasets. In addition, CUL3 mutation was not included as the genomic signature in this study. CUL3 belonged to the ubiquitin-proteasome system, which was involved in many oncogenic processes, and could not be considered as a specific KEAP1/NFE2L2 pathway component [58].
Variations in the KEAP1-NFE2L2 pathway were detected in more than 20% patients with lung cancer, which represented one of the major molecular subtypes [11,12]. Goeman et al revealed that KEAP1/NFE2L2 mutations represented a negative factor of survival, which defined a rapidly progressing molecular subtype [57,59]. The mutant type showed heterogeneities, and one subset was associated with significantly worse survival. Cai et al performed a similar study and divided KEAP1/NFE2L2-mutant lung adenocarcinoma into three subsets based on gene profiling. The present study integrated multi-omics datasets, such as somatic mutation, methylation, and miRNA, to cluster into two subsets. P2/C2 subset displayed active immune pathways compared with the P1/C1 subgroups. The controversies of the prognosis regarding patients with KEAP1/NFE2L2 mutations treated with immunotherapy may be associated with the distinct immune microenvironment of P1 and P2 subgroups [25]. Ricciuti B et al revealed that lung adenocarcinoma harboring concurrent KRAS/STK11 and KRAS/KEAP1 mutations display distinct immune profiles [30]. In this work, we also observed different patterns of concurrent mutations between mutant subsets. Clinical features, somatic mutation signatures and methylation levels showed potential associations with patients' smoking history. Previous studies demonstrated that smoking led to significant nuclear translocation of NFE2L2, which might be potentially fatal in smoking-related lung tumorigenesis [60,61]. These findings might also be potential evidence of distinct KEAP1/NFE2L2 subtypes.
Furthermore, drug sensitivities of cell lines from public datasets were analyzed and several subgroup-specific drugs were discovered in our study. Best et al observed that synergy between KEAP1/NFE2L2 and PI3K pathways promoted lung cancer progression with the altered immune milieu, which supported the compound screening results of inhibitors of PI3K/Akt pathways in this study [13]. Several studies revealed possible associations between the two pathways [62,63]. The pathway analyses of this study also revealed that PI3K/Akt pathway was enriched in the P2 subgroup. Vartanian et al identified alternative pathways critical for NFE2L2-dependent growth in KEAP1-mutant cell lines, including IGF1R [64]. The findings in this study suggested that inhibitors of IGF1R signaling were effective in the C2 subtype. Only one alternative compound existed, which inhibited Rac signaling to mediate the actin cytoskeleton. Wu et al demonstrated that KEAP1 stabilized F-actin cytoskeleton structures and inhibited focal adhesion, thereby restraining migrations and invasions of lung cancers [65]. KEAP1/NFE2L2/CUL3 represented a mechanism of resistance to tyrosine kinase inhibitor in patients with EGFR-mutant non-small cell lung cancer [32]. Most identified compounds in our study were sensitive to the C2 subgroup which represented a subset with a worse prognosis. However, only one compound showed better efficacy to the C1 group with a revised statistical threshold, revealing difficulties in selecting appropriate drugs. However, the intrinsic differences in immune infiltrations suggested distinct immunotherapy strategies, especially developing drugs for the C2/P2 group. Also, concurrent alterations, like STK11 and TP53, could also be potential targets in KEAP1/NFE2L2-mutant diseases.
There were also limitations that should be mentioned in this study. First, it had a small sample size of mutant cell lines and patients. The study explored intrinsic heterogeneities of KEAP1/NFE2L2-mutant lung adenocarcinoma. However, further studies are required to better characterize and precisely differentiate each mutant subtype. Although LN(IC50) was adopted from GDSC to measure compound sensitivities, more experiments should be conducted to test drug efficacy.
Conclusion
Two subtypes of KEAP1/NFE2L2-mutant lung adenocarcinoma were identified based on both patient and cell line samples, and genomic and clinicopathological features of KEAP1/NFE2L2 mutations were characterized. The intrinsic heterogeneities of KEAP1/NFE2L2 mutations was found to be associated with immune microenvironment and smoking-related genomic aberrations.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to thank International Science Editing Co. for language editing service of this manuscript.
A previous version of this study has been placed on an online preprint archive. (https://www.researchsquare.com/article/rs-182066/v1).
Funding
This work was supported by Shanghai Sailing Program (21YF1438600).
This work was supported by the Shanghai Medical Innovation Research Project (Grant No. 20Y11908200).
This work was supported by the National Natural Science Foundation of China (81970092).
Data Availability Statement
All data could be downloaded from public databases (TCGA, GEO, CCLE, GDSC and XENA databases) and previous literatures in the reference.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7-34
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
3. Lu T, Yang X, Huang Y, Zhao M, Li M, Ma K. et al. Trends in the incidence, treatment, and survival of patients with lung cancer in the last four decades. Cancer Manag Res. 2019;11:943-53
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
5. Tubio-Perez RA, Torres-Duran M, Perez-Rios M, Fernandez-Villar A, Ruano-Ravina A. Lung emphysema and lung cancer: what do we know about it? Ann Transl Med. 2020;8:1471
6. Wu G, Zhao Z, Yan Y, Zhou Y, Wei J, Chen X. et al. CPS1 expression and its prognostic significance in lung adenocarcinoma. Ann Transl Med. 2020;8:341
7. Ali J, Liu W, Duan W, Liu C, Song J, Ali S. et al. METTL7B (methyltransferase-like 7B) identification as a novel biomarker for lung adenocarcinoma. Ann Transl Med. 2020;8:1130
8. Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327-43
9. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW. et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-39
10. Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol. 2013;31:1105-11
11. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489: 519-25.
12. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543-50.
13. Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D. et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018;27:935-43
14. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE. et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362-8
15. Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA. et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743-53
16. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411-21
17. Tian Y, Liu Q, He X, Yuan X, Chen Y, Chu Q. et al. Emerging roles of Nrf2 signal in non-small cell lung cancer. J Hematol Oncol. 2016;9:14
18. Best SA, Sutherland KD. "Keaping" a lid on lung cancer: the Keap1-Nrf2 pathway. Cell Cycle. 2018;17:1696-707
19. Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM. et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097-102
20. Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO. et al. Dysfunctional KEAP1-NRF2 Interaction in Non-Small-Cell Lung Cancer. Plos Med. 2006;3:e420
21. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694-8
22. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106-9
23. Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158-68
24. Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H. et al. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016;76:3088-96
25. Hellyer JA, Padda SK, Diehn M, Wakelee HA. Clinical Implications of KEAP1-NFE2L2 Mutations in NSCLC. J Thorac Oncol. 2021;16:395-403
26. Xu X, Yang Y, Liu X, Cao N, Zhang P, Zhao S. et al. NFE2L2/KEAP1 Mutations Correlate with Higher Tumor Mutational Burden Value/PD-L1 Expression and Potentiate Improved Clinical Outcome with Immunotherapy. Oncologist. 2020;25:e955-63
27. Zhang C, Zhang C, Li J, Wang H. KEAP1-NFE2L2-Mutant NSCLC and Immune Checkpoint Inhibitors: A Large Database Analysis. J Thorac Oncol. 2020;15:e85-6
28. Papillon-Cavanagh S, Doshi P, Dobrin R, Szustakowski J, Walsh AM. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open. 2020;5:e706
29. Marinelli D, Mazzotta M, Scalera S, Terrenato I, Sperati F, D'Ambrosio L. et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann Oncol. 2020;31:1746-54
30. Ricciuti B, Arbour KC, Lin JJ, Vajdi A, Vokes N, Hong L. et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J Thorac Oncol. 2021;2:S1556-864
31. Scalera S, Mazzotta M, Corleone G, Sperati F, Terrenato I, Krasniqi E. et al. KEAP1 and TP53 Frame Genomic, Evolutionary, and Immunologic Subtypes of Lung Adenocarcinoma With Different Sensitivity to Immunotherapy. J Thorac Oncol. 2021;16:2065-77
32. Hellyer JA, Stehr H, Das M, Padda SK, Ramchandran K, Neal JW. et al. Impact of KEAP1/NFE2L2/CUL3 mutations on duration of response to EGFR tyrosine kinase inhibitors in EGFR mutated non-small cell lung cancer. Lung Cancer. 2019;134:42-5
33. Jeong Y, Hellyer JA, Stehr H, Hoang NT, Niu X, Das M. et al. Role of KEAP1/NFE2L2 Mutations in the Chemotherapeutic Response of Patients with Non-Small Cell Lung Cancer. Clin Cancer Res. 2020;26:274-81
34. Frank R, Scheffler M, Merkelbach-Bruse S, Ihle MA, Kron A, Rauer M. et al. Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clin Cancer Res. 2018;24:3087-96
35. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C. et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018;6:271-81
36. Bairoch A. The Cellosaurus, a Cell-Line Knowledge Resource. J Biomol Tech. 2018;29:25-38
37. Robin T, Capes-Davis A, Bairoch A. CLASTR: The Cellosaurus STR similarity search tool - A precious help for cell line authentication. Int J Cancer. 2019:1299-306
38. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW. et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics-Us. 2013;8:203-9
39. Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M. et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11:333-7
40. Wang T, Lee C, Lee T, Huang H, Hsu JB, Chang T. Biomarker Identification through Multiomics Data Analysis of Prostate Cancer Prognostication Using a Deep Learning Model and Similarity Network Fusion. Cancers. 2021;13:2528
41. Jacobs GR, Voineskos AN, Hawco C, Stefanik L, Forde NJ, Dickie EW. et al. Integration of brain and behavior measures for identification of data-driven groups cutting across children with ASD, ADHD, or OCD. Neuropsychopharmacol. 2021;46:643-53
42. Zhao L, Zhang J, Liu Z, Wang Y, Xuan S, Zhao P. Comprehensive Characterization of Alternative mRNA Splicing Events in Glioblastoma: Implications for Prognosis, Molecular Subtypes, and Immune Microenvironment Remodeling. Front Oncol. 2020;10:555632
43. Narayana JK, Mac AM, Ali N, Tsaneva-Atanasova K, Chotirmall SH. Similarity network fusion for the integration of multi-omics and microbiomes in respiratory disease. Eur Respir J. 2021 58
44. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572-3
45. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. Bmc Bioinformatics. 2013;14:7
46. Saltz J, Gupta R, Hou L, Kurc T, Singh P, Nguyen V. et al. Spatial Organization and Molecular Correlation of Tumor-Infiltrating Lymphocytes Using Deep Learning on Pathology Images. Cell Rep. 2018;23:181-93
47. Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612
48. Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J. et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174
49. Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O'Hagan HM. et al. Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations. Cancer Cell. 2017;32:360-76
50. Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ. et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 2016;48:600-6
51. Rydzewski NR, Peterson E, Lang JM, Yu M, Laura CS, Sjostrom M. et al. Predicting cancer drug TARGETS - TreAtment Response Generalized Elastic-neT Signatures. Npj Genom Med. 2021;6:76
52. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S. et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955-61
53. Zhang M, Wang Y, Jiang L, Song X, Zheng A, Gao H. et al. LncRNA CBR3-AS1 regulates of breast cancer drug sensitivity as a competing endogenous RNA through the JNK1/MEK4-mediated MAPK signal pathway. J Exp Clin Cancer Res. 2021;40:41
54. Wang H, Wu Y, Chen S, Hou M, Yang Y, Xie M. Construction and Validation of a Ferroptosis-Related Prognostic Model for Endometrial Cancer. Front Genet. 2021;12:729046
55. Wang K, Feng X, Zheng L, Chai Z, Yu J, You X. et al. TRPV4 is a Prognostic Biomarker that Correlates with the Immunosuppressive Microenvironment and Chemoresistance of Anti-Cancer Drugs. Front Mol Biosci. 2021;8:690500
56. Malik V, Kalakoti Y, Sundar D. Deep learning assisted multi-omics integration for survival and drug-response prediction in breast cancer. Bmc Genomics. 2021;22:214
57. Goeman F, De Nicola F, Scalera S, Sperati F, Gallo E, Ciuffreda L. et al. Mutations in the KEAP1-NFE2L2 Pathway Define a Molecular Subset of Rapidly Progressing Lung Adenocarcinoma. J Thorac Oncol. 2019;14:1924-34
58. Scott DC, Rhee DY, Duda DM, Kelsall IR, Olszewski JL, Paulo JA. et al. Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell. 2016;166:1198-214
59. Takahashi T, Sonobe M, Menju T, Nakayama E, Mino N, Iwakiri S. et al. Mutations in Keap1 are a potential prognostic factor in resected non-small cell lung cancer. J Surg Oncol. 2010;101:500-6
60. Chang WH, Thai P, Xu J, Yang DC, Wu R, Chen CH. Cigarette Smoke Regulates the Competitive Interactions between NRF2 and BACH1 for Heme Oxygenase-1 Induction. Int J Mol Sci. 2017;18:2386
61. Muller T, Hengstermann A. Nrf2: friend and foe in preventing cigarette smoking-dependent lung disease. Chem Res Toxicol. 2012;25:1805-24
62. Kaufman JM, Amann JM, Park K, Arasada RR, Li H, Shyr Y. et al. LKB1 Loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. J Thorac Oncol. 2014;9:794-804
63. Dai B, Yoo SY, Bartholomeusz G, Graham RA, Majidi M, Yan S. et al. KEAP1-dependent synthetic lethality induced by AKT and TXNRD1 inhibitors in lung cancer. Cancer Res. 2013;73:5532-43
64. Vartanian S, Lee J, Klijn C, Gnad F, Bagniewska M, Schaefer G. et al. ERBB3 and IGF1R Signaling Are Required for Nrf2-Dependent Growth in KEAP1-Mutant Lung Cancer. Cancer Res. 2019;79:4828-39
65. Wu B, Yang S, Sun H, Sun T, Ji F, Wang Y. et al. Keap1 Inhibits Metastatic Properties of NSCLC Cells by Stabilizing Architectures of F-Actin and Focal Adhesions. Mol Cancer Res. 2018;16:508-16
Author contact
![]() Corresponding authors: Liang Wu, Email: wuliang198209com. Department of Thoracic Surgery, Shanghai General Hospital, Shanghai, China. Chen Zhan, Email: czhan10edu.cn. Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai, China.
Corresponding authors: Liang Wu, Email: wuliang198209com. Department of Thoracic Surgery, Shanghai General Hospital, Shanghai, China. Chen Zhan, Email: czhan10edu.cn. Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai, China.