Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(5):1640-1651. doi:10.7150/jca.69210 This issue Cite
Research Paper
Copy number amplification-activated long non-coding RNA LINC00662 epigenetically inhibits BIK by interacting with EZH2 to regulate tumorigenesis in non-small cell lung cancer
Chunluan Yuan1#, Yue Ding2#, Yan Zhuang3#, Chongguo Zhang4, Liang Han5 ![]() , Wei Li6,7
, Wei Li6,7 ![]() , Renhua Guo6
, Renhua Guo6 ![]() , Erbao Zhang2,8
, Erbao Zhang2,8 ![]()
1. Department of Oncology, Lianyungang Clinical College of Nanjing Medical University, the First People's Hospital of Lianyungang, PR China
2. Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China
3. Department of Oncology, The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210000, PR China
4. Department of Oncology, The second Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210000, PR China
5. Department of Oncology, Xuzhou Central Hospital, Xuzhou, Jiangsu, PR China
6. Department of Oncology, The first Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, 210000, PR China
7. Department of Oncology, Sir Run Run Hospital, Nanjing Medical University, Nanjing, Jiangsu, 210000, PR China
8. Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing 211166, China
#Chunluan Yuan, Yue Ding and Yan Zhuang contributed equally to this work and should be regarded as joint first authors.
Received 2021-11-18; Accepted 2022-2-25; Published 2022-3-14
Abstract

Recently, studies have shown that lncRNAs play important roles in regulation of cancer cells proliferation, apoptosis and metastasis. Here, through systematic bioinformatics analysis and screening, we identified a long noncoding RNA LINC00662 with high copy number amplification in NSCLC. High expression of LINC00662 predicted a poorer survival. The exact sequence full-length of LINC00662 was determined by rapid amplification of cDNA ends (RACE). We also found that LINC00662 could regulate lung cancer cell proliferation both in vitro and in vivo. Mechanically, we obtained global expression profile that respond to LINC00662 knockdown through RNA-Seq analysis. And we found that LINC00662 could bind to EZH2 and recruit EZH2 to the promoter regions of BIK, regulating the level of H3K27me3 in the BIK promoter, thus epigenetically repressing BIK expression. Our results shown that lncRNA LINC00662, driven by copy number amplification, promotes tumorigenesis by EZH2/BIK cell axis, indicating that it was a potential molecular target of NSCLC.
Keywords: Gene amplification, LINC00662, tumorigenesis, NSCLC
Introduction
Lung cancer is the main cause of the high incidence of cancer and high mortality worldwide [1, 2]. NSCLC accounts for about 80-85% of all lung cancers [3, 4]. Although a combination of chemotherapy, radiation treatment, surgical operation, and molecularly targeted therapy approach can be adopted in lung cancer treatment [5, 6]. The survival rate of NSCLC patients is remaining poor [7, 8]. So, it is necessary to further understand the molecular mechanism of NSCLC to improve the problems in the diagnosis and treatment of NSCLC.
Long noncoding RNA (>200 nt) has gained extensive attention because of the deepening research for noncoding RNAs (ncRNAs) [9]. Although lncRNAs were previously considered to be "noise" generated in transcription, now lncRNAs considered to be regulators that play a key role in biological processes including cellular development, differentiation, etc. [9, 10]. Recently, much research has indicated that the lncRNAs aberrant expressions can affect many types of tumorigenesis including lung cancer [11-14]. For instance, LncRNA HOTAIR could lead to the transcriptional activation of c-MYC target genes in breast cancer cells, thus promoting the occurrence of breast cancer [15]. Long noncoding RNA NEAT1 promoted proliferation and invasion of NSCLC cells via targeting miR-181a-5p [16]. Our previous work also found that lncRNAs could play key roles in tumorigenesis[14, 17, 18].
LINC00662 was a lncRNA that was located in the chromosome 19q11. LINC00662 have revealed its carcinogenesis in oral squamous cell carcinoma, gastric cancer and prostate cancer etc. [19-22]. For instance, LINC00662 could increase the abilities of OSCC cell proliferation and invasion by regulating the Wnt/beta-catenin pathway [19]. LINC00662 promoted gastric cancer cell proliferation by LINC00662-miR-497-5p-YAP1 axis [20]. Published research revealed that the dysregulation of LINC00662 could involve in tumorigenesis. However, the cause for LINC00662 activation and its biological mechanism in lung cancer tumorigenesis, especially, the global genes mediated by LINC00662 has not been established. In addition, the exact sequence of LINC00662 has not been determined. Therefore, we pay attention to its specific role in tumorigenesis of NSCLC.
In the research, we discovered LINC00662 was significantly elevated in NSCLC samples and evaluated poor survival. The activation of LINC00662 is driven by the copy number amplification. Then we first identified the full-length sequence of LINC00662. And we found that knockdown LINC00662 could decrease the ability of cell growth and migration both in vitro and in vivo. Mechanically, we obtained global expression profile that respond to LINC00662 knockdown through RNA-Seq analysis. And we found that LINC00662 could interact with EZH2 and recruit EZH2 to the promoter regions of BIK, regulating the level of H3K27me3 in the BIK promoter, thus epigenetically repressing BIK expression and promoting tumorigenesis of NSCLC. These results indicated that LINC00662 may play important role in the tumorigenesis of NSCLC and could supply a certain theoretical basis of the diagnosis and treatment for NSCLC.
Materials and Methods
Lung cancer lncRNA expression profiling data retrieval and analysis
Microarray gene expression data was retrieved from the GEO dataset. The data sets from GSE51852 and GSE43767 were added to our study. We retrieved the original files from the GEO database. Next, the normalization and z-score transformation were performed to the raw data. Then we get the differentially expressed lncRNA.
The copy number variations and expression analysis of LINC00662
NSCLC copy number variation data obtained from whole genome sequencing and transcriptome sequencing of The Cancer Genome Atlas (TCGA) database. We obtained the expression data of samples from the UCSC Xena website (https://xenabrowser.net/datapages/) and downloaded the copy number data of NSCLC samples from https://gdac.broadinstitute.org/. A threshold of 1 in values was considered an amplification and calculated the ratio of copy amplification for LINC00662.
Cell Cultures
Two human NSCLC cell lines were purchased from the Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences (Shanghai, China). A549 cells and SPCA1 cells were cultured in RPMI 1640 and DMEM (Gibco-BRL) containing 10% fetal bovine serum (FBS), respectively, supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA, USA). Both A549 and SPCA1 cells were cultured in an environment with a temperature of 37°C and 5% CO2.
RNA isolation and qRT-PCR Analyses
The cultured cells were lysed with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) to extract total RNA. Then the RNA was reverse transcribed with a reverse transcription kit (Takara, China) to obtain cDNA. SYBR Premix Ex Taq (TaKaRa, China) was used to perform real-time PCR analyses, following the manufacturer's instructions. The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for calibration. Specific primers are in Supplementary Table S2.
RACE (rapid amplification of cDNA ends)
Total RNA was extracted using RNA Purification Kit (Invitrogen) followed the manufacturer's instructions. 5' RACE and 3' RACE was performed using SMART RACE cDNA Amplification Kit (Cat. 634858, Clontech, Palo Alto, CA, USA) according to the manufacturer's instructions.
Cell transfection
Followed the instructions and used Lipofectamine 2000 (Invitrogen) reagent to transfect LUAD cells together with siRNA. Negative control was siRNA (si-NC) (Invitrogen, CA, USA). All sequences of siRNAs are listed in Supplementary Table S2.
Flow Cytometric Analysis
After the medium supernatant was collected, the transfected cells were digested with trypsin, and then re-suspended and collected together with the medium supernatant. Annexin V FITC Annexin V Apoptosis assay kit (BD Biosciences) was subsequently used for double staining of luciferin isothiocyanate-Annexin V and propidium iodide. Results were analyzed using FACScan (BD Biosciences) with Cell Quest software. The cells were divided into living cells, dead cells, early apoptotic cells and apoptotic cells. Then calculated and compared the proportion of apoptotic cells in each experimental group. All experiments were performed three parallel.
EdU Analysis
Cultured the cells in a 24-well plate, the number of cells per well is 5×104, after transfection 48 hours, reagent A from EdU assay kit (Ribobio, Guangzhou, China) prepared in a certain proportion with culture medium and then added to 24-well plate for incubation 2 hours at a temperature of 37°C and a CO2 concentration of 5%. The cells were treated with 4% paraformaldehyde followed by 0.5% Triton X-100, after which the cells were stained with anti-EdU working solution, and the nuclei were labeled with DAPI. After taking pictures through a fluorescence microscope, count and calculate the percentage of EDU-positive cells. Three fields of view were randomly selected for each group.
Tumor formation assay in mouse model
Athymic BALB/ C nude mice aged 5 weeks were raised under certain pathogen-free conditions. After NC or si-LINC00662 transfected A549 cells, the suspension cells were subcutaneously injected to either side of the rear flank of the nude mice. Tumor volume and weight of mice in control group and si-LINC00662 group were measured. Tumor volume was measured. A few days after the injection, the mice were killed, and the tumors were taken out and the tumors were weighed. Our proposal was approved by the Ethics Committee of Animal Experiments of Nanjing Medical University.
Transcriptome sequencing
Total RNA was extracted from LINC00662 knockdown A549 cells and control cells. NanoDrop 2000 (Thermo Scientific, USA) was used to mensurate the concentration of each sample. Quality was evaluated by Agilent2200 (Agilent, USA). Based on a solution provided by the manufacturer (Life Technologies, USA), Ion Proton Total RNA-SEQ Kit V2 was used to prepare sequencing libraries for each RNA sample. Data are provided in supplementary Table S1.
Western blot assays
Transfected cells were harvested 48 h after transfection. Then the protein was extracted and electrophoresed. 10% SDS-PAGE was used to segregate the protein, then incubated together with specific antibodies, and lastly densitometry (Bio-Rad) was used to quantify the electrophoretic bands. The EZH2 antibody (1:1000) were acquired from Abcam (Cat#: ab191250), and the GAPDH served as control (CST, Cat#: 5174).
RNA immunoprecipitation (RIP) assays
RNA immunoprecipitation (RIP) experiments were performed using a Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA) according to the manufacturer's instructions. Antibody for RIP assays of EZH2 (Abcam, Cat#: ab191250) were from Abcam.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed using EZ-CHIP KIT according to the manufacturer instruction (Millipore). Antibody of EZH2 (Abcam, Cat#: ab191250) and H3K27me3 (Abcam, Cat#: ab6002) were from Abcam. Quantification of immunoprecipitated DNA was performed using qPCR. Result was calculated as a percentage relative to input DNA by equation 2[Input Ct- Target Ct] ×100 (%).
Statistical analyses
All statistical analyses were performed using SPSS software. Student's t test, χ2 test, or Wilcoxon test, as appropriate were used for the difference between the two groups. The OS rates were calculated by the Kaplan-Meier method and the log-rank test was used for comparison. A bilateral P value of 0.05 was considered statistically significant.
Results
Copy number amplification could lead to activation of LINC00662 in NSCLC
Raw microarray data were obtained from GEO, containing GSE51852 (n=4vs4) and GSE43767 (n=19vs69). The normalization was performed to the signal data. As shown in Figure 1A, we obtained activated lncRNA in these two data sets (fold change ≥ 1.5). We can see that there are 4 activated lncRNAs in lung cancer (LINC00578, LINC00467, AFAP1-AS1, LINC00662), among these differentially expressed lncRNAs, there also have many other well-known lncRNAs in lung cancer, such as LINC00578[23]. Our previous studies also found that AFAP1-AS1 could display important regulatory roles in lung cancer tumorigenesis[14]. Based on the basic expression level of in lung cancer, we choose LINC00662 for further in-depth study.
Copy number amplification drives LINC00662 high expression in NSCLC. A: Raw microarray data including GSE51852 and GSE43767 were downloaded from GEO and then normalization. B: The expression of LINC00662 was activated in NSCLC tissues with paired and unpaired TCGA data. C: Exact full length of LINC00662 by RACE. D and E: LINC00662 expression was correlated with its copy number level. F: Higher expression of LINC00662 was indicated a poorer overall survival.
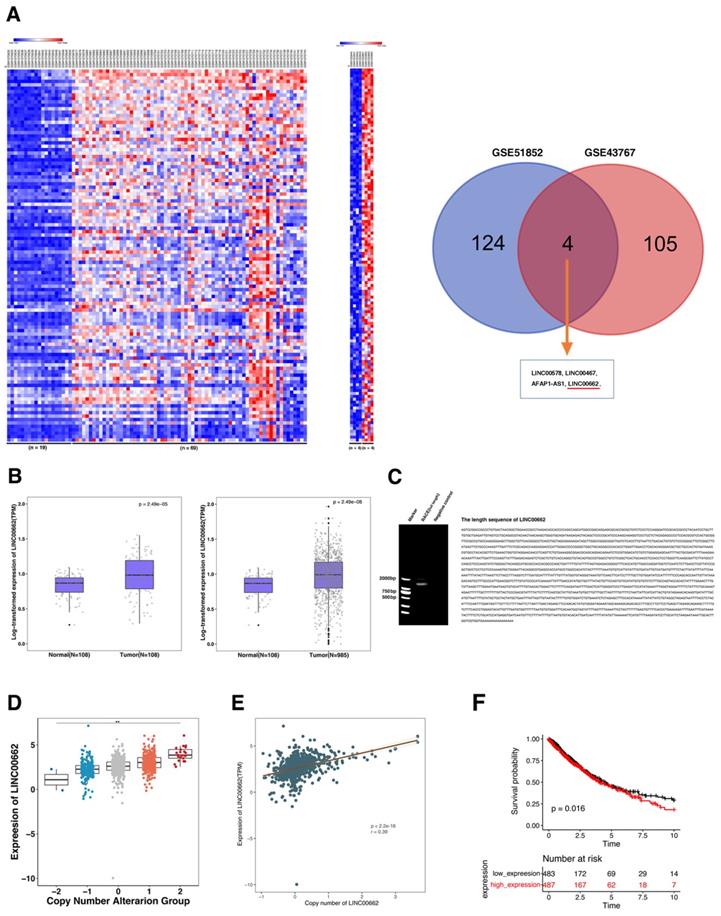
Then we used TCGA database of NSCLC to evaluate the expression of LINC00662 in tumors and normal samples. In Figure 1B, the results indicated that LINC00662 significantly higher expression in NSCLC, which compared with their matched adjacent normal tissues, whether in a matched and unmatched tissues. Then we preformed rapid amplification of cDNA ends (RACE) of LINC00662 in A549 cells. The result showed the exact complete sequence of LINC00662 and its length is 2064nt (Figure 1C). Furthermore, we found that as the copy level increased, the expression of LINC00662 gradually increased (Figure 1D). Moreover, LINC00662 copy number amplification is indeed positively correlated with expression (Figure 1E). This suggested that the increase of LINC00662 expression in NSCLC may partly be due to gene amplification and play an important role in NSCLC tumorigenesis. Further analysis found that the high expression of LINC00662 suggested poor prognosis in NSCLC (Figure 1F).
LINC00662 regulates lung cancer cell proliferation, migration and cell apoptosis in vitro.
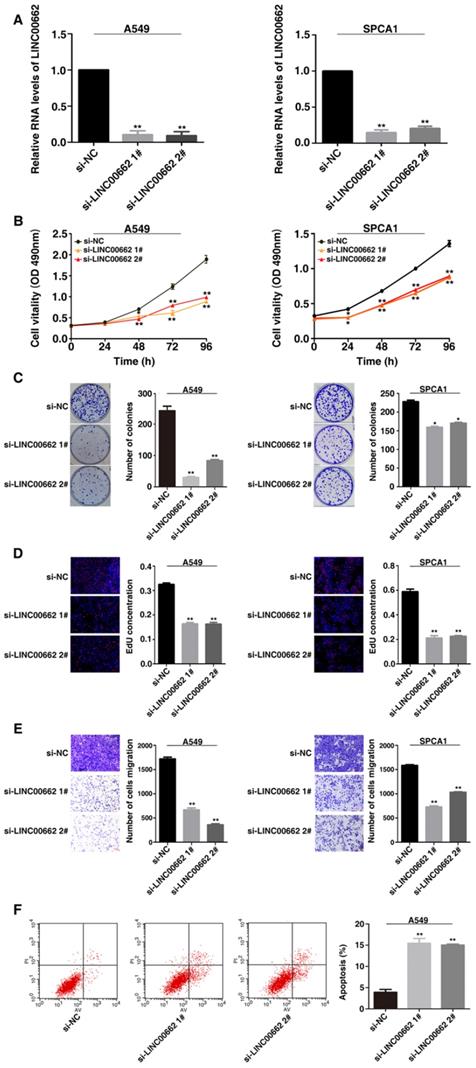
This study selected human lung cancer cell lines A549 and SPCA1 to determine the biological role of LINC00662. Then siRNA was used to mediate silencing of LINC00662 expression and plasmid-mediated overexpression in lung cancer cells (Figure 2A and 3A). Methylthiazol tetrazolium (MTT) assays showed that cell proliferation was inhibited after LINC00662 knockdown in A549 and SPCA1 cells (Figure 2B). And the clone formation experiment results showed that the clone survival were decreased after silencing LINC00662 in A549 and SPCA1 cells (Figure 2C). Consistently, ethynyldeoxyuridine (EdU) staining assays also proved that after silencing LINC00662, the proliferation rate of A549 and SPCA1 were inhibited (Figure 2D). Next, transwell assays revealed the ability of migration of NSCLC cells. As showed in Figure 2E, after silencing of LINC00662, the migration both in A549 and SPCA1 cells were significantly repressed.
Knockdown LINC00662 inhibits NSCLC cell proliferation and migration in vitro. A: The expression levels of LINC00662 in siRNA-mediated A549 and SPCA1 cells were detected by qRT-PCR. B: After siRNA-mediated LINC00662 knockdown, the proliferation of A549 and SPCA1 cells were detect by MTT assays.C: Colony formation assays were detected in LINC00662 silenced A549 and SPCA1 cells. D: 48 h after transfection, the proliferation of siRNA-mediated A549 and SPCA1 cells were detected use the EdU staining kit. E: After siRNA transfection the altered migratory capacity of A549 and SPCA1 cells were performed to investigate by transwell assays. F: A549 cells were used flow cytometry to be stained and analyzed. LR: early apoptotic cells. UR: terminal apoptotic cells. *P<0.05, **P<0.01.
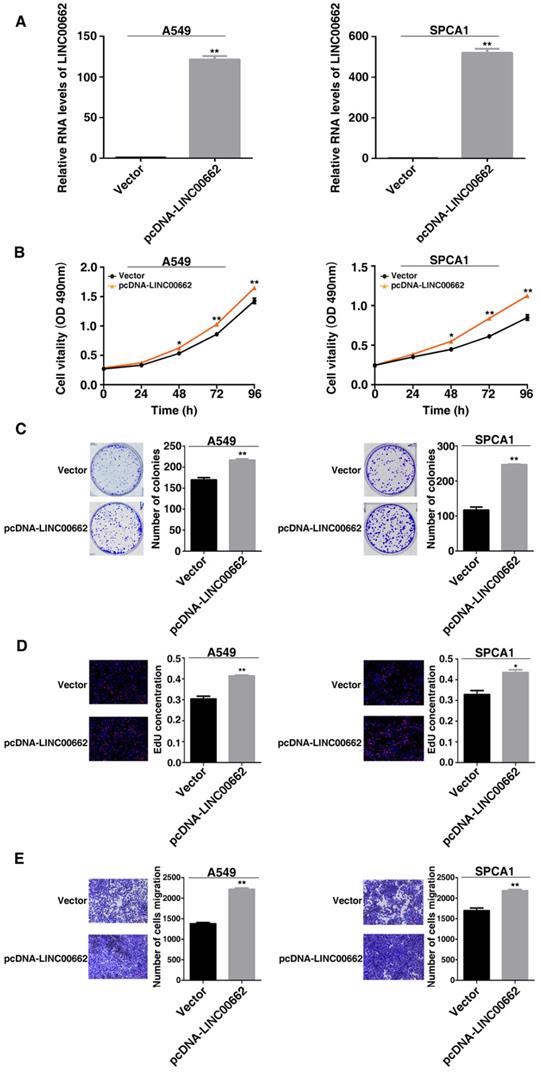
Overexpression of LINC00662 promotes NSCLC cell proliferation and migration in vitro. A: Analysis of LINC00662 expression levels in A549 and SPCA1 by qRT-PCR. B: After LINC00662 overexpression cell proliferation both in A549 and SPCA1 cells were determine by MTT assays. C: Colony formation ability were detected in A549 and SPCA1 cells after LINC00662 was overexpression. D: The proliferation of A549 and SPCA1 cells were determined after overexpression of LINC00662 by EdU assays. E: The alterations in migratory capacity of A549 and SPCA1 cells were used transwell assays to investigate after transfection. *P<0.05, **P<0.01.
Apoptosis plays an important role in the proliferation of various cancer cells. To determine whether apoptosis affects the apoptosis of NSCLC cells, flow cytometry analysis was performed. In Figure 2F, the apoptotic percentage of A549 cells was observably higher than the control group after silencing LINC00662. Therefore, LINC00662 may regulate NSCLC cell proliferation by influencing cell apoptosis.
By contrast, overexpressed LINC00662 promoted cell proliferation and increased the cell clone numbers (Figure 3B and 3C). Similarly, EdU staining assays found the proliferation ability both in A549 and SPCA1 were increased after overexpression of LINC00662 (Figure 3D). And overexpression of LINC00662 promoted cell migration (Figure 3E). The above results indicate that LINC00662 may play a key role in the proliferation and migration of NSCLC cells.
LINC00662 regulates NSCLC cell proliferation in vivo
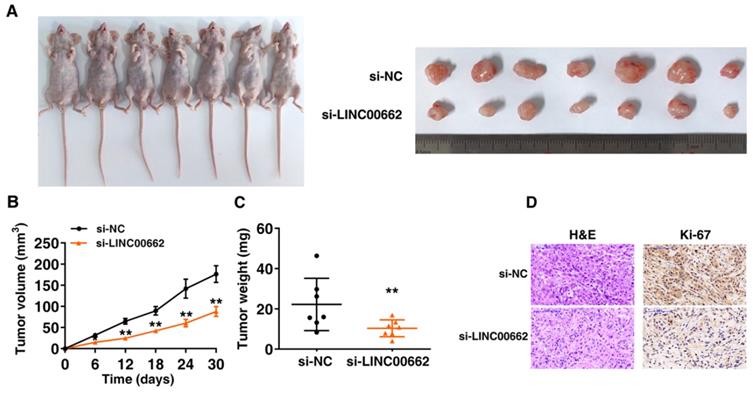
Further research is to confirm whether LINC00662 affects lung cancer cell proliferation in vivo, nude mice were inoculated with the A549 transfected with control and si-LINC00662 (Figure 4A). After continuous measurement, the tumor volume of the si-LINC00662 group was significantly smaller than that of the control group (Figure 4B). In addition, at the end of experiment, the average tumor weight of the LINC00662 knockdown group was significantly lower than that of the control group (Figure 4C). The positive rate of Ki-67 staining for tumors formed by si-LINC00662-transfected A549 cells was significantly lower than that of the control group (Figure 4D). These above results indicated that LINC00662 knockdown can repress tumor growth in vivo. The experiments in vivo are consistent with those of in vitro functional studies which mediated by LINC00662.
LINC00662 regulates NSCLC cell proliferation and migration in vivo. A: Transfected A549 cells were injected into nude mice. B: Tumor volume was calculated every 6 days after injection. C: Tumor weights were presented as mean tumor weight ±S.D. (standard deviation). D: H&E staining and IHC staining (antibodies against Ki-67) were performed to detect tumor sections, and it was found that the tumor developed from the siLINC00662 transfection group was down-regulated. *P<0.05, **P<0.01.
The global downstream genes regulated by LINC00662
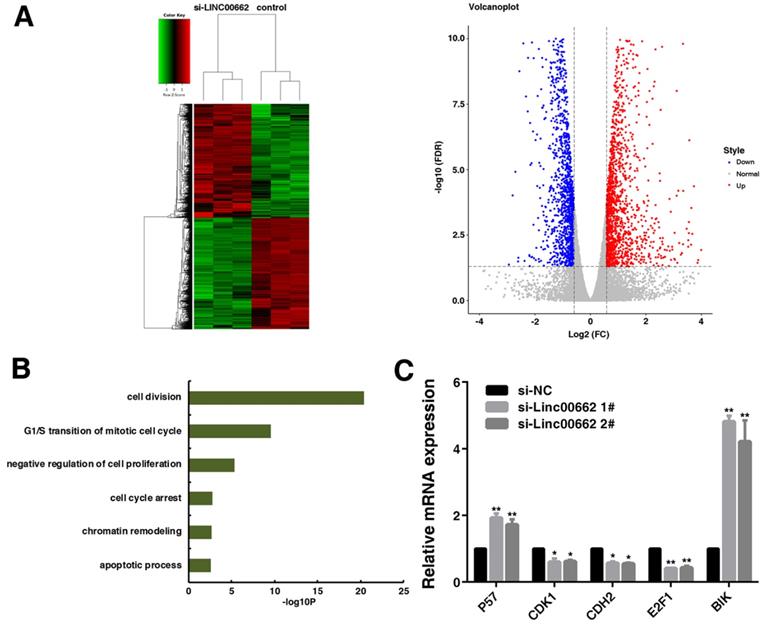
To determine changes in global downstream gene expression of LINC00662, we assessed the global effects of LINC00662 knockdown by RNA transcriptomic sequencing in A549 cells (Figure 5A). We totally identified 2934 differentially expressed genes (fold change ≥ 1.5), 1483 genes were up-regulated while 1451 genes were down-regulated in LINC00662 knockdown A549 cells (Supplementary Table S1). The results of GO terms analysis showed that the majority of these genes were involved in cell division, cell cycle and apoptotic process (Figure 5B). We selected five genes (P57, CDK1, CDH2, E2F1 and BIK) that are quite relevant to cancer cell proliferation and apoptosis [24-28]. We confirmed their change expression by qRT-PCR after LINC00662 knockdown in A549. After LINC00662 knockdown, CDK1, CDH2 and E2F1 were inhibited, while the expressions of P57 and BIK were induced (Figure 5C). These results suggested that LINC00662 could affect tumorigenesis by regulating these key genes related to tumorigenesis.
The global downstream target genes of LINC00662. A: After LINC00662 was knockdown from A549 cells, the mean center and grading clustering of gene transcripts were changed (≥1.5-fold change), with a total of 3 repeats. This was a volcanic map depicting the up-regulated and down-regulated genes knockdown by LINC00662 in RNA-seq. B: Gene ontology analysis of the whole gene expression changes after LINC00662 gene knockdown. C: qRT-PCR selectively confirmed the changes in mRNA levels after LINC00662 knockdown. *P<0.05, **P<0.01.
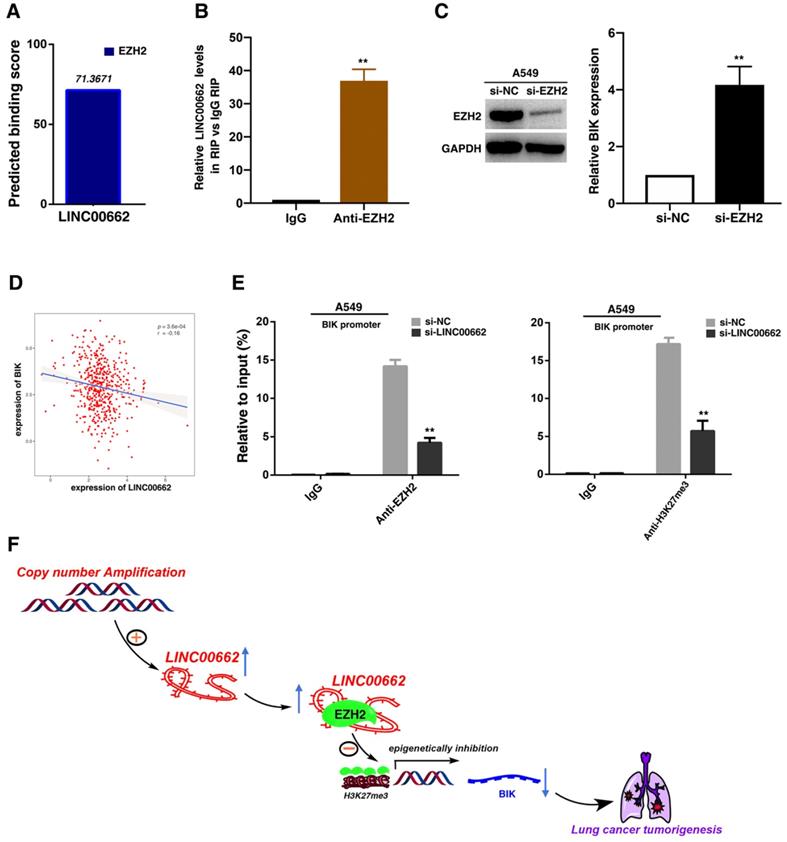
LINC00662 could bind to EZH2 and recruit EZH2 to the promoter regions of BIK, thus epigenetically repressing BIK expression and promoting tumorigenesis of NSCLC
Recent studies have reported that a significant number of lncRNAs have been shown to function in cooperation with chromatin modifying enzymes to exert epigenetic regulation of gene expression[29]. Approximately 20% of human lncRNAs have been shown to interaction with PRC2, indicating that lncRNAs may have a general role in recruiting PRC2 proteins to their target genes[30]. PRC2, a methyltransferase that is composed of EZH2, SUZ12 and EED, can catalyze the trimethylation of lysine residue 27 of histone 3 (H3K27me3), thus epigenetically repressing gene expression[31]. And abnormal expression of PRC2 is closely related to the occurrence and development of tumors [32]. Thus, we hypothesized that LINC00662 may regulate gene expression in such a manner. To test this, we used bioinformatics to predict this interaction possibility (http://bioinfo. bjmu.edu.cn/lncpro/). As shown in Figure 6A, the predicted binding scores between LINC00662 and EZH2 were fairly high. To validate this interaction, RNA immunoprecipitation (RIP) assays showed that the endogenous LINC00662 was enriched in the anti-EZH2 RIP fraction compared to the IgG fraction (Figure 6B). Next, through analysis of sequencing results, we found that BIK was significantly induced after knockdown of LINC00662. Then the role of EZH2 in coregulating suppression of LINC00662-suppressed BIK was investigated by EZH2 knockdown, and both were induced in cells after knockdown of EZH2 (Figure 6C). Further analysis demonstrated that LINC00662 was negatively correlated with BIK expression (Figure 6D). To address whether LINC00662 is involved in epigenetically transcriptional repression by enrichment of EZH2 to target gene promoters, we conducted ChIP analysis afrer LINC00662-knockdown. ChIP assays demonstrated that LINC00662 decreased the binding of EZH2 and H3K27me3 levels across the BIK promoters (Figure 6E). These results indicated that LINC00662 promotes tumorigenesis of NSCLC via epigenetically repressing BIK expression.
LINC00662 could bind to EZH2 and recruit EZH2 to the promoter regions of BIK, thus epigenetically repressing BIK expression and promoting tumorigenesis of NSCLC. A: Bioinformatics were used to predict the possible interaction of LINC00662 and EZH2. B: RIPs experiments were performed, and the coprecipitated RNA was subjected to qRT-PCR for LINC00662. C: The expression of EZH2 after knockdown of EZH2, and the expression of BIK after knockdown of EZH2. D: The level of LINC00662 and BIK showed a statistically negative correlation in TCGA. E: ChIP-qPCR of H3K27me3 and EZH2 of the promoter region of BIK loci after siRNA treatment targeting si-NC or si-LINC00662. Antibody enrichment was quantified relative to the amount of input DNA. An antibody directed against IgG was used as a negative control. F: A proposed model which is mediated by LINC00662.
Discussion
Recent research reported that lncRNA was a vital component in regulating the tumorigenesis of lung cancer. In this research, we focused on lncRNA LINC00662 in NSCLC. LINC00662 have revealed its oncogenic property in many tumor types [19-21, 33], however, the reason for how LINC00662 activated in tumorigenesis remain unclear. As an important reason for activation of lncRNA, at the genomic level, copy number amplification has attracted widespread attention. For example, lncRNA PVT1 which located in 8q24 region is activated by amplification in multiple tumor types, and PVT1 could participate in the regulation of tumorigenesis[34]. Our results showed that the activation of LINC00662 in NSCLC part of the reason may be the copy number amplification. In addition, the exact sequence of LINC00662 is still unclear. We firstly used RACE to get its exact sequence in NSCLC.
We also shown that knockdown of LINC00662 could significantly inhibit NSCLC cell proliferation, migration and induce apoptosis. Moreover, we obtained the global downstream genes which mediated by LINC00662. Several of them were related to cell division, cell cycle and apoptosis processes. Then qRT-PCR was used to identify the five potential downstream target genes P57, CDK1, CDH2, E2F1 and BIK after knockdown of LINC00662. These genes may be potential targets of LINC00662 in lung cancer because they could contribute an important role to tumorigenesis [24, 26-28, 35]. As an inhibitor of cyclin-dependent kinase, P57 is considered to be a tumor suppressor gene in various tumors [36]. It can also inhibit the development of NSCLC by LINC00511-mediated epigenetic silencing [37]. Our study confirmed that LINC00662-knockdown can mediate up-regulation of P57. E2F1 could mediate the cell cycle and apoptosis of tumor cells [38]. Reports suggested that E2F1 played a key role in the proliferation of tumors cells [39]. Its expression was markedly reduced after LINC00662 knockdown. BIK is a member of the BCL-2 family and is considered as a tumor suppressor due to its vital function in promoting apoptosis including lung cancer [38]. Studies have proven that activation of BIK can result in cancer cell death. [40]. In addition, it has been found that BIK can be induced to increase cell cancer apoptosis, and these regulatory mechanisms may serve as important targets for tumor therapy drugs [41]. However, the mechanism by which BIK expression is inhibited is unclear. Our results demonstrated that knockdown of LINC00662 could induce the expression of BIK by EZH2-dependent manner. Many lncRNAs regulate the level of H3K27me3 of specific gene promoter loci via recruiting and binding to EZH2, and EZH2-mediated epigenetically regulation plays a crucial role in process of tumor development[42].
In summary, we found that LINC00662 was upregulated by copy number amplification in NSCLC, and its exact sequence was determined. LINC00662 is crucial for maintaining NSCLC cell proliferation, apoptosis and migration. In addition, our research also revealed the global downstream genes of LINC00662 for the first time. And LINC00662 could interact with EZH2 and recruit EZH2 to the promoter regions of BIK, thus epigenetically repressing BIK expression and promoting tumorigenesis of NSCLC. Therefore, it is believed that targeting LINC00662 may be an effective strategy to inhibit the development of NSCLC. Our studies have further deepened our understanding of the occurrence of lung cancer and may provide potential molecular targets for NSCLC.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China grants (82172992, 81972188, 81772475 and 81772479), and the Natural Science Foundation of Jiangsu Province (BK20211253, BK20191355). This work was supported by six peak talents of Jiangsu Province (SWYY-200).
Author Contributions
CLY, YD, YZ and CGZ: contributed to designing and organizing the experiments, carrying out the data analysis, and writing of the manuscript. YZ contributed to laboratory measurements, data analysis. EBZ, RHG, WL and LH: contributed to conceiving the ideas, supervising the study, and writing the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68:394-424
2. Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nature genetics. 2016;48:607-16
3. Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2015;10:1240-2
4. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB. et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2015;10:1243-60
5. Jeong Y, Hellyer JA, Stehr H, Hoang NT, Niu XM, Das M. et al. Role of KEAP1/NFE2L2 Mutations in the Chemotherapeutic Response of Patients with Non-Small Cell Lung Cancer. Clin Cancer Res. 2020;26:274-81
6. Zhou W, Sun GG, Zhang Z, Zhao LB, Xu L, Yuan HY. et al. Proteasome-Independent Protein Knockdown by Small-Molecule Inhibitor for the Undruggable Lung Adenocarcinoma. J Am Chem Soc. 2019;141:18492-9
7. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature reviews Cancer. 2002;2:563-72
8. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annual review of pathology. 2011;6:49-69
9. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes & development. 2009;23:1494-504
10. Huarte M. The emerging role of lncRNAs in cancer. Nature medicine. 2015;21:1253-61
11. Yu H, Xu Q, Liu F, Ye X, Wang J, Meng X. Identification and validation of long noncoding RNA biomarkers in human non-small-cell lung carcinomas. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2015;10:645-54
12. Chen Z, Li JL, Lin S, Cao C, Gimbrone NT, Yang R. et al. cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J Clin Invest. 2016;126:2267-79
13. Castellano JJ, Navarro A, Vinolas N, Marrades RM, Moises J, Cordeiro A. et al. LincRNA-p21 Impacts Prognosis in Resected Non-Small Cell Lung Cancer Patients through Angiogenesis Regulation. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2016;11:2173-82
14. Yin D, Lu X, Su J, He X, De W, Yang J. et al. Long noncoding RNA AFAP1-AS1 predicts a poor prognosis and regulates non-small cell lung cancer cell proliferation by epigenetically repressing p21 expression. Mol Cancer. 2018;17:92
15. Li Y, Wang Z, Shi H, Li H, Li L, Fang R. et al. HBXIP and LSD1 Scaffolded by lncRNA Hotair Mediate Transcriptional Activation by c-Myc. Cancer research. 2016;76:293-304
16. Li S, Yang J, Xia Y, Fan Q, Yang KP. Long Noncoding RNA NEAT1 Promotes Proliferation and Invasion via Targeting miR-181a-5p in Non-Small Cell Lung Cancer. Oncol Res. 2018;26:289-96
17. Zhang E, Han L, Yin D, He X, Hong L, Si X. et al. H3K27 acetylation activated-long non-coding RNA CCAT1 affects cell proliferation and migration by regulating SPRY4 and HOXB13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res. 2017;45:3086-101
18. Zhang E, He X, Zhang C, Su J, Lu X, Si X. et al. A novel long noncoding RNA HOXC-AS3 mediates tumorigenesis of gastric cancer by binding to YBX1. Genome Biol. 2018;19:154
19. Xu D, Chen Y, Yuan C, Zhang S, Peng W. Long non-coding RNA LINC00662 promotes proliferation and migration in oral squamous cell carcinoma. OncoTargets and therapy. 2019;12:647-56
20. Liu Z, Yao Y, Huang S, Li L, Jiang B, Guo H. et al. LINC00662 promotes gastric cancer cell growth by modulating the Hippo-YAP1 pathway. Biochemical and biophysical research communications. 2018;505:843-9
21. Li N, Zhang LY, Qiao YH, Song RJ. Long noncoding RNA LINC00662 functions as miRNA sponge to promote the prostate cancer tumorigenesis through targeting miR-34a. European review for medical and pharmacological sciences. 2019;23:3688-98
22. Zhang L, Chen S, Wang B, Su Y, Li S, Liu G. et al. An eight-long noncoding RNA expression signature for colorectal cancer patients' prognosis. Journal of cellular biochemistry. 2019;120:5636-43
23. Wang L, Zhao H, Xu Y, Li J, Deng C, Deng Y. et al. Systematic identification of lincRNA-based prognostic biomarkers by integrating lincRNA expression and copy number variation in lung adenocarcinoma. Int J Cancer. 2019;144:1723-34
24. Zhang P, Kawakami H, Liu W, Zeng X, Strebhardt K, Tao K. et al. Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer. Molecular cancer research: MCR. 2018;16:378-89
25. Mebratu YA, Tesfaigzi Y. Does the BCL-2 family member BIK control lung carcinogenesis? Molecular & cellular oncology. 2018;5:e1435182
26. Miao J, Wang W, Wu S, Zang X, Li Y, Wang J. et al. miR-194 Suppresses Proliferation and Migration and Promotes Apoptosis of Osteosarcoma Cells by Targeting CDH2. Cell Physiol Biochem. 2018;45:1966-74
27. Dyer MA, Cepko CL. p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development. 2000;127:3593-605
28. Feng L, Jing L, Han J, Wang G, Liu Y, Zhang X. et al. MicroRNA 486-3p directly targets BIK and regulates apoptosis and invasion in colorectal cancer cells. OncoTargets and therapy. 2018;11:8791-801
29. Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics. 2014;9:21-6
30. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667-72
31. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039-43
32. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-9
33. Gong W, Su Y, Liu Y, Sun P, Wang X. Long non-coding RNA Linc00662 promotes cell invasion and contributes to cancer stem cell-like phenotypes in lung cancer cells. Journal of biochemistry. 2018;164:461-9
34. Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H. et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82-6
35. Espada L, Udapudi B, Podlesniy P, Fabregat I, Espinet C, Tauler A. Apoptotic action of E2F1 requires glycogen synthase kinase 3-beta activity in PC12 cells. J Neurochem. 2007;102:2020-8
36. Avrahami D, Li CH, Yu M, Jiao Y, Zhang J, Naji A. et al. Targeting the cell cycle inhibitor p57(Kip2) promotes adult human beta cell replication. J Clin Invest. 2014;124:670-4
37. Sun CC, Li SJ, Li G, Hua RX, Zhou XH, Li DJ. Long Intergenic Noncoding RNA 00511 Acts as an Oncogene in Non-small-cell Lung Cancer by Binding to EZH2 and Suppressing p57. Mol Ther-Nucl Acids. 2016 5
38. Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B. et al. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. Embo J. 2003;22:3580-90
39. Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer cell. 2008;13:11-22
40. Chinnadurai G, Vijayalingam S, Rashmi R. BIK, the founding member of the BH3-only family proteins: mechanisms of cell death and role in cancer and pathogenic processes. Oncogene. 2008;27:S20-S9
41. Hur J, Bell DW, Dean KL, Coser KR, Hilario PC, Okimoto RA. et al. Regulation of expression of BIK proapoptotic protein in human breast cancer cells: p53-dependent induction of BIK mRNA by fulvestrant and proteasomal degradation of BIK protein. Cancer research. 2006;66:10153-61
42. Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071-6
Author contact
![]() Corresponding authors: Erbao Zhang, Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China; Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing 211166, China; E-mail: erbaozhangedu.cn; Tel: +86-025-86868460. Renhua Guo, Department of Oncology, First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China; E-mail: rhguoedu.cn. Wei Li, Department of Oncology, First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China; Department of Oncology, Sir Run Run Hospital, Nanjing Medical University, Nanjing, Jiangsu, 210000, PR China; E-mail: liwei_realcom. Liang Han, Department of Oncology, Xuzhou Central Hospital, Xuzhou, Jiangsu, PR China; E-mail: hanliang_njmucom.
Corresponding authors: Erbao Zhang, Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, China; Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing 211166, China; E-mail: erbaozhangedu.cn; Tel: +86-025-86868460. Renhua Guo, Department of Oncology, First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China; E-mail: rhguoedu.cn. Wei Li, Department of Oncology, First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China; Department of Oncology, Sir Run Run Hospital, Nanjing Medical University, Nanjing, Jiangsu, 210000, PR China; E-mail: liwei_realcom. Liang Han, Department of Oncology, Xuzhou Central Hospital, Xuzhou, Jiangsu, PR China; E-mail: hanliang_njmucom.