Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(6):1745-1757. doi:10.7150/jca.63609 This issue Cite
Review
Cholesterol metabolism and its implication in glioblastoma therapy
Xuyang Guo1, Shaolong Zhou1,2, Zhuo Yang1, Zi-An Li1, Weihua Hu2, Lirui Dai1, Wulong Liang1, Xinjun Wang1,2 ![]()
1. Department of Neurosurgery, The Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, 450052, China.
2. International Joint Laboratory of Glioma Metabolism and Microenvironment Research, Zhengzhou, Henan, China.
Received 2021-6-7; Accepted 2022-1-7; Published 2022-3-14
Abstract

Glioblastoma (GBM) is the most lethal malignant tumor in the central nervous system, with a median survival of only 14 months. Cholesterol, which is the main component of cell membrane and the precursor of many hormones, is one of the most important lipid components in human body. Since reprogramming of the cholesterol metabolic profile has been discovered in many cancers including GBM, cholesterol metabolism becomes a promising potential target for therapy. Since GBM cells rely on external cholesterol to survive and accumulate lipid droplets to meet their rapid growth needs, targeting the metabolism of cholesterol by different strategies including inhibition of cholesterol uptake and promotion of cholesterol efflux by activating LXRs and disruption of cellular cholesterol trafficking, inhibition of SREBP signaling, inhibition of cholesterol esterification, could potentially oppose the growth of glial tumors. In this review, we discussed the above findings and describe cholesterol synthesis and homeostatic feedback pathways in normal brain tissues and brain tumors, statin use in GBM and the role of lipid rafts and cholesterol precursors and oxysterols in the treatment and pathogenesis of GBM are also summarized.
Introduction
Cholesterol, synthesized by all mammalian cells [1], is one of the most important lipid components and is widely distributed in various tissues of the human body. Cholesterol predominantly localizes to cellular membranes, where it maintains membrane integrity and fluidity and forms membrane microstructures [2]. Increasing evidence has shown that cholesterol metabolism disorders are not only associated with cardiovascular disease and atherosclerosis but are also closely related to the pathogenesis and progression of cancer. On the one hand, cholesterol and its precursors or metabolites are involved in a variety of biological processes, including the cell immune response, posttranslational modification of proteins, and cell signal transduction, which may contribute to the malignant behavior of tumors. On the other hand, the immortal proliferation of cancer cells is accompanied by an increased requirement for cholesterol [3].
Glioblastoma (GBM) is the most common central nervous system (CNS) malignant tumor, and the prognosis for patients remains devastating despite surgical resection combined with radiotherapy and chemotherapy. Studies have found that metabolic disorders of cholesterol occur in many kinds of malignant tumors, including GBM [4], which indicates that reprogramming the cholesterol metabolic profile is a novel hallmark of cancer. Consistently, abundant preclinical experiments have demonstrated the anticancer effect of metabolic therapy targeting cholesterol in various tumors, including GBM, breast cancer, prostate cancer, and colorectal cancer. Cholesterol is unable to cross the blood-brain barrier from the periphery to the CNS, which maintains an isolated cholesterol metabolism microenvironment. Due to this distinct cholesterol pool and the reprogrammed cholesterol metabolic profile found in GBM, therapy for glioblastoma targeting cholesterol metabolism has recently received wide interest. In this review, we discussed the regulation of cholesterol homeostasis in the brain and advances in the treatment of GBM by metabolic therapy targeting cholesterol through several different mechanisms.
Regulation of Cholesterol homeostasis
There are two main pathways providing cholesterol for cells: 1. Cells take up low- density lipoprotein (LDL) by low-density lipoprotein receptor (LDLR)-mediated endocytosis from the periphery. After entering the cell, LDL is then transported to the lysosome, where the cholesterol ester in LDL is hydrolyzed to release unesterified cholesterol. 2. Cells utilize acetyl-CoA and NADPH as raw materials to biosynthesize cholesterol via de novo synthesis (also known as the mevalonate pathway). HMG-CoA synthase (HMGCS) catalyzes the condensation of acetyl-CoA and acetoacetyl-CoA into HMG-CoA. HMG-CoA is reduced and catalyzed by HMGCR to mevalonic acid, which is subsequently decarboxylated and phosphorylated to isopentenyl pyrophosphate (IPP) and 3,3-dimethylallyl pyrophosphate (DPP). IPP and DPP are catalyzed by farnesyl pyrophosphate synthase (FPPS, also known as farnesyl diphosphate synthase, FDPS) to synthesize farnesyl pyrophosphate (FPP), and then FPP is catalyzed by squalene synthase to synthesize squalene. Squalene undergoes a series of catalytic reactions to finally generate cholesterol. The key rate-limiting enzymes are 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (HMGCR) and squalene epoxidase (SQLE), which reduce HMG-CoA to mevalonate and catalyze the oxidation of squalene to 2,3-epoxysqualene, respectively.
The brain can hardly take up cholesterol from the periphery due to the blood-brain barrier; thus, de novo synthesis predominantly by astrocytes and oligodendrocytes is the main source of cholesterol present in this organ. The myelin sheaths formed by oligodendrocytes and surrounding axons contain a large amount of cholesterol, which explains why the brain is rich in cholesterol [5]. Cholesterol enrichment of myelin leads to reduced permeability to ions, thus ensuring the speed and accuracy of electrical signal conduction in neural activities [6]. The excretion of cholesterol also plays an important role in maintaining cholesterol homeostasis in the brain, 24(S)-hydroxycholesterol, one of the cholesterol metabolites, is the major hydroxylated sterol excreted from brain [7]. The hydroxylated cholesterol can cross the BBB and go to the liver to be converted to bile acids and excreted from the body [8]. Addtionally, a study has found that glioma cells can convert cholesterol into corticosteroids such as progesterone, androstanedione, androstenediol, androstenedione, which may conribute to the progresssion of glioma [9].
Unesterified cholesterol can also be condensed with fatty acyl-CoA by sterol O-acyltransferase (SOAT) (also known as acyl-CoA cholesterol acyl-transferase, ACAT) to form cholesteryl ester, which is stored in lipid droplets. Astrocytes synthesize apolipoprotein E-containing cholesterol and excrete it via ATP-binding cassette transporter 1 (ABCA1). Neurons take up cholesterol-containing apo-E by endocytosis. Since the synthesis of cholesterol requires a diverse array of enzymes and consumes a large amount of energy, neurons, which specialize in the generation of electrical activity, may reduce or even abandon cholesterol synthesis [6]. Thus, neurons rely on outsourcing of cholesterol that is delivered from nearby astrocytes. The esterified cholesterol that enters the cell is hydrolyzed in the lysosome by cholesterol ester hydrolase, and then the unesterified cholesterol is transported out of the lysosome to the cell membrane or endoplasmic reticulum by Niemann-Pick type C protein 1 (NPC1).
Cholesterol homeostasis is predominantly regulated by two major signaling pathways: liver X receptors (LXRs) and the transcription factors sterol regulatory element-binding proteins (SREBPs). When the intracellular cholesterol level decreases, SREBPs enter the nucleus to activate the transcription of adipose and cholesterol synthesis-related genes to promote the elevation of cellular lipids and steroids. When the cellular cholesterol level rises, the level of the cholesterol metabolite oxysterol rises and activates LXRs. The LXR transcriptional network drives cholesterol efflux and reduces cholesterol influx and synthesis. LXRs and SREBPs constitute a negative feedback loop to regulate the homeostasis of cellular cholesterol metabolism, and numerous studies have found that a variety of drugs alter the cholesterol levels in GBM cells by acting on these two pathways (Figure 1). There is also an interaction between LXRs and SREBPs. When cellular cholesterol levels increase to activate LXRα, activated LXRα can activate SREBP-1c to promote fat synthesis [10-12]. On the one hand, activated LXRα shifts acetyl-CoA from cholesterol synthesis to fatty acid synthesis. On the other hand, the increased fatty acids can be esterified with unesterified cholesterol into cholesterol esters, and then they are stored in cell lipid droplets. The network regulation mechanism of LXRα ultimately decreases the level of unesterified cholesterol.
Metabolic Therapy targeting SREBP and LXRs in GBM. SREBPs enter the nucleus to activate the transcription of adipose synthase (ACC, FAS, SCD-1) and cholesterol synthesis-related genes (HMGCS, HMGCR, SQS) and promote uptake of cholesterol (LDLR) to promote the elevation of cellular lipids and steroids. SREBP is upregulated by S1P, SOAT1, SIRT1 and downregulated by quercetin, phytol, retinol and miR-29. PF-429242, Avasimibe, MicroRNA-132 can inhibit the SERBP pathway by inhibiting S1P, SOAT1, and SIRT1, respectively. The LXR transcriptional network drives cholesterol efflux by ABCA1, ABCG1 and reduces cholesterol influx by mediating the degradation of LDLR through the induction of IDOL. LXR is activated by oxysterols, 24,25-epoxycholesterol and compounds LXR-623, GW3965,4-7rr, 4-13, 4-13rr. YTHDF2 upregulated by EGFR/SRC/ERK signaling suppresses LXR. Efavirenz inhibits LXR by promoting CYP46A1.
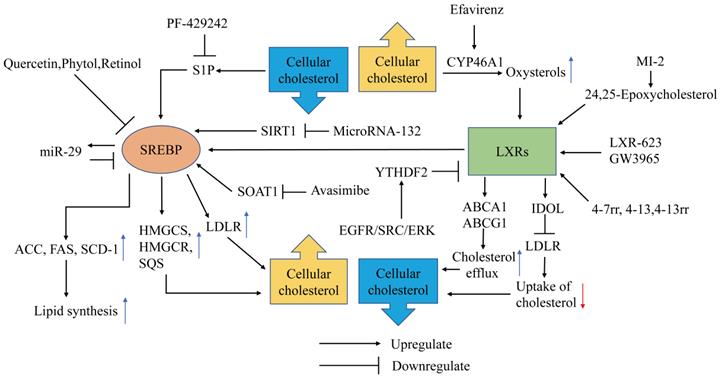
SREBPs are transcription factors with basic-helix-loop-helix-leucine zipper (bHLH-LZ) structures [13], and the SREBP family has three subtypes: SREBP-1a, SREBP-1c, and SREBP-2. SREBP-1 regulates fatty acid and cholesterol metabolism, and SREBP-2 mainly regulates cholesterol metabolism. The SREBP precursor needs to be cleaved by a protease into the mature form containing NH2-terminal (nSREBP-1a, nSREBP-1c, nSREBP-2) to exert transcription factor activity. SREBP processing can be controlled by the cellular sterol content. The precursors of SREBP-1a and SREBP-2 bind to the SREBP cleavage activating protein (SCAP) on the endoplasmic reticulum (ER) [14]. When the endoplasmic reticulum cholesterol concentration decreases, the SCAP configuration changes and it falls off of the ER, transporting the SREBP precursor to the Golgi apparatus, where site 1 protease (S1P) and site 2 protease (S2P) successively cleave the SREBP precursor into nSREBPs, which are then released into the cytoplasm [15,16]. The mature form of SREBPs containing NH2-terminal transactivation domain enters the nucleus to activate the transcription of genes related to cholesterol synthesis and fatty acid synthesis. These enzymes include HMG-CoA synthetase (HMGCS), HMGCR, squalene synthase, acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS) and stearoyl-CoA desaturase-1 (SCD-1). SREBP-1 in GBM can also promote the expression of LDLR to increase cholesterol uptake, which is upregulated by epidermal growth factor receptor/phosphoinositide 3-kinase EGFR/PI3K signaling [17].
Inhibition of SREBP signaling
SREBP plays an essential role in the regulation of cholesterol homeostasis and may function as an oncogene in GBM. Lewis et al. found that the expression of SREBP increases and activates the expression of related lipid synthesis genes, such as SCD, FAS, and acid-binding protein 3-7 (FABP3 and -7), and activates the expression of oxidative stress-related genes to maintain the activity of tumor cells in a lipid- and oxygen-deprived environment. This study also found that poor prognosis genes of GBM highly overlapped with genes regulated by SREBP activation [18]. At present, research on blocking SREBP-related pathways has made progress in preclinical experiments.
S1P is responsible for the activation of SREBP. Amino-pyrrolidineamide PF-429242 inhibits the activation of the SREBP pathway and induces GBM cell apoptosis by inhibiting S1P proteolytic processing of SREBP-2 [19]. Some fat-soluble small molecule compounds, such as quercetin, phytol and retinol, can also inhibit the viability of GBM cells by inhibiting the expression of SREBP-1 in vitro [20,21]. MicroRNAs have emerged as novel regulators of SREBP in recent years, providing a new perspective for the metabolic treatment of GBM. SREBP is a downstream target gene of silencing information regulator 2-related enzyme 1 (SIRT1). Li et al. found that microRNA-132 can induce U87 and U251 cell apoptosis by inhibiting the expression of SREBP-1c by downregulating the transcription of SIRT1 in vitro [22]. Ru et al. demonstrated that miR-29 plays an essential role in the negative feedback regulation of SREBP-1/SCAP; specifically, elevation of the expression of SREBP-1/SCAP promotes the expression of miR-29, and miR-29 attenuates the expression of SREBP-1/SCAP [23]. Thus, microRNA analogs have become a potential treatment for disrupting GBM cholesterol metabolism. The mechanism of the SREBP signaling pathway and its role in tumor metabolism treatment need to be further explored.
Inhibition of cholesterol esterification
GBM cells accumulate lipid droplets (LDs) to meet their rapid growth needs, and the LDs correlated with poor prognosis in glioma patients [24]. Thus, SOAT1, which is responsible for cholesterol esterification and LD formation, promotes the malignant behavior of GBM.
Inhibition of cholesterol esterification by targeting SOAT1 blocks LD formation, and the elevated unesterified cholesterol in the ER inhibits SREBP-1-regulated lipogenesis, leading to the suppression of GBM growth. The SOAT1 inhibitor avasimibe can selectively inhibit the viability of the GBM cell line EGFRvIII U87 without affecting astrocytes, and avasimibe can inhibit the growth of intracranial gliomas in xenograft model mice and prolong mouse survival [24]. A subsequent adult Drosophila glioma model elucidated that ACAT1 (SOAT1) is involved in gliomagenesis and presents a potential therapeutic target for GBM [25]. Avasimibe can also inhibit GBM cell growth by inducing cell cycle arrest and caspase-8-dependent apoptotic pathways [26]. Moreover, Luo Y et al. found avasimibe can inhibit the proliferation, migration and invasion of neoplasm cells by inhibiting the expression of linc00339 [27]. Additionally, Avasimibe elevates plasma membrane cholesterol concentrations, which, in turn, promote T cell receptors (TCRs) clustering and thus improve effector function of T cells [28]. Hao et al. designed a cell-surface anchor-engineered T cells which connected with tetrazine (Tre) and bicyclononyne (BCN)-containing liposomal avasimibe on its cell membrane, showing superior antitumor efficacy in mouse models of GBM [29], providing evidences for immunotherapy combined with metabolic treatment of glioma. K604, another SOAT1 inhibitor, can also suppress the proliferation of U251-MG cells and downregulate the activation of Protein kinase B (Akt) and extracellular signal‑regulated kinase in proliferating glioblastoma cells [30]. Paillasse et al. discovered that cholesterol esterification is also upregulated by activated cholecystokinin 2 receptor (CCK2R) and Protein kinase C/extracellular signal-regulated kinase1/2 (PKC/ERK1/2) signaling, and antagonists of CCK2R significantly reduced cell proliferation and invasion by inhibiting cholesterol esterification [31]. Avasimibe can penetrate the blood-brain barrier and exhibited anti-cancer effect in vitro and vivo and has been adopted to clinical trials for the treatment of atherosclerotic lesions [32], while currently there is insufficient clinical evidence to show patients can benefit from SOAT1 inhibitors such as avasimibe, further clinical randomized controlled study needs to be investigated.
Inhibition of cholesterol uptake and promotion of cholesterol efflux by activating LXRs
Liver X receptors (LXRs) are critical nuclear receptor transcription factors that maintain cellular cholesterol homeostasis. Activation of LXRs promotes the expression of ABCA1 and ABCG1 to induce cholesterol efflux and mediates the degradation of LDLR through the induction of the E3 ubiquitin ligase inducible degrader of LDLR (IDOL also known as Myosin regulatory light chain interacting protein MYLIP) to reduce the uptake of cholesterol. The activator of LXR is oxysterols, the metabolites of cholesterol. The expression of cytochrome P450 46A1 (CYP46A1), an enzyme that catalyzes cholesterol into 24OHC, significantly declines in GBM as do endogenous LXR ligand levels [33]. These results indicate that uncoupling LXRs is a crucial transformation of the GBM cholesterol metabolic profile compared to normal glial cells.
Additionally, the expression of 3-hydroxy-3-methylglutaryl-CoA synthase 1 (HMGCS1), HMGCR, and 3β-hydroxysteroid-Δ24 reductase (DHCR24), which play a key role in cholesterol de novo synthesis, is reduced in GBM cells [34], indicating that the de novo synthesis pathway of cholesterol in GBM is suppressed. Lipid-removed medium induces the death of a large number of GBM cells but has no effect on the viability of normal human astrocytes (NHAs). GBM cells take up three to fourfold more LDL than NHAs [34]. The overactivated EGFR/PI3K pathway, which is a common molecular feature in GBM, promotes the expression of LDLR through SREBP-1 signaling. Lipoprotein-deficient serum inhibits the viability of EGFRvIII U87 cells more significantly than the viability of U87 cells [17]. The above evidence shows that GBM mainly depends on the outsourcing of cholesterol for growth rather than de novo synthesis. Since the mevalonate pathway consumes 26 reducing equivalents of NADPH, it is tempting to speculate that the reliance of GBM cells on CNS-derived cholesterol enables them to direct their cellular NADPH, a key reducing agent in relatively short supply, towards buffering reactive oxygen species (ROS) and synthesizing other macromolecules [34,35]. GBM inhibits the production of oxysterols, which uncouples LXR, leading to increased cholesterol uptake and reduced cholesterol efflux, meeting the robust proliferation needs of tumors. Therefore, disturbing cholesterol uptake by activating LXR has become a promising approach for the treatment of GBM.
LXR activators inhibit the uptake of GBM cholesterol and promote cholesterol efflux by activating LXRs, thus disturbing cholesterol homeostasis in GBM. LXR-623 and GW3965 are two kinds of LXR activators that have been intensively studied in GBM therapy. LXR-623 and GW3965 selectively kill GBM cells in vitro, relieve tumor progression and prolong the survival of tumor-bearing mice. LXR-623 may have a better effect on GBM patients than GW3965 because LXR-623 can cross the blood-brain barrier. The study also found that the enhanced cellular cholesterol efflux of LXR-623 and GW3965 destabilizes the respiratory complexes within the inner mitochondrial membrane, which leads to inhibition of cellular oxidative phosphorylation. Energy starvation drives an integrated stress response that upregulates proapoptotic Noxa in an Activating transcription factor 4 (ATF4)-dependent manner. Hence, the combination treatment of BH3 mimetics and LXR623 has a synergistic antitumor effect [36]. The three LXRβ agonists 4-7rr, 4-13 and 4-13rr discovered by machine learning and structural analysis have lethal effects on GBM cells in vitro and in vivo [37]. Han et al. demonstrated that overexpression of CYP46A1 or 24OHC inhibited the growth of GBM cells in vitro. Efavirenz, a CYP46A1 activator capable of penetrating the blood-brain barrier, significantly inhibits the growth of tumors and prolongs survival in orthotopic xenograft mice [33]. LXRs are also agonistically regulated by oxysterols such as 24,25-epoxycholesterol produced by the cholesterol shunt synthesis pathway. The cholesterol precursor squalene produced in mevalonate pathway can be catalyzed to squalene-2,3-epoxide or 2,3,22,23-dioxidosqualene by SQLE, and squalene-2,3-epoxide can be catalyzed to lanosterol or 24(s),25-epoxylanosterol by LSS, the pathway that generates Lanosterol is the way to biosynthesize cholesterol, the process that generates 2,3,22,23-dioxidosqualene and 24(s),25-epoxylanosterol is called the shunt pathway. In the shunt pathway, 2,3,22,23-dioxidosqualene is catalyzed to 24(s),25-epoxylanosterol by LSS, and 24(s),25-epoxylanosterol is ultimately catalyzed to 24,25-epoxycholesterol through a series of catalytic reactions. Menin is a scaffold protein that functions in histone modification and epigenetic gene regulation. Inhibition of the Menin (MEN1) and MLL (MLL1, KMT2A) interaction is a potential therapeutic strategy for MLL-rearranged (MLL-r) leukemia [38]. A study found that MI-2, a small molecule menin inhibitor, inhibits the viability of GBM cells in vitro. The underlying mechanism is that MI-2 inhibits LSS and causes the cholesterol synthesis pathway to shift toward the shunt pathway. The accumulation of 24,25-epoxycholesterol produced by the shunt pathway stimulates LXR, which prompts the clearance of cellular cholesterol. MI-2 disrupts cholesterol homeostasis in GBM, eventually leading to cell death [39]. LXRα is also regulated by signals other than oxysterols in GBM. Fang et al. found that the activation of the EGFR/SRC/ERK pathway in GBM promotes the expression of YT521-B homology (YTH) domain-containing family protein 2 (YTHDF2), and YTHDF2 promotes tumor invasion by downregulating LXRα in an m6A-dependent mRNA decay manner, suggesting that YTHDF2 is a potential target for GBM therapy [40].
Although many studies have shown that activating LXR is an effective target for killing GBM cells, Patel et al. have found that LXRβ maintains the cholesterol homeostasis of GBM cells during high-density growth in vitro by upregulating ABCA1 and inhibiting the mevalonate pathway and that LXRβ maintains the cell viability during high-density growth in an ABCA1-independent manner. These results may be related to the ability of LXRβ to regulate the cell immune response and lipid metabolism [41]. LXRs may exhibit oncogenic functions in conditions lacking nutrients and oxygen. Thus, the complex mechanisms of LXR in GBM need to be further investigated.
Oxysterols
Oxysterols are produced by enzymatic or nonenzymatic oxidation of cholesterol. Oxysterols can be roughly divided into two categories: those oxygenated on the sterol ring at the 7 position such as 7α/β-hydroperoxy-cholesterol (7α/βOOHC), 7-ketocholesterol (7KC), and 7α/β hydroxycholesterol (7α/βHC) with a nonenzymatic origin, and products that are oxidized on side chains such as 24S-hydroxycholesterol (24OHC), 25-hydroxycholesterol (25OHC), 27-hydroxycholesterol (27OHC), and 22-hydroxycholesterol (22OHC), which are mainly produced by enzymes [42]. 27OHC, which is catalyzed by CYP27A1, is the most abundant oxysterol in cell membranes and blood. CYP46A1 is the most highly expressed oxysterol synthase in the brain; thus, the 24OHC produced by the catalysis by CYP46A1 is called “cerebrosterol” [43]. Oxysterols are major activators of LXR. Thus, oxysterols are considered to exert antitumor effects on GBM (Figure 2). In addition, oxysterols such as 24OHC/25OHC can also inhibit cholesterol synthesis by inhibiting SREBP [33,44].
Role of lipid rafts, cholesterol precursors and oxysterols in the treatment and pathogenesis of GBM. FDFT1: farnesyl-diphosphate farnesyltransferase 1, LSS: Lanosterol synthase, EBI2: G protein-coupled receptor EBV-induced gene 2. The first step of de novo synthesis of cholesterol is HMG-CoA converted to mevalonate by HMGCR. Mevalonate converted to IPP by a series of catalytic reactions, IPP is converted to FPP by FDPS, FPP is converted to squalene by FDFT1, squalene is converted to 2,3-epoxysqualene by SQLE, 2,3-epoxysqualene is converted to lanosterol by LSS. LSS is converted to cholesterol by a series of catalytic reactions. FDPS is involved in maintenance of GBM stemness. Precursors of cholesterol such as FPP and GGPP and play a role in tumorigenesis. IPP activates γδ T cells, which may enhance tumor immunotherapy. Oxysterols are metabolites of cholesterol and have anti-tumor effects on GBM such as activate LXRs, trigger energy stress, induce ROS overproduction, activate apoptosis. However, 25OHC promotes tumor progression by acting as a chemokine to promote the recruitment of tumor-related macrophages. Cholesterol maintains the integrity of Lipid raft. On the one hand, Lipid raft promote invasion and migration of GBM by CD44, connexin43 and TRPC1 and promote progression by spread of microvesicles. On the other hand, lipid rafts act as a channel for delivering drugs and mediate apoptosis signal transduction.
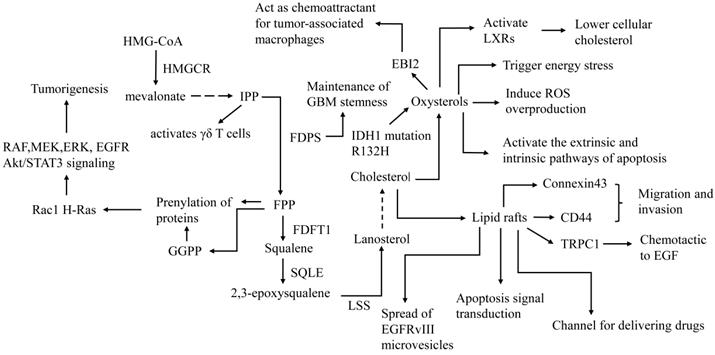
27OHC inhibits cell viability and induces apoptosis by reducing cholesterol in C6 glioma cells [45]. Zhu et al. found that the R132H mutation of IDH1, which is a common feature of a major subset of human gliomas, promotes U87 cell proliferation, cell growth, and cell migration. Expression of R132H mutational IDH1 upregulates SREBP1a and its several downstream genes [46]. Yang et al. further confirmed that mutant IDH1 enhances 24OHC production, which activates LXR and leads to the inhibition of GBM cholesterol uptake, and cellular cholesterol reduction activates the SREBP pathway, thus stimulating cholesterol de novo synthesis, which endowed IDH1-mutant glioma cells with sensitivity to statins [47]. This suggests that mutant IDH1 may be a biomarker for sensitivity to statin treatment.
In addition to lowering cellular cholesterol, oxysterols inhibit GBM growth through multiple mechanisms. A study found that 7β-OHC induces ROS overproduction in C6 glioma cells, resulting in apoptotic death [48]. The cholesterol metabolite pregnenolone, which is a precursor of various important steroid hormones, induces GBM cell death in a caspase-dependent manner in vitro, which is mediated by activation of the extrinsic and intrinsic apoptotic pathways [49]. Clarioin et al. demonstrated that 7β-HC exerts cytotoxicity in GBM cells via the accumulation of 7β-HC esters in lipid rafts, which triggers energy stress, activates a variety of signaling pathways, such as ERK, AMP-activated protein kinase (AMPK) and PI3K/Akt, and finally activates the P38 signaling pathway, leading to cell death [50]. Nevertheless, Eibinger et al. found that 25OHC acts as a chemokine to promote the recruitment of tumor-related macrophages [51], suggesting that 25OHC may be related to tumorigenesis and tumor progression. Oxysterols are involved in various cellular biological processes, such as cholesterol metabolism, cell immunity, cell injury, and tumorigenesis. Due to the ability to traverse the blood-brain barrier, oxysterols act as a link between the periphery and the CNS in cholesterol metabolism and represent an important target in metabolic therapy.
Lipid rafts
Lipid rafts are microdomains on the cell membrane with a variety of functions, such as being involved in transmembrane cell signaling pathways, mediating cell endocytosis/exocytosis, and providing membrane scaffolding for protein interactions. Cholesterol plays an important role in maintaining the integrity of lipid rafts. Lipid rafts are involved in the migration and invasion of GBM. Murai et al. demonstrated that CD44, which is located in lipid rafts, promotes the migration of GBM. Methyl-β-cyclodextrin (MCD) (a membrane cholesterol depletor that is widely used to disrupt the integrity of lipid rafts) induces CD44 shedding from the cell membrane, thus inhibiting cell migration. Simvastatin, an HMGCR inhibitor, inhibits the migration of GBM cells by destroying cell membrane lipid rafts by reducing cellular cholesterol [52]. Strale et al. found that Connexin43 promotes the invasion of GBM via lipid raft-dependent gap-junctional intercellular communication (GJIC) between cancer and normal parenchymal cells [53]. Bomben et al. demonstrated that transient receptor potential canonical 1 (TRPC1) colocalizes with lipid rafts in cells. Gliomas are attracted in a chemotactic manner to epidermal growth factor (EGF) via the TRPC1 channel, which depends on the integrity of lipid rafts [54]. Lipid rafts also mediate the spread of oncogenic receptor EGFRvIII microvesicles (“oncosomes”) between tumor cells and promote the malignant transformation of tumor cells lacking EGFRvIII [55]. The above studies suggest that destroying the integrity of cell lipid rafts is an effective way to suppress the migration and invasion of GBM.
However, cancer-targeted drugs such as liposome-packaged drugs and nanoparticles rely on lipid raft-mediated endocytosis to exert lethal effects on GBM [56-61]. Moreover, antitumor drugs such as arachidonoylethanolamide (AEA) may cross the membrane through cholesterol-rich lipid rafts. The accumulation of anandamide may lead to an increase in cellular ROS, which in turn triggers the apoptosis-inducing signaling cascade. Disruption of lipid rafts prevents anandamide-induced apoptosis [62]. Lipid rafts also mediate apoptosis signaling. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) activates death receptor 5 (DR5) and recruits Fas-associated death domain (FADD) and caspase-8 for the formation of a death-inducing signaling complex (DISC). Cleavage of caspase-8 in DISC then initiates downstream effector caspases such as caspase-3 to mediate GBM apoptosis, and the formation of DISC requires the integration of lipid rafts [63]. Y Yamamoto et al. further found that the cellular cholesterol content of a temozolomide (TMZ)-resistant GBM cell line was lower than that of a TMZ-sensitive GBM cell line. Increasing cellular cholesterol enhances TMZ-induced GBM cell death through the DR5-mediated extrinsic apoptotic pathway, and clinical statin concentrations may weaken TMZ-induced GBM cell death [64,65]. Intriguingly, disruption of lipid rafts can also trigger apoptosis in GBM. Wu et al. discovered that simvastatin promotes GBM tumor cell apoptosis and inhibits cell proliferation and migration. Mechanistically, simvastatin reduces the cholesterol level of the cell membrane, which destroys the integrity of lipid rafts, promotes Fas translocation into lipid raft fractions, leads to downregulation of the PI3K/Akt signaling pathway, and results in caspase-3-dependent apoptosis of GBM in vitro [66]. Lipid rafts not only promote cancer progression by mediating migration and invasion but also mediate apoptosis signal transduction (Figure 2). Otherwise, lipid rafts act as a channel for delivering drugs. Although cholesterol-lowering drugs may inhibit the invasion of GBM by disrupting lipid rafts, they can also reduce the efficacy of liposome-packaged and nanoparticle drugs and increase the risk of tumor resistance. The complex regulatory network of lipid rafts and its mechanism in GBM are not fully understood at present, and more research is needed to clarify the mechanism.
Disruption of cellular cholesterol trafficking
Cellular trafficking of cholesterol is of great significance for maintaining cholesterol homeostasis. Cholesterol transport disorder is closely associated with autophagy and apoptosis. Many studies have demonstrated that impairing the release of cholesterol from lysosomes effectively induces GBM cell antiproliferative autophagy (Figure 3). Loperamide and pimozide, an opioid receptor agonist and an antipsychotic agent, respectively, induce autophagy-dependent cell death in MZ-54 GBM cells in an autophagy related 5 and autophagy related 7 (ATG5 and ATG7)-dependent manner. Sphingomyelin phosphodiesterase 1 (SMPD1) is the enzyme that catalyzes sphingomyelin to phosphorylcholine and ceramide. Loperamide and pimozide impair lysosomals' function and induce accumulation of ceramides in lysosomal by inhibiting SMPD1. Ceramides and their hexosylmetabolites contribute to the disruption of lysosomal degradation. The accumulation of cholesterol in the dysfunctional lysosomes caused by these drugs leads to lysosomal membrane damage due to increased oxidative stress, thus resulting in the induction of lysosomal membrane permeabilization (LMP) and the release of CTSB (cathepsin B) into the cytosol, which eventually promotes autophagy and cell death [67].
Metabolic therapy targeting cholesterol trafficking in GBM. Cholesterol transport disorder is closely associated with autophagy and apoptosis. Archazolid B inhibits NPC1, which hinders the release of cholesterol from lysosomes, resulting in inhibition of cell viability. Loperamide and pimozide impair lysosomals' function and induce accumulation of ceramides in lysosomal by inhibiting SMPD1. Ceramides and their hexosylmetabolites contribute to the disruption of lysosomal degradation. The accumulation of cholesterol in the dysfunctional lysosomes caused by these drugs leads to lysosomal membrane damage due to increased oxidative stress, resulting in the induction of lysosomal membrane permeabilization and the release of cathepsin B into the cytosol, which eventually promotes autophagy and cell death. Alkylphospholipids such as perifosine, edelfosine, erucylphosphocholine interfere with cholesterol trafficking from the plasma membrane to the endoplasmic reticulum, hindering cholesterol esterification, Accumulation of unesterified cholesterol in the cell leads to autophagy, which inhibits the viability of GBM cells. SCP2 binds to cholesterol with high affinity and is involved in transporting cytoplasmic cholesterol to the plasma membrane. Itraconazole interferes with the transport of cholesterol from endosomes and lysosomes to the cell membrane by inhibiting the transcription of SCP2.
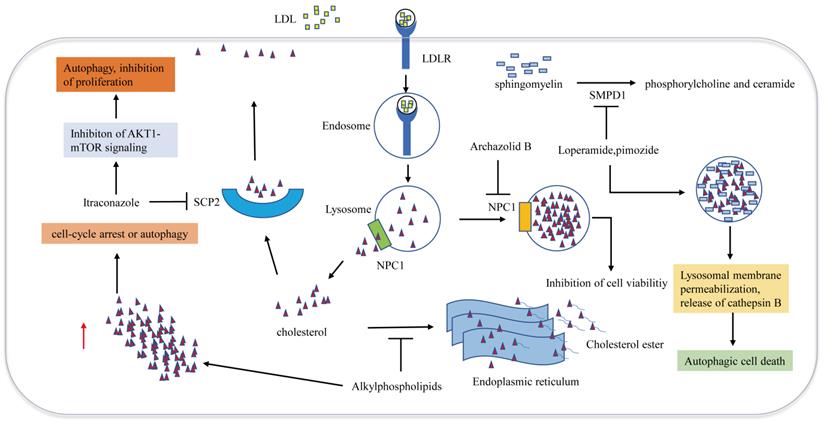
Niemann-Pick disease type 1/2 (NPC1/NPC2) is responsible for the transport of LDL-derived cholesterol out of the lysosome. Archazolid B is a highly cytotoxic vacuolar H+-ATPase (V-ATPase) inhibitor that inhibits NPC1 by impairing proton transport and elevating lysosomal pH levels, resulting in disturbances in the trafficking of plasma membrane-derived cholesterol to the endoplasmic reticulum. LDL-derived cholesterol trapped in lysosomes imitates the absence of cholesterol uptake, thus inducing the inhibition of cell viability [68]. Since a study has demonstrated that NPC2 is an unfavorable prognostic biomarker in GBM [69], NPC2 may also become a potential target for the treatment of GBM. Ríos-Marco et al. found that alkylphospholipids such as perifosine, edelfosine, erucylphosphocholine (ErPC) and hexadecylphosphocholine (HePC) interfere with cholesterol trafficking from the plasma membrane to the endoplasmic reticulum, hindering cholesterol esterification. Unesterified cholesterol in the cell leads to autophagy, which inhibits the viability of GBM cells [70]. Cleaved sterol carrier protein 2 (SCP2) binds to cholesterol with high affinity and is involved in transporting cytoplasmic cholesterol to the plasma membrane. Itraconazole, an antifungal drug, interferes with the transport of cholesterol from endosomes and lysosomes to the cell membrane by inhibiting the transcription of SCP2. AKT1-mTOR (Mechanistic target of rapamycin signaling) is suppressed due to the decreased level of cholesterol in the cell membrane, resulting in elevated antiproliferation of autophagosomes [71]. Another antifungal drug Luliconazole, was found to inhibit sphere growth and viability of glioma-initiating cells (GICs) in vitro and inhibit tumor growth and parenchymal infiltration in brain explants, cholesterol rescued sphere growth in the presence of luliconazole [72].
Cellular transport of cholesterol is involved in cell autophagy. Although the role of autophagy in regulating tumor cell survival or death is still complex and controversial, the above studies suggest that disruption of the cellular trafficking of cholesterol to induce GBM autophagy may be an effective approach for killing tumors (Figure 3).
Precursors of cholesterol
FPP and geranylgeranyl pyrophosphate (GGPP), precursors of cholesterol in the de novo pathway, play an important role in the prenylation of proteins that are known to be involved in the pathogenesis and progression of some cancers (Figure 2) [73]. For example, prenylation of small Rho GTPases such as Rac1 H-Ras with FPP and GGPP enables their localization to membranes, which is essential for activating direct downstream effectors (e.g. rapidly accelerated fibrosarcoma RAF, mitogen-activated protein kinase kinase MEK, and ERK) to promote tumorigenesis [74]. Studies have found that the expression of FDPS in glioma tissue is elevated and that FDPS is positively correlated with the expression of oncogenes such as Signal transducer and activator of transcription 3 (STAT3), ERK and AKT [75]. FPPS attenuates paclitaxel-induced apoptotic cell death in U87MG cells by blocking the c-Jun N-terminal kinase (JNK) signaling cascade and activating mevalonate metabolism [76]. The above studies show that the mevalonate pathway is an oncogenic signaling pathway that may represent a potential therapeutic target for GBM. N6-Benzyladenosine (i6A) derivatives inhibit GBM cell viability by inhibiting FDPS in vitro [77]. The i6A analog CM223 selectively inhibits the activity of U87MG without affecting NHAs. The underlying mechanism is that CM223 disrupts prenylation by inhibiting FPPS, which leads to downregulation of EGFR and Akt/STAT3 signaling [78]. Additionally, FPP is also the precursor of the Coenzyme Q (CoQ), an enzyme that plays a central role in the mitochondrial electron transport chain. I Liparulo et al. found the 4-nitrobenzoate (4-NB), an inhibitor of CoQ biosynthesis, significantly increased the cholesterol content in glioma cell, resulting in decreased plasma membrane fluidity. Furthermore, Reduced level of oxygen content caused by cholesterol overproduction and increased ROS levels caused by CoQ depletion, synergistically stabilized HIF-1α, which driving metabolic switch to glycolysis in glioma [79].
Moreover, enzymes in the mevalonate pathway participate in the maintenance of GBM stemness. Cancer stem-like cells (CSLCs) of GBM possess a unique lipid metabolomic profile. Lanosterol synthas (LSS), SCD and HMGCS1 may be critical for CSLC enrichment and survival [80]. FDPS also has been found to play an essential role in the maintenance of glioblastoma stemness, and the zoledronate, FDPS inhibitors, significantly inhibit the formation of glioblastoma spheres [81].
γδT lymphocytes are innate immune cells that can be found in situ as tumor-infiltrating lymphocytes and are able to recognize and kill several cancer cells in vitro. Cimini et al. demonstrated that zoledronic acid (ZOL) is able to block FPPS, thus inducing the accumulation of IPP, which is able to activate γδ T cells (Figure 2) [82]. These results indicate that FPPS inhibitors represent potential sensitizers for GBM immunotherapy, providing a novel approach of combined immune/chemotherapy for GBM management.
Statins in GBM
Statins inhibit the growth of GBM by inhibiting HMGCR and reducing the production of intermediate products of the mevalonate pathway, such as IPP, FPP, and GGPP. Yanae et al. discovered that statins (mevastatin, fluvastatin, or simvastatin) inhibit GGPP production, leading to inhibition of ERK1/2 and Akt activation and thus inducing apoptosis of C6 glioma cells [83]. Afshordel et al. further confirmed that the reduction in membrane-bound H-Ras and small GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) levels diminishes ERK signaling [84]. Oliveira et al. showed that atorvastatin reduces GBM cell migration and proliferation in vitro, whereas no toxicity was observed in astrocytes. Atorvastatin also exerts cytotoxicity by partly preventing antagonism of ionotropic and metabotropic glutamate receptors [85]. Yi et al. found that atorvastatin suppresses the invasion and migration of GBM cells by inhibiting microglial MT1-MMP expression and that atorvastatin may inhibit microglial MT1-MMP expression by inhibiting the p38 MAPK pathway [86]. A study found that simvastatin increases temozolomide-induced GBM cell death. TMZ induces GBM cell autophagy, and simvastatin blocks the fusion of autophagosomes and lysosomes, which results in the accumulation of autophagosomes. The accumulation of autophagosomes eventually leads to the potentiation of TMZ-induced apoptosis in GBM cells [87]. However, the abovementioned study suggests that the use of statins may increase the resistance to TMZ. Comparing the two studies, they used the same cell line U251 and the same simvastatin concentration of 1 μM but got the opposite outcomes. Thus, whether statins can enhance the TMZ-induced tumor cell death remains controversial.
Although many studies have reported that statins inhibit the growth of GBM cells in vitro, there is still insufficient evidence to prove that statins benefit GBM patients. An analysis of a retrospective study that enrolled a cohort of 810 patients showed that statins could not improve the overall survival (OS) and progression-free survival (PFS) of GBM patients [88]. There is also a retrospective study that enrolled a cohort of 1,093 high-grade glioma patients showing that statins are not associated with improving OS and PFS in HGG patients [89].
In addition, the relationship between statins and the risk of glioma is controversial. According to a year of 2012 matched case-control study that included 517 cases and 400 population-based controls, simvastatin and lovastatin can reduce the risk of glioma [90]. A subsequent matched case-control study from Denmark that included 2,656 cases and 18,480 controls (matched on birth year and sex with population controls) supports this conclusion [91]. However, a case-control study from The Clinical Practice Research Database (CPRD) denied this conclusion. The study included 2,469 cases and 24,690 controls (matched on index date, age, sex, general practice, and number of years of active history in the database prior to the index date). The conclusion showed that compared with the nonuse of statins, the use of statins was not associated with the risk of glioma [92]. Moreover, according to a recent prospective cohort study, the use of statins is significantly associated with an increased risk of glioma compared with no statins [93]. Some studies showed statins exerted antitumor effects on GBM cells in vitro, whereas other studies believe that statins increase tumor resistance and even increase the risk of glioma. Therefore, the effects of statins on GBM need to be further explored.
Conclusion
Cholesterol is a component of cell membranes; thus, the immortal proliferation of malignant tumor cells will inevitably lead to an exuberant demand for cholesterol. GBM cells rely on the external uptake of cholesterol to maintain cholesterol homeostasis; thus, reducing cholesterol uptake has become an effective antitumor mechanism. LXR activators inhibit GBM viability by inhibiting the uptake of cholesterol and increasing the efflux of cholesterol. Inhibiting SREBP signaling and its downstream lipid metabolism genes leads to the disruption of GBM lipid metabolism homeostasis. Cholesterol esterification is an important means of storing cholesterol in cells, and LD accumulation is a prominent feature of the GBM cholesterol metabolism profile; thus, SOAT1 inhibitors, such as avasimibe, exert antitumor effects on GBM by blocking LD formation. New approaches that target cholesterol metabolism in GBM are displayed in Table 1.
New approaches that target cholesterol metabolism in GBM
| Drugs | Targets | Mechanisms | References |
|---|---|---|---|
| PF-429242 Quercetin, phytol, retinol MicroRNA-132 MicroRNA -29 | S1P SREBP-1 SIRT1 SREBP-1/SCAP | Inhibit lipid and cholesterol synthesis | [19-23] |
| Avasimibe K604 L365260, YM022 | SOAT1 | Inhibit the cholesterol esterification, apoptotsis, inhibit Akt signaling | [24,26,30,31] |
| LXR-623, GW3965 4-7rr, 4-13,4-13rr Efavirenz MI-2 | LXRs LXRβ CYP46A1 LSS | Reduce the uptake of cholesterol and promote cholesterol efflux | [33,34,37,39] |
| Pregnenolone | Bcl-2, Fas/FasL | Activate the extrinsic and intrinsic apoptotic pathways | [49] |
| Loperamide, pimozide Archazolid B Itraconazole | SMPD1 NPC1 SCP2 | Disrupt the cellular cholesterol trafficking | [67,68,71] |
| i6A derivatives, CM223, ZOL | FDPS | Activate γδ T cells, inhibit EGFR and Akt/STAT3 signaling | [77,78,82] |
| Statins | HMGCR iGluRs mGluRs p38 MAPK pathway | Inhibit ERK1/2 and Akt signaling, Destroy cell membrane lipid rafts, Apoptosis, inhibit the MT1-MMP expression, Block the fusion of autophagosomes and lysosomes | [52,66,83-87] |
iGluRs: ionotropic glutamate receptors, mGluRs: metabotropic glutamate receptors. B cell lymphoma-2: Bcl-2, Fas: FS7-associated cell surface antigen, FasL: FS7-associated cell surface antigen ligand, LSS: Lanosterol synthase, MT1-MMP: Membrane-type 1 matrix metalloproteinase, CYP46A1: cytochrome P450 46A1, SMPD1: sphingomyelin phosphodiesterase 1.
FPPS and its catalytic products FPP and GGPP are involved in a variety of cancer-promoting signals related to GBM. Inhibiting the mevalonate pathway has made achievements in preclinical experiments. Oxysterols are LXR activators, which are essential negative feedback regulators of cholesterol metabolism. Oxysterols can also exert cytotoxicity by triggering ROS or inducing apoptosis signaling. Oxysterols are also related to the tumor immunity microenvironment. Lipid rafts promote invasion and metastasis of GBM and also play an important role in the transduction of apoptosis signaling and mediate the entry of liposome-packaged drugs into cells. Cholesterol maintains the integrity of lipid rafts; thus, disruption of cholesterol metabolism may impair lipid raft microstructure, which affects the behavior of cancer cells in various aspects. Cellular cholesterol trafficking disorder promotes autophagy, which subsequently induces apoptosis of GBM cells. Statins exert anti-GBM effects in vitro by a diversity of mechanisms, including inhibiting de novo pathways and inducing apoptosis signaling. However, whether GBM patients can benefit from the use of statins or whether statins can reduce the risk of glioma remains controversial.
There are many challenges in GBM therapy targeting cholesterol metabolism. First, the existence of the blood brain barrier limits the efficacy of many drugs. Second, because GBM and neurons have similar cholesterol metabolism features (relying on the external uptake of cholesterol), the side effects of GBM cholesterol metabolism treatment on neurons need to be studied in depth. Finally, individualized therapy needs to be further explored due to the heterogeneity of GBM. Vast Genetic alterations occurring in cholesterol pathways have been identified in cancer cells [4], whether tumors could be classified into subclasses based on genetic abnormalities occurring in cholesterol homeostasis genes is a new question, because the efficacy of inhibitors of cholesterol biogenesis might be more effective for certain patients with characteristic genetic signatures [8].
Cholesterol homeostasis is regulated by complex feedback loops. Inhibiting one pathway of cholesterol metabolism might have little effect on tumor growth, and the combination of different inhibitors that simultaneously block cholesterol synthesis, uptake, esterification, or trafficking in cancer might pave the way for next-generation metabolic therapies [94]. Recently K Bhat et al. found the radiation-treated glioma cells significantly upregulate the expression of cholesterol biosynthesis genes, Combining application of quetiapine (a dopamine receptor antagonist) with atorvastatin and radiation significantly increases the survival of patient-derived orthotopic xenograft mice [95]. Indicating cholesterol metabolism is involved in radiotherapy resistance and radiotherapy combined with metabolic therapy is a promising strategy to prolong patients' survival. In addition, GBM cholesterol metabolism is associated with the tumor immune microenvironment and tumor resistance. Metabolic remodeling profoundly impacts on the tumor microenvironment [96], which promotes tumor progression and immunosuppression [97]. Therefore, the combined use of metabolic therapies with chemotherapy and immunotherapy is also an affirmative approach. In conclusion, emerging experimental results show significant progress in GBM therapy targeting cholesterol metabolism. Continuing to clarify the cholesterol metabolism of GBM and develop a new generation of metabolic therapies may represent a promising path for improving the prognosis of GBM patients.
Abbreviations
GBM: Glioblastoma; CNS: central nervous system; LDL: low-density lipoprotein; LDLR: low-density lipoprotein receptor; HMGCS: HMG-CoA synthase; IPP: isopentenyl pyrophosphate; DPP: 3,3-dimethylallyl pyrophosphate; FPPS: farnesyl pyrophosphate synthase; FPP: farnesyl pyrophosphate; HMG-CoA: 3-hydroxy-3-methylglutaryl-coenzyme A; HMGCR: HMG-CoA reductase; SQLE: squalene epoxidase; SOAT: sterol O-acyltransferase; ACAT: acyl-CoA cholesterol acyltransferase-1; ABCA1: ATP-binding cassette transporter 1; NPC1: Niemann-Pick type C protein 1; LXRs: liver X receptors; SREBPs: sterol regulatory element-binding proteins; bHLH-LZ: basic-helix-loop-helix-leucine zipper; SCAP: SREBP cleavage activating protein; ER: endoplasmic reticulum; S1P: site 1 protease; S2P: site 2 protease; HMGCS: HMG-CoA synthetase; ACC: acetyl-CoA carboxylase; FAS: fatty acid synthase; SCD-1: stearoyl-CoA desaturase-1; EGFR/PI3K: epidermal growth factor receptor/phosphoinositide 3-kinase; FABP3: fatty acid-binding protein 3; SIRT1: silencing information regulator 2-related enzyme 1; LDs: lipid droplets; Akt: activation of Protein kinase B; CCK2R: cholecystokinin 2 receptor; PKC/ERK1/2: Protein kinase C/extracellular signal-regulated kinase1/2; LXRs: Liver X receptors; IDOL: inducible degrader of LDLR; HMGCS1: cytochrome P450 46A1 CYP46A1, 3-hydroxy-3-methylglutaryl-CoA synthase 1; DHCR24: 3β-hydroxysteroid-Δ24 reductase; NHAs: normal human astrocytes; YTH: YT521-B homology; YTHDF2: domain-containing family protein 2; 7α/βOOHC: 7α/β-hydroperoxy-cholesterol; 7KC: 7-ketocholesterol; 7α/βHC: 7α/β hydroxycholesterol; 24OHC: 24S-hydroxycholesterol; 25OHC: 25-hydroxycholesterol; 27OHC: 27-hydroxycholesterol; 22OHC: 22-hydroxycholesterol; ROS: reactive oxygen species; AMPK: AMP-activated protein kinase; MCD: Methyl-β-cyclodextrin; GJIC: gap-junctional intercellular communication; TRPC1: canonical 1; EGF: epidermal growth factor; AEA: arachidonoylethanolamide; TRAIL: factor-related apoptosis-inducing ligand; DR5: death receptor 5; FADD: Fas-associated death domain; DISC: death-inducing signaling complex; TMZ: temozolomide; ATG5 and ATG7: autophagy related 5 and autophagy related 7; LMP: lysosomal membrane permeabilization; CTSB: cathepsin B; NPC1/NPC2: Niemann-Pick disease type 1/2; ErPC: erucylphosphocholine; HePC: hexadecylphosphocholine; SCP2: sterol carrier protein 2; mTOR: Mechanistic target of rapamycin signaling; GGPP: geranylgeranyl pyrophosphate; STAT3: Signal transducer and activator of transcription 3; RAF: rapidly accelerated fibrosarcoma; MEK: mitogen-activated protein kinase kinase; JNK: c-Jun N-terminal kinase; i6A: N6-Benzyladenosine; CSLCs: Cancer stem-like cells; LSS: Lanosterol synthase; ZOL: zoledronic acid; RAC1: small GTPase Ras-related C3 botulinum toxin substrate 1; GTPase H-Ras: HRas Proto-Oncogene; OS: overall survival; PFS: progression-free survival; CPRD: Clinical Practice Research Database.
Acknowledgements
Thanks to the Fifth Affiliated Hospital of Zhengzhou University.
Funding
This study was supported by the National Natural Science Foundation of China: 81972361.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. Apr. 2020;21(4):225-245
2. Espinosa G, López-Montero I, Monroy F, Langevin D. Shear rheology of lipid monolayers and insights on membrane fluidity. Proc Natl Acad Sci U S A. Apr 12. 2011;108(15):6008-6013
3. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. Feb. 2011;11(2):85-95
4. Murai T. Cholesterol lowering: role in cancer prevention and treatment. Biological chemistry. Jan. 2015;396(1):1-11
5. Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biological chemistry. Apr. 2009;390(4):287-293
6. Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arteriosclerosis, thrombosis, and vascular biology. May. 2004;24(5):806-815
7. Lütjohann D, Breuer O, Ahlborg G. et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A. Sep 3. 1996;93(18):9799-9804
8. Ahmad F, Sun Q, Patel D, Stommel JM. Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers (Basel). Jan 26. 2019 11(2)
9. Pinacho-Garcia LM, Valdez RA, Navarrete A, Cabeza M, Segovia J, Romano MC. The effect of finasteride and dutasteride on the synthesis of neurosteroids by glioblastoma cells. Steroids. Mar. 2020;155:108556
10. Schultz JR, Tu H, Luk A. et al. Role of LXRs in control of lipogenesis. Genes & development. Nov 15. 2000;14(22):2831-2838
11. Repa JJ, Liang G, Ou J. et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes & development. Nov 15. 2000;14(22):2819-2830
12. Laffitte BA, Chao LC, Li J. et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci U S A. Apr 29. 2003;100(9):5419-5424
13. Hua X, Sakai J, Ho YK, Goldstein JL, Brown MS. Hairpin orientation of sterol regulatory element-binding protein-2 in cell membranes as determined by protease protection. The Journal of biological chemistry. Dec 8. 1995;270(49):29422-29427
14. Nohturfft A, Brown MS, Goldstein JL. Topology of SREBP cleavage-activating protein, a polytopic membrane protein with a sterol-sensing domain. The Journal of biological chemistry. Jul 3. 1998;273(27):17243-17250
15. Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. Apr 8. 1994;77(1):53-62
16. Sakai J, Rawson RB, Espenshade PJ. et al. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell. Oct. 1998;2(4):505-514
17. Guo D, Reinitz F, Youssef M. et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer discovery. Oct. 2011;1(5):442-456
18. Lewis CA, Brault C, Peck B. et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. Oct 1. 2015;34(40):5128-5140
19. Caruana BT, Skoric A, Brown AJ, Lutze-Mann LH. Site-1 protease, a novel metabolic target for glioblastoma. Biochem Biophys Res Commun. Aug 26. 2017;490(3):760-766
20. Facchini G, Ignarro RS, Rodrigues-Silva E. et al. Toxic effects of phytol and retinol on human glioblastoma cells are associated with modulation of cholesterol and fatty acid biosynthetic pathways. J Neurooncol. Feb. 2018;136(3):435-443
21. Damiano F, Giannotti L, Gnoni GV, Siculella L, Gnoni A. Quercetin inhibition of SREBPs and ChREBP expression results in reduced cholesterol and fatty acid synthesis in C6 glioma cells. The international journal of biochemistry & cell biology. Dec. 2019;117:105618
22. Li Y, Zhang J, He J, Zhou W, Xiang G, Xu R. MicroRNA-132 cause apoptosis of glioma cells through blockade of the SREBP-1c metabolic pathway related to SIRT1. Biomed Pharmacother. Mar. 2016;78:177-184
23. Ru P, Hu P, Geng F. et al. Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth. Cell Rep. Aug 9. 2016;16(6):1527-1535
24. Geng F, Cheng X, Wu X. et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin Cancer Res. Nov 1. 2016;22(21):5337-5348
25. Chi KC, Tsai WC, Wu CL, Lin TY, Hueng DY. An Adult Drosophila Glioma Model for Studying Pathometabolic Pathways of Gliomagenesis. Molecular neurobiology. Jun. 2019;56(6):4589-4599
26. Bemlih S, Poirier MD, El Andaloussi A. Acyl-coenzyme A: cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer biology & therapy. Jun 15. 2010;9(12):1025-1032
27. Luo Y, Liu L, Li X, Shi Y. Avasimibe inhibits the proliferation, migration and invasion of glioma cells by suppressing linc00339. Biomed Pharmacother. Oct. 2020;130:110508
28. Yang W, Bai Y, Xiong Y. et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. Mar 31. 2016;531(7596):651-655
29. Hao M, Hou S, Li W. et al. Combination of metabolic intervention and T cell therapy enhances solid tumor immunotherapy. Sci Transl Med. Nov 25. 2020 12(571)
30. Ohmoto T, Nishitsuji K, Yoshitani N. et al. K604, a specific acyl-CoA:cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251-MG glioblastoma cells. Molecular medicine reports. Oct. 2015;12(4):6037-6042
31. Paillasse MR, de Medina P, Amouroux G, Mhamdi L, Poirot M, Silvente-Poirot S. Signaling through cholesterol esterification: a new pathway for the cholecystokinin 2 receptor involved in cell growth and invasion. Journal of lipid research. Nov. 2009;50(11):2203-2211
32. Tardif JC, Grégoire J, L'Allier PL. et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. Nov 23. 2004;110(21):3372-3377
33. Han M, Wang S, Yang N. et al. Therapeutic implications of altered cholesterol homeostasis mediated by loss of CYP46A1 in human glioblastoma. EMBO Mol Med. Jan 9. 2020;12(1):e10924
34. Villa GR, Hulce JJ, Zanca C. et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell. Nov 14. 2016;30(5):683-693
35. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. May 22. 2009;324(5930):1029-1033
36. Nguyen TTT, Ishida CT, Shang E. et al. Activation of LXRbeta inhibits tumor respiration and is synthetically lethal with Bcl-xL inhibition. EMBO Mol Med. Oct. 2019;11(10):e10769
37. Chen H, Chen Z, Zhang Z. et al. Discovery of new LXRbeta agonists as glioblastoma inhibitors. Eur J Med Chem. May 15. 2020;194:112240
38. Krivtsov AV, Evans K, Gadrey JY. et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell. Dec 9. 2019;36(6):660-673.e611
39. Phillips RE, Yang Y, Smith RC. et al. Target identification reveals lanosterol synthase as a vulnerability in glioma. Proc Natl Acad Sci U S A. Apr 16. 2019;116(16):7957-7962
40. Fang R, Chen X, Zhang S. et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun. Jan 8. 2021;12(1):177
41. Patel D, Ahmad F, Kambach DM. et al. LXRbeta controls glioblastoma cell growth, lipid balance, and immune modulation independently of ABCA1. Sci Rep. Oct 29. 2019;9(1):15458
42. Bovenga F, Sabbà C, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell metabolism. Apr 7. 2015;21(4):517-526
43. Björkhem I, Lütjohann D, Diczfalusy U, Ståhle L, Ahlborg G, Wahren J. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. Journal of lipid research. Aug. 1998;39(8):1594-1600
44. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. Jan 13. 2006;124(1):35-46
45. An Y, Zhang DD, Yu HL. et al. 27-Hydroxycholesterol regulates cholesterol synthesis and transport in C6 glioma cells. Neurotoxicology. Mar. 2017;59:88-97
46. Zhu J, Cui G, Chen M. et al. Expression of R132H mutational IDH1 in human U87 glioblastoma cells affects the SREBP1a pathway and induces cellular proliferation. Journal of molecular neuroscience: MN. May. 2013;50(1):165-171
47. Yang R, Zhao Y, Gu Y. et al. Isocitrate dehydrogenase 1 mutation enhances 24(S)-hydroxycholesterol production and alters cholesterol homeostasis in glioma. Oncogene. Oct. 2020;39(40):6340-6353
48. Sassi K, Nury T, Zarrouk A. et al. Induction of a non-apoptotic mode of cell death associated with autophagic characteristics with steroidal maleic anhydrides and 7β-hydroxycholesterol on glioma cells. The Journal of steroid biochemistry and molecular biology. Jul. 2019;191:105371
49. Xiao X, Chen L, Ouyang Y. et al. Pregnenolone, a cholesterol metabolite, induces glioma cell apoptosis via activating extrinsic and intrinsic apoptotic pathways. Oncol Lett. Aug. 2014;8(2):645-650
50. Clarion L, Schindler M, de Weille J. et al. 7β-Hydroxycholesterol-induced energy stress leads to sequential opposing signaling responses and to death of C6 glioblastoma cells. Biochem Pharmacol. Jan 1. 2012;83(1):37-46
51. Eibinger G, Fauler G, Bernhart E. et al. On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines. Implications for chemotactic monocyte recruitment. Experimental cell research. Jul 15. 2013;319(12):1828-1838
52. Murai T, Maruyama Y, Mio K, Nishiyama H, Suga M, Sato C. Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. The Journal of biological chemistry. Jan 21. 2011;286(3):1999-2007
53. Strale PO, Clarhaut J, Lamiche C, Cronier L, Mesnil M, Defamie N. Down-regulation of Connexin43 expression reveals the involvement of caveolin-1 containing lipid rafts in human U251 glioblastoma cell invasion. Mol Carcinog. Nov. 2012;51(11):845-860
54. Bomben VC, Turner KL, Barclay TT, Sontheimer H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J Cell Physiol. Jul. 2011;226(7):1879-1888
55. Al-Nedawi K, Meehan B, Micallef J. et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nature cell biology. May. 2008;10(5):619-624
56. Han B, Xie W, Zhang Y. et al. The influx/efflux mechanisms of d-peptide ligand of nAChRs across the blood-brain barrier and its therapeutic value in treating glioma. J Control Release. Nov 10. 2020;327:384-396
57. Shao K, Hou Q, Go ML. et al. Sulfatide-tenascin interaction mediates binding to the extracellular matrix and endocytic uptake of liposomes in glioma cells. Cell Mol Life Sci. Feb. 2007;64(4):506-515
58. Gu G, Gao X, Hu Q. et al. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials. Jul. 2013;34(21):5138-5148
59. Yang L, Li W, Huang Y, Zhou Y, Chen T. Rational Design of Cancer-Targeted Benzoselenadiazole by RGD Peptide Functionalization for Cancer Theranostics. Macromol Rapid Commun. Sep. 2015;36(17):1559-1565
60. Melamed JR, Ioele SA, Hannum AJ, Ullman VM, Day ES. Polyethylenimine-Spherical Nucleic Acid Nanoparticles against Gli1 Reduce the Chemoresistance and Stemness of Glioblastoma Cells. Mol Pharm. Nov 5. 2018;15(11):5135-5145
61. Gu G, Hu Q, Feng X. et al. PEG-PLA nanoparticles modified with APTEDB peptide for enhanced anti-angiogenic and anti-glioma therapy. Biomaterials. Sep. 2014;35(28):8215-8226
62. Sarker KP, Maruyama I. Anandamide induces cell death independently of cannabinoid receptors or vanilloid receptor 1: possible involvement of lipid rafts. Cell Mol Life Sci. Jun. 2003;60(6):1200-1208
63. Bellail AC, Tse MC, Song JH. et al. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J Cell Mol Med. Jun. 2010;14(6A):1303-1317
64. Yamamoto Y, Sasaki N, Kumagai K. et al. Involvement of Intracellular Cholesterol in Temozolomide-Induced Glioblastoma Cell Death. Neurol Med Chir (Tokyo). Jul 15. 2018;58(7):296-302
65. Yamamoto Y, Tomiyama A, Sasaki N. et al. Intracellular cholesterol level regulates sensitivity of glioblastoma cells against temozolomide-induced cell death by modulation of caspase-8 activation via death receptor 5-accumulation and activation in the plasma membrane lipid raft. Biochem Biophys Res Commun. Jan 1. 2018;495(1):1292-1299
66. Wu H, Jiang H, Lu D. et al. Effect of simvastatin on glioma cell proliferation, migration, and apoptosis. Neurosurgery. Dec. 2009;65(6):1087-1096 discussion 1096-1087
67. Meyer N, Henkel L, Linder B. et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy. Jan 19. 2021:1-20
68. Hamm R, Zeino M, Frewert S, Efferth T. Up-regulation of cholesterol associated genes as novel resistance mechanism in glioblastoma cells in response to archazolid B. Toxicology and applied pharmacology. Nov 15. 2014;281(1):78-86
69. Wei D, Shen S, Lin K. et al. NPC2 as a Prognostic Biomarker for Glioblastoma Based on Integrated Bioinformatics Analysis and Cytological Experiments. Frontiers in genetics. 2021;12:611442
70. Ríos-Marco P, Martín-Fernández M, Soria-Bretones I, Ríos A, Carrasco MP, Marco C. Alkylphospholipids deregulate cholesterol metabolism and induce cell-cycle arrest and autophagy in U-87 MG glioblastoma cells. Biochimica et biophysica acta. Aug. 2013;1831(8):1322-1334
71. Liu R, Li J, Zhang T. et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking. Autophagy. Jul. 2014;10(7):1241-1255
72. Nagashima H, Koike N, Yoshida K, Saya H, Sampetrean O. Antifungal Agent Luliconazole Inhibits the Growth of Mouse Glioma-initiating Cells in Brain Explants. The Keio journal of medicine. Dec 25. 2020;69(4):97-104
73. McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. Feb. 2006;63(3):255-267
74. Lo HW. Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Current cancer drug targets. Dec. 2010;10(8):840-848
75. Abate M, Laezza C, Pisanti S. et al. Deregulated expression and activity of Farnesyl Diphosphate Synthase (FDPS) in Glioblastoma. Sci Rep. Oct 26. 2017;7(1):14123
76. Woo IS, Eun SY, Kim HJ. et al. Farnesyl diphosphate synthase attenuates paclitaxel-induced apoptotic cell death in human glioblastoma U87MG cells. Neurosci Lett. Apr 26. 2010;474(2):115-120
77. Grimaldi M, Randino R, Ciaglia E. et al. NMR for screening and a biochemical assay: Identification of new FPPS inhibitors exerting anticancer activity. Bioorganic chemistry. May. 2020;98:103449
78. Ciaglia E, Grimaldi M, Abate M. et al. The isoprenoid derivative N(6) -benzyladenosine CM223 exerts antitumor effects in glioma patient-derived primary cells through the mevalonate pathway. British journal of pharmacology. Jul. 2017;174(14):2287-2301
79. Liparulo I, Bergamini C, Bortolus M. et al. Coenzyme Q biosynthesis inhibition induces HIF-1α stabilization and metabolic switch toward glycolysis. The FEBS journal. Mar. 2021;288(6):1956-1974
80. Song M, Lee H, Nam MH. et al. Loss-of-function screens of druggable targetome against cancer stem-like cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. Feb. 2017;31(2):625-635
81. Kim HY, Kim DK, Bae SH. et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Experimental & molecular medicine. Oct 17. 2018;50(10):1-12
82. Cimini E, Piacentini P, Sacchi A. et al. Zoledronic acid enhances Vδ2 T-lymphocyte antitumor response to human glioma cell lines. International journal of immunopathology and pharmacology. Jan-Mar. 2011;24(1):139-148
83. Yanae M, Tsubaki M, Satou T. et al. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J Exp Clin Cancer Res. Aug 10. 2011;30(1):74
84. Afshordel S, Kern B, Clasohm J. et al. Lovastatin and perillyl alcohol inhibit glioma cell invasion, migration, and proliferation-impact of Ras-/Rho-prenylation. Pharmacological research. Jan. 2015;91:69-77
85. Oliveira KA, Dal-Cim T, Lopes FG, Ludka FK, Nedel CB, Tasca CI. Atorvastatin Promotes Cytotoxicity and Reduces Migration and Proliferation of Human A172 Glioma Cells. Molecular neurobiology. Feb. 2018;55(2):1509-1523
86. Yongjun Y, Shuyun H, Lei C, Xiangrong C, Zhilin Y, Yiquan K. Atorvastatin suppresses glioma invasion and migration by reducing microglial MT1-MMP expression. J Neuroimmunol. Jul 15. 2013;260(1-2):1-8
87. Shojaei S, Koleini N, Samiei E. et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. The FEBS journal. Mar. 2020;287(5):1005-1034
88. Happold C, Gorlia T, Nabors LB. et al. Do statins, ACE inhibitors or sartans improve outcome in primary glioblastoma? J Neurooncol. May. 2018;138(1):163-171
89. Seliger C, Schaertl J, Gerken M. et al. Use of statins or NSAIDs and survival of patients with high-grade glioma. PloS one. 2018;13(12):e0207858
90. Ferris JS, McCoy L, Neugut AI, Wrensch M, Lai R. HMG CoA reductase inhibitors, NSAIDs and risk of glioma. Int J Cancer. Sep 15. 2012;131(6):E1031-1037
91. Gaist D, Andersen L, Hallas J, Sørensen HT, Schrøder HD, Friis S. Use of statins and risk of glioma: a nationwide case-control study in Denmark. British journal of cancer. Feb 19. 2013;108(3):715-720
92. Seliger C, Meier CR, Becker C. et al. Statin use and risk of glioma: population-based case-control analysis. European journal of epidemiology. Sep. 2016;31(9):947-952
93. Cote DJ, Rosner BA, Smith-Warner SA, Egan KM, Stampfer MJ. Statin use, hyperlipidemia, and risk of glioma. European journal of epidemiology. Nov. 2019;34(11):997-1011
94. Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochimica et biophysica acta. Reviews on cancer. Aug. 2020;1874(1):188394
95. Bhat K, Saki M, Cheng F. et al. Dopamine Receptor Antagonists, Radiation, and Cholesterol Biosynthesis in Mouse Models of Glioblastoma. J Natl Cancer Inst. Aug 2. 2021;113(8):1094-1104
96. Qiu R, Zhong Y, Li Q, Li Y, Fan H. Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct From Peripheral Tumors. Frontiers in cell and developmental biology. 2021;9:693215
97. Wegiel B, Vuerich M, Daneshmandi S, Seth P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front Oncol. 2018;8:284
Author contact
![]() Corresponding author: Xinjun Wang, M.D, Ph.D. Department of Neurosurgery, The Fifth Affiliated Hospital of Zhengzhou University, International Joint Laboratory of Glioma Metabolism and Microenvironment Research, Zhengzhou, Henan, 450052, China. E-mail: wangxjedu.cn.
Corresponding author: Xinjun Wang, M.D, Ph.D. Department of Neurosurgery, The Fifth Affiliated Hospital of Zhengzhou University, International Joint Laboratory of Glioma Metabolism and Microenvironment Research, Zhengzhou, Henan, 450052, China. E-mail: wangxjedu.cn.