Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(7):2126-2137. doi:10.7150/jca.69375 This issue Cite
Research Paper
BET inhibitor JQ1 enhances anti-tumor immunity and synergizes with PD-1 blockade in CRC
Huijin Wang1,2#, Guangyao Liu1,2#, Xinghan Jin1,2, Shenglei Song1,2, Songyao Chen1,2, Peiqing Zhou1, Huan Li1,2, Jianming Liang1,2, Bo Li1 ![]() , Changhua Zhang1,2
, Changhua Zhang1,2 ![]() , Yulong He1,2
, Yulong He1,2 ![]()
1. Digestive Diseases Center, The Seventh Affiliated Hospital of Sun Yat-Sen University, No.628, Zhenyuan Road, Guangming District, Shenzhen, 518107, China
2. Department of Gastrointestinal Surgery, The First Affiliated Hospital, Sun Yat-Sen University, 58 Zhongshan 2nd Road, Guangzhou, Guangdong, 510080, China
# These authors contributed equally to this work.
Received 2021-11-23; Accepted 2022-3-21; Published 2022-3-28
Abstract

Most colorectal cancer (CRC) patients are insensitive to immune checkpoint inhibitors (ICIs) due to the immunosuppressive tumor microenvironment (TME). Epigenetic factors such as the bromo-and extraterminal domain (BET) family proteins may be responsible for the immunosuppressive microenvironment. Previous studies have shown that inhibitors of BET family proteins have the potential to remodel the immunosuppressive TME. However, data on the role of BET inhibitors in immune microenvironment in CRC remains unclear. Here, we evaluated the immunoregulatory role of JQ1, a BET inhibitor, in CRC. Transcriptome sequencing data showed that JQ1 decreased CD274 expression and increased H2Kb expression in MC38 cells. Flow cytometry assays demonstrated that JQ1 decreased cell-surface PD-L1 expression in MC38 and HCT116 cells. Moreover, JQ1 significantly increased cell-surface expression of major histocompatibility complex class I (MHC-I) in MC38 cells and HCT116 cells. Antigen-specific cytotoxic T lymphocytes (CTLs) assay demonstrated that JQ1 enhanced the MHC-I-mediated cytotoxicity of CTLs. Mouse colon cancer cell line MC38 was used to establish the syngeneic mouse tumor model. Compared with the control, JQ1 significantly inhibited tumor growth and prolonged the overall survival of the mice. Besides, JQ1 did not only inhibit tumor growth by enhancing anti-tumor immunity, but also promoted the anti-tumor effect of PD-1 antibody. In addition, our data showed that JQ1 reduced infiltration of intratumoral regulatory T cells (Treg), thus remodeling the immunosuppressive TME. Taken together, these results highlight a new approach that enhances anti-PD-1 sensitivity in CRC.
Keywords: Colorectal cancer, JQ1, immunotherapy, immunosuppression.
Introduction
Despite advances in treatment, colorectal cancer (CRC) remains the second leading cause of cancer-related deaths worldwide [1]. Programmed cell death ligand 1 (PD-L1), which is expressed on the surface of cancer cells, binds to programmed cell death 1 (PD-1) and inhibits T cell activity, which results in immunosuppression [2, 3]. Immunotherapy targeting the PD-L1/PD-1 immune checkpoint has shown significant efficacy in solid tumors, including melanoma and non-small-cell lung cancer (NSCLC) [4, 5]. However, PD-L1/PD-1 blockade is effective in only CRC patients with mismatch repair-deficient and microsatellite instability-high (dMMR-MSI-H) phenotypes [6]. Most CRC patients are resistant to immune checkpoint inhibitors (ICIs), probably due to low tumor mutation burden and immunosuppressive TME [7, 8].
In addition to the PD-L1-mediated immunosuppression, suppression of MHC-I-mediated tumor antigen presentation is another key factor in immune escape [9]. CD8+ T cells are activated by recognition of specific antigens presented by MHC-I. Previous data has shown that downregulation of MHC-I molecules in cancer leads to dysfunction and even exhaustion of cytotoxic T cells [10]. Therefore, expression of MHC-I in cancer cells plays a crucial role in anti-tumor immunity [11]. Besides, previous studies have demonstrated that loss of MHC-I molecules in cancer reduces sensitivity to PD-L1/PD-1 blockade [12]. Correspondingly, upregulation of MHC-I molecules synergizes with PD-L1/PD-1 blockade to enhance anti-tumor activity [13]. Several studies have confirmed loss of MHC-I molecules in CRC [14, 15]. Thus, we hypothesized that the loss of MHC-I molecules mediates the suppression of anti-tumor immunity in CRC. Upregulation of MHC-I may be a viable strategy to enhance anti-tumor immunity in CRC.
Epigenetic modifications regulate gene expression without altering DNA sequences [16]. The epigenetic alterations are important drivers of cancer development and are associated with tumor immune escape [17, 18]. Some epigenetic drugs have been shown to remodel the TME and reverse immunosuppression in tumor-bearing animals [19, 20]. In addition, epigenetic drugs may strengthen the anti-tumor effects of ICIs [21]. The bromo- and extraterminal domain (BET) family proteins are epigenetic factors that regulate gene transcription through reading chromatin histone acetylation [22]. For instance, BRD4 is a BET family protein that affects tumor immunity by regulating transcription of the CD274 (PD-L1) gene in certain cancer cells. JQ1, a BRD4 inhibitor, downregulates the expression of PD-L1, which leads to reduced immunosuppression [23-25]. Besides, JQ1 inhibits tumor growth by enhancing anti-tumor immunity. Moreover, JQ1 facilitates anti-tumor effects of ICIs. For instance, combination of JQ1 and PD-1 blockade enhances the anti-tumor effect in Kras-mutant NSCLC [26]. JQ1 has also been shown to potentiate anti-tumor immunity with anti-PD-L1 therapy in hepatocellular carcinoma [27].
Since CRC is insensitive to immunotherapy due to the immunosuppressive TME, BET inhibitors could reverse the immunosuppressive TME. However, the role of BET inhibitors in the immune microenvironment of CRC remains unclear. Here, we evaluated the immunoregulatory role of the BET inhibitor, JQ1, in CRC.
Materials and methods
Cell lines and lentivirus infection
Mouse colon cancer cell line MC38 and human CRC cell lines HCT116, DLD1, LoVo, LS174T were purchased from the Cell Bank of the Chinese Academy of Sciences. MC38 BRD4 knockdown cell line and MC38-OVA cell line were generated by lentivirus infection. Knockdown lentivirus of BRD4 was acquired from GeneCopoeia (China). The sequence for mouse BRD4 shRNA was 5ʹ-GGTACCAAACACAACTCAAGC-3ʹ; The sequence for human BRD4 shRNA was 5ʹ-GCTCAAGACACTATGGAAACA-3ʹ. Overexpression lentivirus of ovalbumin was obtained from Syngentech (Beijing, China). Cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin and streptomycin.
Colony formation assay
500 MC38 cells per well were cultured in 6-well plates containing RPMI 1640 medium with 1 μM or 0.1 μM JQ1. The medium was changed every 3 days for 14 days. Cells were washed with PBS and fixed with 4% paraformaldehyde. The cells were stained with 0.1% crystal violet for 30 minutes prior to analysis.
Western blot analysis
We resolved the protein samples in SDS-PAGE. The samples were then transferred into 0.45-μm polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% bovine serum albumin (BSA) at room temperature for 1 hour and then incubated with primary antibodies overnight at 4°C. Thereafter, the blots were incubated with secondary antibodies conjugated with horseradish peroxidase (HRP). The HRP signal for protein expression was detected by electrochemiluminescence (ECL). Target protein signals were quantified and analyzed by ImageJ software. The primary antibodies included: anti-BRD4 antibody: catalog number ab128874, 1:1000, Abcam; and anti-ovalbumin antibody: catalog number ab181688, 1:1000, Abcam.
Quantitative real-time PCR and RNA sequencing
TRIzol (Invitrogen, USA) was used to extract total RNA from the cells. cDNA was then synthesized from 1 µg of the RNA using the Evo M-MLV RT Premix for qPCR Kit (Accurate Biology, China) using oligo (dT) primers. Quantitative real-time PCR (qPCR) was performed using a SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, China), following the manufacturer's instructions. RNA-sequencing was evaluated by Lc-bio Technologies co., ltd. (Hangzhou, China). In Brief, the MC38 cells were treated with the JQ1 inhibitor for 48 hours. TRIzol (Invitrogen, USA) was used to extract total RNA (Invitrogen, USA), which was then purified and used to synthesize cDNA. The cDNA libraries were sequenced on an Illumina Novaseq™ 6000 (Lc-bio Technologies co., ltd. Hangzhou, China).
Antigen-specific CTLs assay
MC38-OVA cells were incubated with JQ1 for 48 hours. The cells were then labeled with PKH26 (MINI26, Sigma, USA) according to the manufacturer's instructions. After removal of residual PKH26, the cells were incubated with 100 ng/ml Ovalbumin (OVA) peptide (AnaSpec, USA) for 12 hours. We harvested and washed the cells, and then plated them at a density of 5×106 cells /cm2, followed by incubation for 6 hours. OT-I transgenic mice were obtained from The Jackson Laboratory (003831, USA). The mice were sacrificed and then spleens and lymph nodes were prepared into single-cell suspensions. The cells were cultured in complete medium, supplemented with 10 ng/ml murine IL-2 (PeproTech, USA) for an additional 24 hours. CD8+ T cells were enriched using mouse CD8a+ T cell magnetic microbeads (Miltenyi Biotec, Germany). The CD8+ T cells were incubated in a medium containing 100 ng/ml OVA peptide for 24 hours. After washing with PBS to remove the OVA peptide, the cells were expanded in a medium containing murine 10 ng/ml IL-2 for 48 hours. OT-1 CD8+ T cells were harvested and counted, followed by coculture with MC-38OVA cells, labeled with PKH26 for 12 hours. On the other hand, dead cells were labeled with FVS780 (catalog number 565388, BD). PKH26+FVS780+ double-positive MC38-OVA cells were lysed by CD8+ T cells.
Animal models and treatments
Animal experiments were approved by the Experimental Animal Care Commission of Shenzhen Top Biotechnology Co.,Ltd (Number: TOP-IACUC-2021-0103). Six- to eight-week old female BALB/c nude mice and C57BL/6 mice were purchased from BesTest Biotechnology company (Zhuhai, China). MC38 cells (1×106) were injected subcutaneously into the right flank of the C57BL/6 or nude mice. The C57BL/6 tumor-bearing mice were randomly divided into four groups (n=5). Tumor size was measured every 2-3 days using electronic vernier calipers. Tumor volumes were calculated using the following formula: longest diameter ×shortest diameter2/2. Starting from day 8, mice were injected intraperitoneally with 50 mg/kg JQ1 (5% DMSO in 2-hydroxypropyl-β-cyclodextrin, Selleck, USA) daily for 7 days. For the anti-PD-1 and the combination therapy group, 100 μg of PD-1 antibody (catalog number BP0146, clone RMP1-14, Bioxcell, USA) was intraperitoneally injected on days 9 and 12. The mice were euthanized when they reached endpoints. Humane endpoints were defined as tumors that have 2 cm longest diameter or 2000 mm3 volume. The nude mice were randomly distributed into control and experimental group (five mice in each group). The experimental group mice were intraperitoneally injected with 50 mg/kg JQ1 daily. The control group mice were intraperitoneally injected with DMSO. Tumors from both groups were collected and measured on day 8 after treatment.
Cell harvesting and flow cytometry analysis of Treg cells
Approximately 1×106 MC38 cells were injected into the C57BL/6 mice. The mice were intraperitoneally injected with JQ1 daily for 7 days, starting on day 8 after cell implantation. The tumors were harvested and minced into 1-mm3 pieces, and then soaked in a digestion solution containing collagenase P (2 mg/ml, Sigma) and DNase I (50 μg/ml, Sigma) for 30 minutes at 37°C. The digested tumor tissues were filtered through a 40-μm cell strainer to obtain a cell suspension. Dead cells were labeled using Zombie Aqua (Biolegend). The cells were washed with PBS and then blocked with an anti-CD16/32 antibody for 20 minutes on ice. Thereafter, the cells were blocked and incubated with surface antibody for 20 minutes. The cells were then fixed and permeabilized for staining of intracellular antigens. We resuspended the cells in 200 µl of staining buffer (2% FBS in PBS) and then analyzed by flow cytometry (Beckman Coulter Cytoflex LX; Beckman, USA). The data were analyzed by FlowJo10 software.
Antibodies and related reagents for flow cytometry
Human TruStain FcX: clone 93, catalogue number 422302, Biolegend. TruStain FcX (anti-mouse CD16/32) Antibody: clone 93, catalogue number 101320, Biolegend. PE anti-human CD274 (B7-H1, PD-L1) Antibody: clone 29E.2A3, catalogue number 329706, Biolegend. FITC anti-human HLA-ABC Antibody: clone W6/32, catalogue number 311404, Biolegend. PE anti-mouse CD274 (B7-H1, PD-L1) Antibody: clone 10F.9G2, catalogue number 124308, Biolegend. FITC anti-mouse H2Kb Antibody: clone AF6-88.5, catalogue number 116506, Biolegend. FITC anti-mouse CD45: clone 30-F11, catalogue number 103108, Biolegend. Pacific blue anti-mouse CD3ε: clone 145-2c11, catalogue number 100334, Biolegend. Alexa Fluor647 anti-mouse CD4: clone GK1.5, catalogue number 100424, Biolegend. PE anti-mouse Foxp3: clone MF-14, catalogue number 126403, Biolegend. Zombie Aqua Fixable Viability Kit: catalogue number 423102, Biolegend. True-Nuclear Transcription Factor Buffer Set: catalogue number 424401, Biolegend. FVS780: catalogue number 565388, BD.
Statistical analysis
All in vitro experiments were conducted in triplicates. Comparison between two independent data sets was performed by two-tailed unpaired t-test. Statistical significance of tumor growth curves was analyzed using paired two-tailed t-test. Differences in the overall survival curves between two groups were analyzed with Log-rank test. Data were expressed as mean ± standard error of the mean (SEM). p< 0.05 indicates statistical significance.
Results
JQ1 downregulates PD-L1 expression and upregulates MHC-I expression in CRC
RNA was extracted and sequenced from the MC38 cells treated with JQ1. The results showed that CD274 gene was significantly suppressed, while H2Kb (MHC-I) was upregulated following JQ1 treatment (Fig. 1A). The results were confirmed by qPCR (Fig. 1B). We then employed flow cytometry to analyze the effect of JQ1 on the expression of PD-L1 at indicated doses. The data showed that JQ1 significantly decreased the expression of PD-L1 in a dose-dependent manner (Fig. 1C). To exclude the bias caused by cytotoxicity of JQ1. We selected a drug concentration (0.1 μM), with little effect on cell activity for subsequent experiments based on cell viability assays (Fig. S1A). Use of 0.1 μM JQ1 did not inhibit cell colony formation (Fig. S1B). Thereafter, we assessed PD-L1 expression at indicated times. As expected, JQ1 inhibited PD-L1 expression in a time-dependent manner (Fig. 1D). However, JQ1 increased IFN-γ-induced PD-L1 expression (Fig. 1E). These data demonstrated that there may be multiple mechanisms by which JQ1 regulates PD-L1 expression.
BET inhibitor JQ1 regulates the expression of PD-L1 in MC38 cells. (A) mRNA sequencing results; (B) Confirmation of mRNA sequencing results by qPCR; The surface expression of PD-L1 were determined by flow cytometry: (C) Cells were stimulated by different doses of JQ1 for 48 hours; (D) Cells were incubated with JQ1 (0.1 μM) for indicated times; (E) cells were treated with or without 0.1 μM JQ1 for 48 hours in the absence or presence of IFN-γ (20 ng/ml). *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
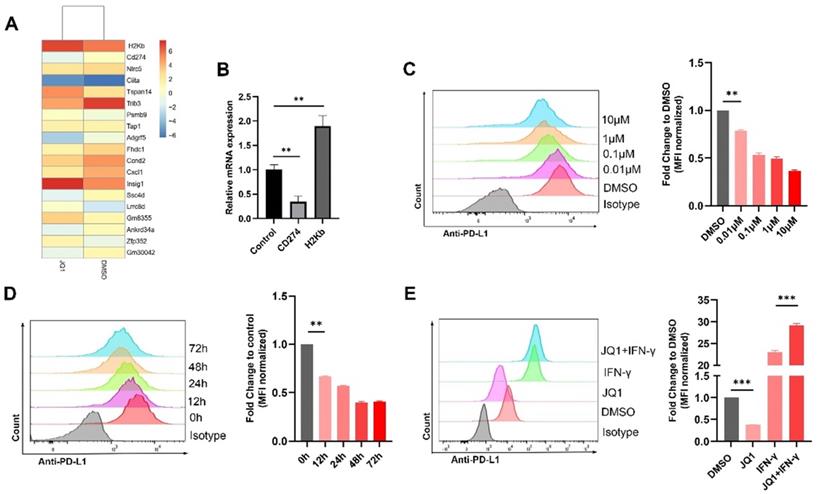
We also explored regulation of H2Kb by JQ1. JQ1 at 0.1 μM significantly increased the expression of H2Kb in the MC38 cells (Fig. 2A). JQ1 increased the expression of H2Kb in a time-dependent manner (Fig. 2B). In addition, JQ1 facilitated IFN-γ-induced H2Kb expression (Fig. 2C). These results demonstrated that JQ1 promotes both constitutive and IFN-γ-induced expression of H2Kb in the MC38 cells. We then analyzed the expression of PD-L1 and HLA-ABC (MHC-I) in human CRC cell lines treated with JQ1. The data showed that JQ1 significantly decreased the cell surface expression of PD-L1, but increased the cell surface expression of HLA-ABC in HCT116 cells (Fig. 3A,3B).
JQ1 increases the expression of H2Kb in MC38 cells. The surface expression of H2Kb were evaluated by flow cytometry. (A) Cells were stimulated by indicated doses of JQ1 for 48 hours; (B) Cells were treated with JQ1 (0.1 μM) for different times; (C) cells were incubated with or without 0.1μM JQ1 for 48 hours in the absence or presence of IFN-γ (20 ng/ml). *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
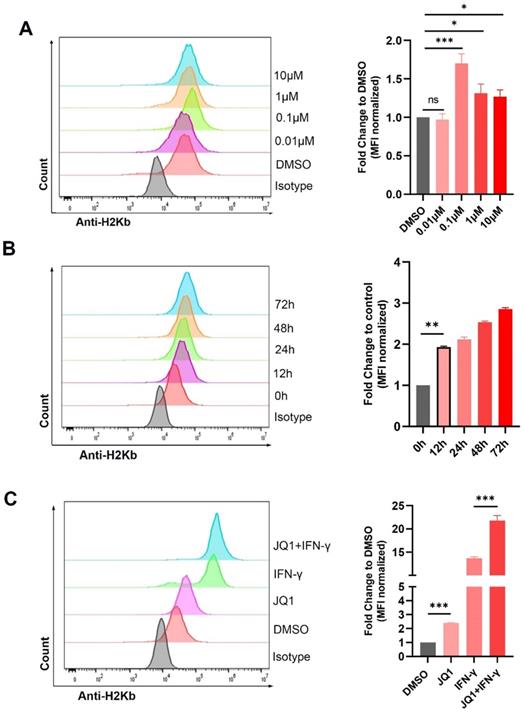
Expression of PD-L1 and HLA-ABC were examined in the 4 human colorectal cancer cell lines by flow cytometry. (A) The PD-L1 expression of cells treated with 0.1 μM JQ1; (B) The HLA-ABC expression of cells treated with 0.1 μM JQ1; *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
JQ1 regulates the expression of PD-L1 and MHC-I through BRD4
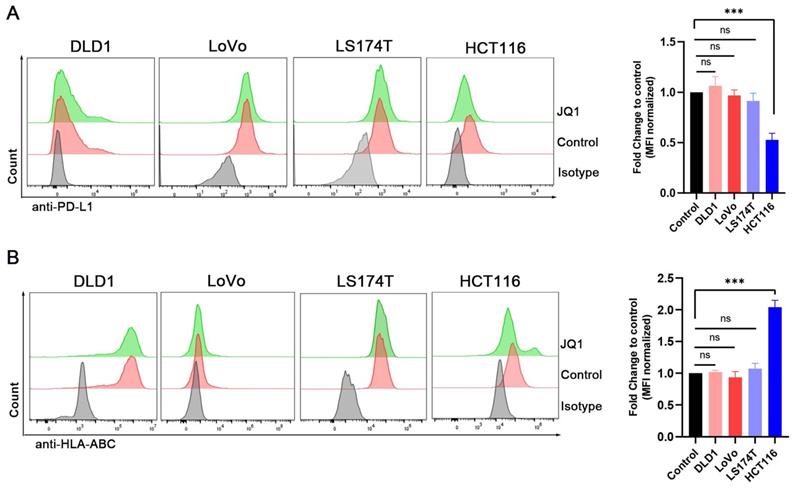
It has been reported that JQ1 blocks binding of BRD4 to the CD274 promoter, thereby repressing transcription of CD274. To evaluate whether JQ1 affects PD-L1 and MHC-I expression through BRD4, we knocked down the BRD4 gene in MC38 and HCT116 cells using short hairpin RNAs (shRNA). Western blot (Fig. 4A) and qPCR (Fig. 4B, 4C) analysis demonstrated down regulation of BRD4. Compared with the control, silencing of BRD4 reduced PD-L1 expression (Fig. 4D, E). In addition, BRD4 knockdown elevated the expression of MHC-I (Fig. 4F, G). These findings show that BRD4 regulate both PD-L1 and MHC-I genes in CRC.
JQ1 regulates the expression of PD-L1 and MHC-I through BRD4. The BRD4 shRNA knockdown efficiency was evaluated by western blot (A) and qPCR (B) (C). MC38/HCT116 cells transfected with shBRD4, wild-type cells treated with JQ1 (0.1 μM, 48 hours) were determined for PD-L1 expression (D) (E) and MHC-I expression (F) (G) by flow cytometry. *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
JQ1 enhances the cytotoxicity of CTLs by promoting tumor antigen presentation
An antigen-specific model was used to evaluate the regulation of CD8+ CTLs cytotoxicity by elevated H2Kb expression. It is well known that CTLs are activated by recognition of antigen/MHC-I complexes. Since ovalbumin (OVA) peptides are not expressed in mammals, they can mimic tumor-specific antigens [28]. CD8+ CTLs from OT-I mice express T cell receptors that specifically recognize OVA peptides. Theoretically, JQ1 promotes antigen presentation by increasing the expression of H2Kb, thereby enhancing killing efficiency of CTLs against cancer cells (Fig. 5A).
JQ1 enhances CD8+T cell cytotoxicity by promoting tumor antigen presentation. (A) Illustration of antigen-specific CTLs assay. (B) Identification of MC38-OVA cells. (C) Percent lysis of MC38-OVA cells pretreated with JQ1 or DMSO in the absence of OT-I CD8+ T cells. (D) Specific lysis percentage of MC38-OVA cells pretreated with DMSO or with JQ1 by CD8+ T cells. *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
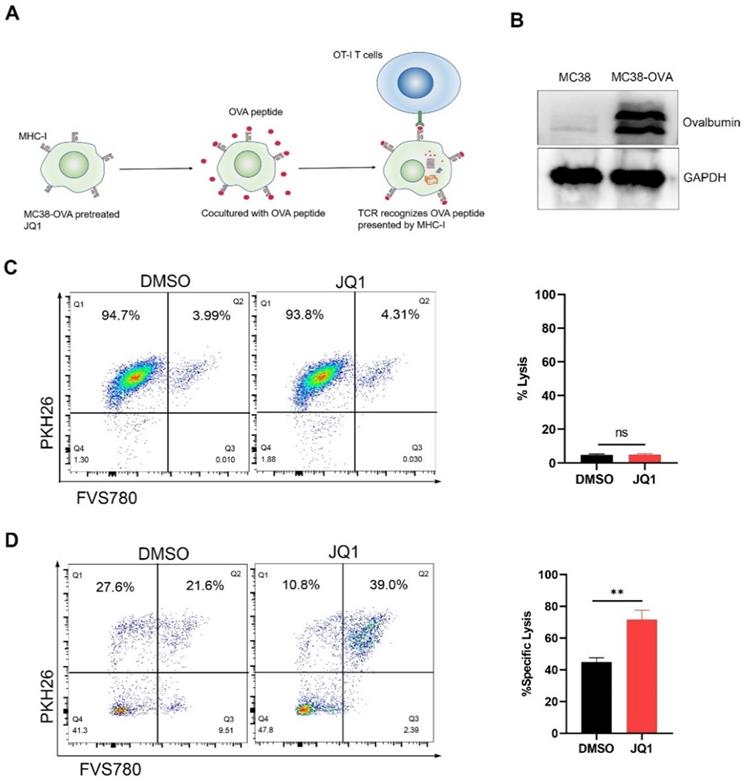
We used the lentivirus to infect MC38 cells to construct OVA-expression cells. Western blot analysis demonstrated that MC38-OVA cells express OVA (Fig. 5B). To define cell killing by JQ1 toxicity, we compared cytolysis by JQ1 or DMSO in the absence of T cells. Compared with DMSO, incubation with 0.1 μM JQ1 did not enhance cytolysis in the absence of T cells (Fig. 5C). Therefor, 0.1 μM JQ1 treatment had no significant cytotoxicity. For the CTLs assay, MC38-OVA cells were treated with OVA peptides for 24 hours following pretreatment with JQ1. The MC38-OVA cells were then cocultured with CD8+ T cells from OT-I mice. PKH26/FVS780 double-positive MC38-OVA cells were lysed by CD8+ T cells. These data revealed that the proportion of cytolysis in cells treated with JQ1 was notably higher than that in the control group (Fig. 5D). These data demonstrate that JQ1 enhances the killing capacity of CD8+ T cells against MC38-OVA cells. Our results confirm the observation that JQ1 enhances cytotoxicity of CD8+ T cells by promoting MHC-I-mediated antigen presentation.
JQ1 combined with anti-PD-1 enhances anti-tumor immunity in CRC
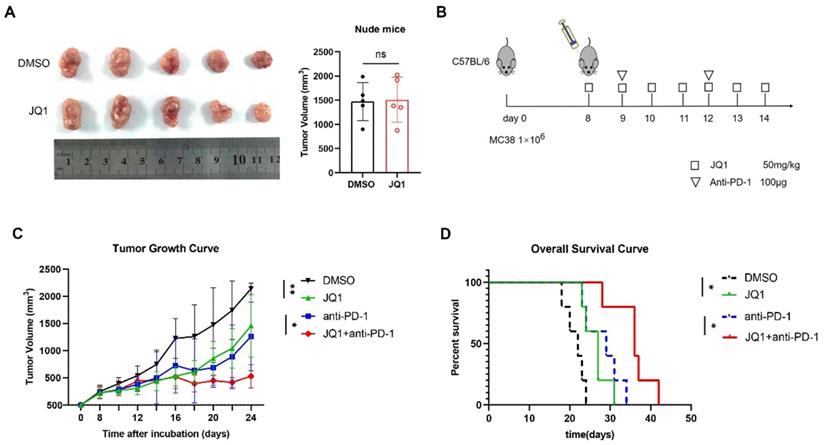
Since JQ1 promotes antigen presentation and enhances CD8+ T cell cytotoxicity, we hypothesized that JQ1 could enhance anti-tumor immunity in vivo. Moreover, JQ1 upregulates IFN-γ-induced PD-L1 expression, thus showing that JQ1 may synergistically fuel the anti-tumor ability of the PD-1 antibodies. To exclude JQ1 from inhibiting tumor growth through nonadaptive immunity, we established an animal model of T-cell-deficient BALB/c nude mice bearing MC38 cells. Compared with the control, JQ1 did not inhibit tumor growth in nude mice (Fig. 6A). MC38 cells were then subcutaneously injected into C57BL/6 mice to establish a transplantation tumor model with intact immune systems. The mice were then randomly divided into 4 groups: control, JQ1, anti-PD-1, and JQ1+anti-PD-1 (Fig. 6B). As expected, JQ1 treatment markedly inhibited tumor growth compared with the control (Fig. 6C). In comparison with the anti-PD-1 monotherapy, combination of JQ1 and anti-PD-1 exhibited more potent anti-tumor efficacy. Besides, the overall survival of mice treated with JQ1 was prolonged compared to that of the control group (Fig. 6D). Compared with the anti-PD-1 monotherapy, a combination treatment with JQ1 resulted in increased overall survival (Fig. 6D). These results demonstrate that JQ1 enhances anti-tumor immunity in tumor-bearing mice. In addition, JQ1 synergistically augments the anti-tumor capacity of the PD-1 antibody.
JQ1 combined with anti-PD-1 enhances anti-tumor immunity in vivo. (A) Comparison of tumor volumes from nude mice. n=5 mice per group; (B) The animal experimental protocol. n=5 mice per group; (C)Tumor growth curve. Statistical significance was analysed using paired two-tailed t-test; (D) Overall survival curve. p value tested using Log-rank test. *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
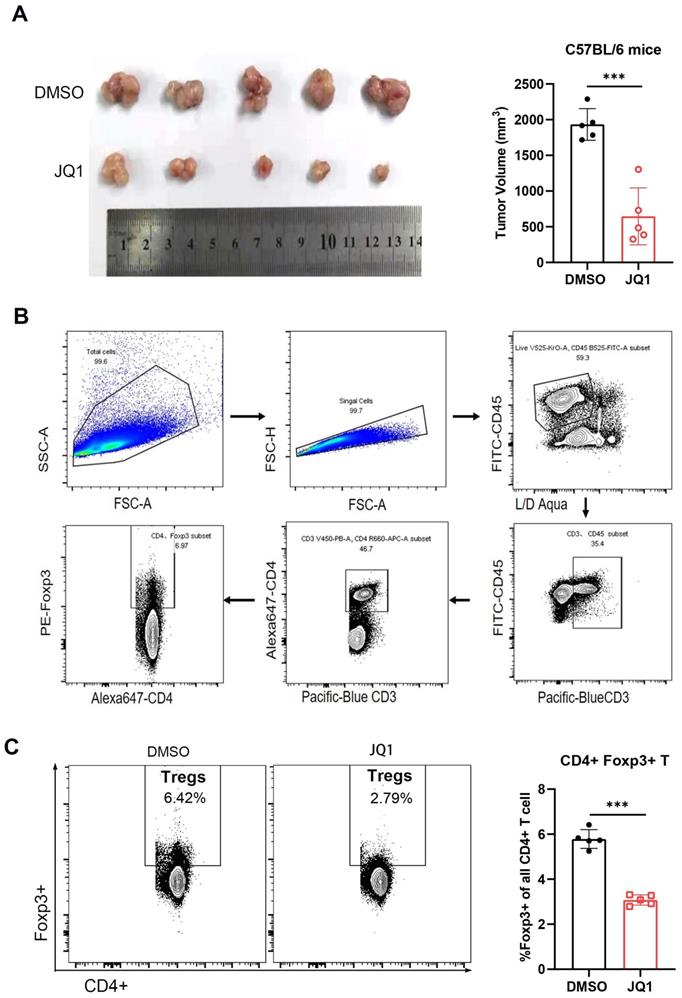
Presence of Treg cells is a major cause of immunosuppressive microenvironment in CRC [29]. To determine the effect of JQ1 on tumor infiltrating Treg cells, tumor tissues were harvested at the same time point. The results showed that the tumor volume of JQ1-treated mice was significantly smaller compared to that of the control mice (Fig. 7A). The anti-tumor effect of JQ1 was demonstrated again. The cells were then isolated from the tumor tissues for flow cytometric analysis. The data showed that the number of intratumoral Foxp3+ Treg cells in the JQ1 group was less than that in the control group (Fig. 7C). These results demonstrated that JQ1 inhibits the infiltration of Treg cells, leading to enhanced anti-tumor immunity.
JQ1 reduces tumor-infiltrating Foxp3+ regulatory T cells. (A) Comparison of tumor volumes from C57BL/6 mice. n=5; (B) Flow cytometry gating strategy for analyzing Treg cells; (C) Percentage of Foxp3+ Treg cells among CD4+ T cells. n=5. *p<0.05, **p<0.01, ***p<0.001, ns, no significant difference.
Discussion
Despite FDA approval of ICI drugs for use in dMMR-MSI-H metastatic CRC patients, not all the patients respond to the ICIs [30]. Patients with MSI-stable CRC, who account for the majority of CRC cases, do not respond to ICIs treatment [31]. Many studies have tried to elucidate the causes of CRC insensitivity to immunotherapy. However, to date, there is no new approach developed to improve immunotherapy outcomes in these patients.
Expression of PD-L1 by cancer cells significantly suppresses the function of T cells, leading to immunosuppression [2]. JQ has been shown to inhibit PD-L1 expression in various cancer cells [24, 25]. Our data showed that JQ1 inhibits constitutive PD-L1 expression but promotes IFN-γ-induced PD-L1 expression in MC38 cells. Some studies have shown that the level of PD-L1 expression in cancer cells is directly proportional to the effect of PD-1 blockade [32, 33]. However, patients with low PD-L1 expression may also respond to PD-1 blockade [31]. For CRC, there is no clear relationship between the effect of PD-1 antibody therapy and the expression of PD-L1 [34]. We observed that the regulation of PD-L1 by JQ1 contributes to enhanced the efficacy of PD-1 blockade. We considered the initial stage of adaptive immunity, during which JQ1 decreases the level of PD-L1 in cancer cells and reduces the inhibition of activated T cells. Activated T cells produce IFN-γ, and JQ1 increases IFN-γ-induced PD-L1 expression in cancer cells, thus enhancing the sensitivity of cancer cells to PD-1 antibody.
MHC-I is a key molecule in tumor antigen presentation. CD8+ T cells recognize only tumor antigens presented by MHC-I. Therefore loss of MHC-I molecules is an important cause of tumor immune escape [9]. MHC-I expression in cancer cells does not only predict prognosis, but also therapeutic response to ICIs [12, 35]. Upregulation of MHC-I expression partially reverses the immunosuppressive TME [36]. In recent years, epigenetic modifications have been shown to play an important role in the regulation of tumor immunity [37]. In addition, histone deacetylase inhibitors have been shown to upregulate the expression of MHC-I in cancer cells and to enhance the therapeutic effect of anti-PD-1 on lymphoma [38, 39]. JQ1, a BRD4 inhibitor, has been reported to upregulate MHC-I expression in prostate cancer [40]. In this study, we found that JQ1 increases MHC-I expression in CRC. However, the mechanism by which JQ1 promotes MHC-I expression remains unclear. It is now clear that JQ1 reduces protein expression by repressing gene transcription [41]. We speculate that MHC-I is not directly regulated by JQ1 and that there may be a target gene that negatively regulates MHC-I. Furthermore, using antigen-specific CTLs assay, we showed that upregulation of MHC-I remarkably strengthened the killing of cancer cells by CD8+ T cells.
We explored the anti-tumor immune effects of JQ1 in the MC38 animal model. The results demonstrated that JQ1 inhibited tumor growth and prolonged survival of the mice. The combination group had better tumor suppression and longer survival compared to the PD-1 antibody monotherapy group. Thus, JQ1 was shown to synergize the anti-tumor effect of PD-1 blockade.
Foxp3+ Treg cells are enriched in the human colorectal cancer TME and are associated with poor prognosis [42]. Foxp3+ Treg cells are an immunosuppressive subset of CD4+ T cells [43]. Treg cells promote tumor progression by suppressing anti-tumor immunity. This is achieved through consumption of IL-2, secretion of immunosuppressive cytokines, and upregulation of immune checkpoint levels in effector T cells [44-46]. These events ultimately impair the function of effector T cells and lead to apoptosis. Treg cells do not only play an important role in tumor progression, but also positively correlates with acquired resistance to ICIs [47]. Furthermore, targeting Treg cells contribute to reduction of the acquired resistance to ICIs [48]. It has been reported that JQ1 reduces the number of Treg cells in the TME of NSCLC, consequently enhances anti-tumor immunity [49]. In this study, we demonstrated that JQ1 significantly reduces intratumoral Treg cells infiltration in MC38 model. Besides, the Treg cells infiltration levels were correlated with tumor progression and poor prognosis. These data demonstrated that JQ1 enhances anti-tumor immunity by remodeling the immunosuppressive microenvironment in CRC.
In conclusion, we revealed that JQ1, a BET inhibitor, regulates PD-L1 and promotes MHCI-I expression in CRC. JQ1 enhances anti-tumor immunity by strengthening CTLs cytotoxicity and reducing Treg cells infiltration. In addition, JQ1 synergizes the anti-tumor effect of PD-1 blockade. These findings highlight a novel approach that improves anti-tumor immunity in CRC and enhances the sensitivity to PD-1 blockade. However, there is need for further studies on the mechanism by which JQ1 regulates MHC-I expression.
Supplementary Material
Supplementary figure.
Acknowledgements
Ethical approval statement
Animal experimental procedures used in this study were obtained with the approval of the ethics committee of Shenzhen Top Biotechnology Co.,Ltd.
Funding
This study was supported by Sanming Project of Medicine in Shenzhen (SZSM201911010), Shenzhen Key Medical Discipline Construction Fund (SZXK016), Shenzhen Sustainable Project (KCXFZ202002011010593), Guangdong Provincial Key Laboratory of Digestive Cancer Research (No. 2021B1212040006).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-a Cancer Journal for Clinicians. 2021;71:209-249
2. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer. 2012;12:252-264
3. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T. et al. IFN-alpha Directly Promotes Programmed Cell Death-1 Transcription and Limits the Duration of T Cell-Mediated Immunity. Journal of Immunology. 2011;186:2772-2779
4. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J. et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. New England Journal of Medicine. 2018;379:2040-2051
5. Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M. et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). 2017; 35: 3924-3933.
6. Goldstein J, Tran B, Ensor J, Gibbs P, Wong HL, Wong SF. et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Annals of Oncology. 2014;25:1032-1038
7. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960-1964
8. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-413
9. Bodmer W, Browning M, Krausa P, Rowan A, Bicknell D, Bodmer JJAotNYAoS. Tumor escape from immune response by variation in HLA expression and other mechanisms. 1993; 690: 42-49.
10. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone SJAii. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. 1999; 74: 181-273.
11. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. In: Littman DR, Yokoyama WM, editors. Annual Review of Immunology Vol 33. 2015 p. 169-200
12. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C. et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Science Translational Medicine. 2018;10:268-281
13. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M. et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity. 2016;44:609-621
14. Smith ME, Bodmer WF, Bodmer JG. Selective loss of HLA-A,B,C locus products in colorectal adenocarcinoma. Lancet (London, England). 1988;1:823-824
15. Lopez-Nevot MA, Esteban F, Ferron A, Gutierrez J, Oliva MR, Romero C. et al. HLA class I gene expression on human primary tumours and autologous metastases: demonstration of selective losses of HLA antigens on colorectal, gastric and laryngeal carcinomas. British journal of cancer. 1989;59:221-226
16. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571:489-499
17. Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G. et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. 2005;37:391-400
18. Cao J, Yan Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends in Cancer. 2020;6:580-592
19. Topper MJ, Vaz M, Chiappinelli KB, Shields CED, Niknafs N, Yen R-WC. et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell. 2017;171:1284-1300
20. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H. et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nature Communications. 2018;9:622-633
21. Olino K, Park T, Ahuja N. Exposing Hidden Targets: Combining epigenetic and immunotherapy to overcome cancer resistance. Seminars in Cancer Biology. 2020;65:114-122
22. Shi J, Vakoc CR. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Molecular Cell. 2014;54:728-736
23. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ. et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Reports. 2016;16:2829-2837
24. Andrieu GP, Shafran JS, Smith CL, Belkina AC, Casey AN, Jafari N. et al. BET protein targeting suppresses the PD-1/PD-L1 pathway in triple-negative breast cancer and elicits anti-tumor immune response. Cancer Lett. 2019;465:45-58
25. Liu K, Zhou Z, Gao H, Yang F, Qian Y, Jin H. et al. JQ1, a BET-bromodomain inhibitor, inhibits human cancer growth and suppresses PD-L1 expression. Cell Biology International. 2019;43:642-650
26. Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S. et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunology Research. 2018;6:1234-1245
27. Liu C, Miao X, Wang Y, Wen L, Cheng X, Kong D. et al. Bromo- and extraterminal domain protein inhibition improves immunotherapy efficacy in hepatocellular carcinoma. Cancer Science. 2020;111:3503-3515
28. Kakimi K. Cancer immunotherapy targeting neoantigens. Cancer Science. 2018;109:937-937
29. Liu Z, Huang Q, Liu G, Dang L, Chu D, Tao K. et al. Presence of FOXP3(+)Treg cells is correlated with colorectal cancer progression. International Journal of Clinical and Experimental Medicine. 2014;7:1781-1785
30. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH. et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nature Reviews Gastroenterology & Hepatology. 2019;16:361-375
31. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD. et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine. 2015;372:2509-2520
32. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Science Translational Medicine. 2016;8:328-342
33. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WEE, Poddubskaya E. et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. New England Journal of Medicine. 2015;373:123-135
34. Andre T, Overman M, Lonardi S, Aglietta M, McDermott R, Wong KYM. et al. Analysis of tumor PD-L1 expression and biomarkers in relation to clinical activity in patients (pts) with deficient DNA mismatch repair (dMMR)/high microsatellite instability (MSI-H) metastatic colorectal cancer (mCRC) treated with nivolumab (NIVO) plus ipilimumab (IPI): CheckMate 142. Annals of Oncology. 2017;28:163-171
35. Simpson JAD, Al-Attar A, Watson NFS, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59:926-933
36. Pulido M, Chamorro V, Romero I, Algarra I, Collado A, Garrido F. et al. Restoration of MHC-I on tumor cells by Fhit transfection promotes immune rejection and acts as an individualized immunotherapeutic vaccine. 2020; 12: 1563-1586.
37. Renaude E, Kroemer M, Borg C, Peixoto P, Hervouet E, Loyon R. et al. Epigenetic Reprogramming of CD4(+) Helper T Cells as a Strategy to Improve Anticancer Immunotherapy. Frontiers in Immunology. 2021;12:669-682
38. Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C. et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. Journal of Immunology. 2000;165:7017-7024
39. Wang X, Waschke BC, Woolaver RA, Chen Z, Zhang G, Piscopio AD. et al. Histone Deacetylase Inhibition Sensitizes PD1 Blockade-Resistant B-cell Lymphomas. Cancer Immunology Research. 2019;7:1318-1331
40. Mao W, Ghasemzadeh A, Freeman ZT, Obradovic A, Chaimowitz MG, Nirschl TR. et al. Immunogenicity of prostate cancer is augmented by BET bromodomain inhibition. Journal for Immunotherapy of Cancer. 2019;7:277-291
41. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. 2011; 146: 904-917.
42. Chaudhary B, Elkord EJV. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. 2016; 4: 28.
43. Sakaguchi S, Miyara M, Costantino CM, Hafler DAJNRI. FOXP3+ regulatory T cells in the human immune system. 2010; 10: 490-500.
44. Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD. et al. Helios+ and Helios- Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. 2019; 49: 398-412.
45. Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CAJI. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-β: the role of T regulatory cells. 2006; 117: 433-442.
46. Sasidharan Nair V, Elkord EJI, biology c. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. 2018; 96: 21-33.
47. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S. et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. 2017; 18: 1332-1341.
48. Saleh R, Elkord EJCl. Treg-mediated acquired resistance to immune checkpoint inhibitors. 2019; 457: 168-179.
49. Adeegbe DO, Liu S, Hattersley MM, Bowden M, Zhou CW, Li S. et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in Kras-mutant non-small cell lung cancer. 2018; 6: 1234-1245.
Author contact
![]() Corresponding authors: Yulong He (heyulongsysu.edu.cn), Changhua Zhang (zhchanghsysu.edu.cn), Bo Li (libocom).
Corresponding authors: Yulong He (heyulongsysu.edu.cn), Changhua Zhang (zhchanghsysu.edu.cn), Bo Li (libocom).