Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(9):2757-2767. doi:10.7150/jca.73143 This issue Cite
Review
Interaction between prostate cancer stem cells and bone microenvironment regulates prostate cancer bone metastasis and treatment resistance
Lu Yao1, Xiangyu Zhang2 ![]()
1. Department of Clinical Medicine, Jining Medical University, Jining 272067, China.
2. Department of Pathology, Jining First People's Hospital, Jining Medical University, Jining 272000, China.
Received 2022-3-22; Accepted 2022-5-31; Published 2022-6-13
Abstract

Prostate cancer (PCa) is one of the most common cancers with increasing rates of incidence. Bone metastasis and drug resistance are the most serious threat faced by patients following a delayed diagnosis of cancer, which might lead to treatment failure and death. The theoretical model of cancer stem cells (CSCs) explains the diverse molecular characteristics of cancer as well as its relapse, metastasis and drug resistance. Prostate cancer involves heterogeneous cells community, including prostate cancer stem cells as an important component. These subtypes of cancer cells are usually monoclonal, expressing specific biomarkers and exhibiting self-renewal and differentiation capacity. Therefore, therapies that target CSCs might be more effective in overcome drug resistance and metastasis. Thus, anti-CSCs therapies differ from the traditional anti-proliferative approach. We focus here on reviewing the effects of prostate CSCs on bone metastasis and resistance to traditional treatment in PCa, and report new clinical strategies that address CSC-based tumorigenesis.
Keywords: Prostate cancer stem cell, CRPC, Bone microenvironment, Metastasis
Introduction
Prostate cancer (PCa) is the second most common cancer and the fifth most common cause of cancer-related mortality among men worldwide [1, 2]. The five-year survival rate in patients with non-metastatic PCa has been consistently growing in recent years, practically approaching 100%. However, advanced PCa often leads to bone metastases, which is currently incurable [3, 4]. Skeletal-related events (SREs) due to PCa bone metastasis often cause pain, which reduces the patients' quality of life. Agents that inhibit osteoclastic activity, including zoledronate and denosumab, are usually used clinically to treat patients with PCa metastatic disease [5].
Early-stage PCa is androgen-dependent and surgical prostate removal is the most common treatment strategy, which can effectively cure localized prostate cancer [6]. However, most patients with PCa eventually become androgen-independent and progress to castration-resistant prostate cancer (CRPC) [7,8]. Castration-resistant prostate cancer usually leads to bone metastasis, which is lethal as no effective treatment currently exists [9-11]. Most currently available therapies usually focus on cancer cells in bone metastasis, rather than the bone microenvironment (BME), which plays an important role in prostate cancer bone metastasis. This might be the limitation of current therapy. It is exciting, however, to know that studies have reported the interactions between cancer cells (including CSCs) and BME [12-15]. CSCs are an important component of prostate cancer cells and usually promote bone metastasis progression. CSCs exhibit high clonogenic activity and potential for cancer initiation. Accordingly, effective treatment of metastatic PCa requires elucidation of the mechanisms underlying the growth of PCa cells in the BME, especially in the context of the important role played by prostate CSCs (PCSCs). This may shed some light on understanding of prostate cancer bone metastasis and pharmacological target selection when treating this disease.
Stem cells
Advances leading to the discovery of hematopoietic stem cells have increased the number of treatment options for cancer [16]. Among cancer cells, CSCs form a small subpopulation characterized by self-renewal, quiescence, and potential for differentiation, with a key role in cancer recurrence, metastasis, drug resistance and heterogeneity [17-19]. CSCs exhibit two types of cell division: 1) symmetric cell division in which one stem cell divides into two stem cells; 2) asymmetric cell division, in which one stem cell forms a new stem cell and a daughter cell [20,21]. Adult stem cells are often in a quiescent state, and are regulated by multiple cell cycle regulatory genes, such as p21, p18, and p63. When they exit the quiescent state, these cells self-renew and differentiate into other types cells [22]. Bonnet and Dick first demonstrated in 1977 that human acute myeloid leukemia originates in CSCs, which express the cell surface markers CD44+ and CD38- [23].
The human prostate is composed of stromal cells and three types of epithelial cells: basal cells, luminal secretory cells, and neuroendocrine cells [24]. Only a small subset of basal cells comply with the stem cell hypothesis [25]. They can replenish the apoptotic and dead cells. They also induce the continuous differentiation from primitive basal cell to secretory cell. Prostate stem cells are most likely located in the basal layer as a component of the prostate basal cell subpopulation, probably in specific niches. One study indicated that the stem cells express the Zeb1 marker [25, 26]. These niches are surrounded by several types of cells, such as mesenchymal stem cells, inflammatory cells, and immune cells. The stromal cells interact with the normal prostate stem cells via multiple biological signals exhibiting paracrine signaling pattern [27, 28]. The prostate CSCs may originate in the normal stem cell of prostate via genetic alternations.
Prostate cancer stem cells
Prostate cancer is a highly heterogeneous entity, comprised of multiple cancer cell subpopulations (including CSCs) and non-tumoral cells such as epithelial cells, stromal cells, immune cells, vascular endothelial cells, tumor-associated macrophages (TAMs) and cancer associated fibroblasts [29-31]. Studies involving CSCs has progressed rapidly, and novel techniques for identification, purification, and characterization of these cells have been developed. It is possible that CSCs are derived from normal stem cells, as they display many phenotypic and functional characteristics of normal prostate stem cells. They express several types of cell surface pluripotency makers, including cell-adhesion molecules (CD24, CD44, CD133), transcription factors (OCT4, SOX2, KLF4, NANOG, c-Myc, and HER2), integrin α2β1, and aldehyde dehydrogenase [32-39]. Prostate CSCs were found to be innately chemo-resistant and highly metastatic, resulting in clinical recurrence, progression to metastatic disease, and cancer-related death in patients diagnosed with prostate cancer [40]. As some CSCs markers are also expressed by normal stem cells, it is necessary to use a combination of several markers to reliably isolate the PCSCs and quantify them. BRCA1 and EZH2 tightly cooperate to regulate the PCSCs phenotype and properties [41]. The number of cells expressing ALDHhi, CD44+, and integrin α2β1+ increases when castration resistance occurs, and such cells display potential self-renewal capacity and colony formation, which indicates that castration resistance increase the number of PCSCs [42]. Meanwhile, cells expressing CD44+, integrin α2β1+, and CD133+ were isolated from PCa patients, and shown to exhibit self-renewal, suggesting that the three markers can be used to define the CSCs phenotype [43]. The use of multiple markers to isolate and characterize CSCs has been widely used; however, some studies propose that the stemness-related transcription factors are also important to identify CSCs.
It is necessary to understand the molecular basis of CSCs stemness, and the mechanism of reprogramming non-CSCs into CSCs. The important transcription regulators, SOX2, OCT4, and NANOG, contribute to the maintenance of the pluripotent state of CSCs. Some studies indicated that inhibition of androgen receptor (AR) function increases the stemness of PCSCs [44, 45]. MDM2 mediates AR degradation to maintain the pluripotency of PCSCs. AR alternative splice variants (AR-Vs) are involved in the progression of prostate cancer bone metastasis. AR-Vs also play a role in the resistance to anti-androgen therapy and radiotherapy. The expression of AR-Vs (including AR-V7, AR-V1, and V567es) increased significantly in CRPC bone metastasis when compared with hormone-naïve prostate cancer. AR-V7 induces prostate cancer cell EMT and impart the PCSCs characteristics to prostate cancer cell. NANOG regulates the pluripotency of cancer cells and plays an important role in the evolution of PCSCs. The SPOP gene interferes with this process by degrading the NANOG protein. It was inferred that tumor suppressor genes also play an important role in the evolution of PCSCs. Mutations involving genes such as TP53 and PTEN usually occur in PCSCs [46]. It is well-known that BRCA1 and BRCA2 are tumor suppressor genes, while the mutant genotypes are closely related to different cancer types, including breast cancer and PCa [47, 48]. The main role of BRCA1 and BRCA2 proteins is to repair damaged DNA, especially double strand DNA breaks. BRCA1 also plays an important role in maintaining the breast CSC population. BRCA1 and EZH2 cooperate tightly in regulating PCSCs phenotype and properties, and the loss of BRCA1 is related to PCSCs phenotype [41, 49].
Bone microenvironment of metastatic prostate cancer
Bone is composed of cortical bone and bone marrow in different proportions [50], with the cortical bone surrounding the bone marrow. Cortical bone is highly mineralized when compared with the trabecular bone, and the main mineral substance of bone is hydroxylapatite. Bone extracellular matrix is composed of organic and inorganic components, with collagen constituting the initial architecture, followed by mineralization of apatite. Bone is in a constant process of remodeling, which is mediated by osteoclasts and osteoblasts; osteoblasts promote bone formation while osteoclast promote bone resorption [51-55]. Most importantly, many growth factors are found in the bone matrix, including insulin-like growth factors (IGFs), bone morphogenetic proteins (BMPs), transforming growth factor-beta 1 (TGF-β1), and platelet-derived growth factors (PDGFs) [56-59]. The bone cells, bone matrix components, and growth factors plays an important roles during PCa metastasis. When prostate cancer cell metastases to the bone, the cancer cell secrete matrix metalloproteinase-9 (MMP-9), urokinase plasminogen activator receptor (uPAR), and cathepsin B (CB), which degrade the bone matrix to release growth factors. Additionally, TGF-β promote bone metastasis via regualtion of prostate cancer cell proliferation, migration and invasion. The Smad protein is phosphorylated and translocated to nucleus after TGF-β stimulation. Interferon-inducible Transmembrane Protein 3 (IFITM3) interacts with Smad 4 to enhance Smad 2 phosphorylation. TGF-β induces EMT and MMP secretion to promote bone mtastasis. TGF-β also activates the Smad-independent pathway, such as the MAPK pathway. Inhibition of TGF-β signaling can attenulating prostate cancer bone mtastasis. TGF-β can also induce immune suppression by regulating immune cell proliferation and differentiation. It can activate the M2 macrophage and inhibit the generation of antigen-presenting dendritic cells. It can also convert neutrophils from CD11b+/Ly6G+ tumor-associated types to pro-tumor types. As for lymphocytes, TGF-β signaling inhibits the generation of effector T lymphocytes and promote Treg production. It can inhibit B lymphocyte maturation and natural killer cells via mTOR signaling inhibition. Myeloid-derived suppressor cells (MDSCs) can suppress the anti-tumor immunity by inhibiting T cell functions. TGF-β activation can recruit the MDSCs to the prostate cancer microenvironment.
Bone microenvironment factors regulate prostate cancer stem cells (PCSCs) during both processes of prostate cancer bone metastasis and drug resistance. When the PCSCs occupy the hematopoietic stem cells (HSCs) niche, the PCSCs interact with bone marrow stromal cells (BMSCs) and osteoblast, BMSCs influence the PCSCs via SHH signaling pathway, osteoblast influence the PCSCs via Notch and Wnt signaling pathway. The endothelial in the bone marrow microenvironment can secrete CXCL12 to induce PCSCs migration and colonization at the metastatic niche. Cancer-associated fibroblasts (CAFs) can maintain the stemness of PCSCs through MMP secretion and pro-inflammatory signaling pathway. As the PCSCs possess various drug-resistance mechanism, such as overexpression of drug-resistance protein, anti-apoptotic capacity to overcome apoptosis, a dormant state is beneficial to prevent drug-induced death.
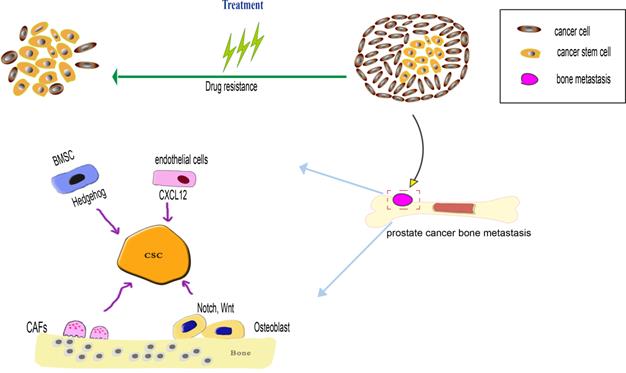
The phenotypic heterogeneity of metastatic prostate cancer cell is established. CSCs are one important component of bone metastasis, and promote bone metastasis progression via various mechanism. Most cancer-related deaths are attributed to metastasis, and PCa is likely to lead to bone metastasis, with devastating consequences for patients' health [60]. Several mechanisms play an important role in this process, including epithelial-mesenchymal transition (EMT), during which the CSCs express several EMT markers (vimentin, fibronectin, N-cadherin, and E-cadherin) [61-63]. The bone metastatic process mainly involves EMT, dissemination and colonization to bone. EMT plays an important role in initiating bone metastasis. Several signaling molecules, released by the tumor microenvironment, might influence the EMT in PCSCs through autocrine and paracrine signaling manners. These signaling molecules include transforming growth factor β (TGF-β), fibroblast growth factor (FGF), interleukin 6 (IL-6), and hypoxia-induced factor (HIF).
Circulating tumor cells (CTCs) have been known for 150 years, since they were first reported by Ashworth in 1869 [64]. The hypothesis of “seed and soil” is an important theory of cancer metastasis, in which the CTCs are the “seeds” and the source of distant metastasis [65-67]. The EMT process plays a role in regulating entry of the CTCs into the circulation. CTCs contain various types of cancer cells, and the CSCs express specific markers and play a key role in metastasis. Upon reaching the bone via blood vessels after bloodstream circulation, they colonize this site and form niches [68]. The bone stromal cells regulate the CSCs, and induce dormancy. The stromal-derived factor 1 (SDF-1, namely CXCL12), and its chemokine receptor CXCR4 play a critical role in PCa bone metastasis. CXCL12, which is produced by osteoblasts in the bone microenvironment, attracts CSCs into the bone environment and occupy the hematopoietic stem cell (HSC) niche [69, 70]. Furthermore, overexpression of CXCR4 was positively correlated with cell stemness in hTERT-immortalized human prostate epithelial cells [71, 72.] In addition, the CXCL12/CXCR4 axis plays an important role in attracting HSCs to bone, and maintain a niche for HSCs [73,74]. The process is similar to that of CSCs, which are transported to the niche via appropriate CXCL12 and ANXA2 signaling, suggesting that CSCs occupy HSC niche when PCa metastasizes to bone. Once in the niche, the CSCs remain dormant for long periods and interact with the bone microenvironment. The disseminated tumor cells (DTCs) occupying the niche of HSCs, and then the DTCs are induced to CSCs via interactions with the niche. HSCs play a key role in maintaining the stem cell function. The stem cell-related genes such as KLF4, Bmi-1 and Nanog are overexpressed in the DTCs after their arrival at HSCs niche. MicroRNAs (miRNAs) play an important role in regulating various genes, and have become a focus of study in recent years. The miR-141 was found to suppress PCSCs properties in PC-3 and DU145 PCa cell lines, and affected PCa metastasis in an orthotopic cancer model [75]. miR-34a was also shown to suppress the function of PCSCs and inhibit cancer metastasis [76,77].
The CSCs and bone marrow microenvironment are closely linked, and signals from bone marrow are very important for the function of CSCs. We will briefly elaborate the most crucial signaling pathway, including those mediated by Notch, Wnt, Hedgehog, FGF, and TGF [78].
Notch pathway
Notch is a transmembrane receptor expressed in CSCs that plays an important role in stem cell self-renewal and differentiation, and inhibition of this signaling pathway appears to be a therapeutic strategy for CSCs elimination [79]. Osteoblasts in the bone microenvironment express the ligand for Notch, and activate the Notch signaling pathway in the CSCs. Notch signaling pathway promotes the EMT process of CSCs and enhances their metastatic ability. It was reported that Notch regulates the EMT process through Snail protein. Notch1 expression in metastatic lesions is substantially higher than in the primary site. When Notch1 is down-regulated, the EMT properties of CSCs are inhibited, while when Notch1 is overexpressed in PCa cells, their migration and invasion ability is enhanced [80]. In docetaxel-resistant PCa cells, a subpopulation of CSC-like cells showed activation of Notch and Hedgehog signaling pathway, while the inhibition of Notch signaling reversed the drug resistance [81]. Another study showed that pharmacological inhibition of Notch signaling pathway, using γ-secretase inhibitor (GSI), increased the efficiency of castration therapy [82]. In yet another study, it was shown that GSI enhanced the antitumor effects of docetaxel in PCa, which might be related to a decrease in PCSCs [83]. It was also reported that miRNAs are closely related to Notch signaling in PCSCs. Overexpression of miR-34a in PCa cells reduced Notch1 expression, and decreased the growth and self-renewal of these cells [84]. Overexpression of miR-199-5p downregulated the CSC-associated markers in PCa cells and inhibited the Notch signaling of PCSCs [85]. One study showed that the CD54-p38-Notch1 axis plays important roles in colony formation, apoptosis, and cancer recurrence [86].
Wnt pathway
Wnt signaling pathway is very important for embryonic development and adult tissue homeostasis. The canonical Wnt signaling pathway also plays an important role in stem cell self-renewal, maintenance, and differentiation, partly via enhancement of human telomerase reverse transcriptase (hTERT) activity [78,91]. Wnt signaling in PCa bone metastasis can enhance osteoblastic activity in the bone microenvironment. PCa cells interact with osteocyte via Wnt signaling to induce differentiation of pre-osteoblasts into mature osteoblasts, while inhibiting the RANKL/OPG signaling pathway to suppress bone resorption [87]. Furthermore, the activation of Wnt3 signaling pathway increased the expression of CSC markers, such as CD133, CD44, keratin 18, and β-catenin [88]. Specifically, it was demonstrated that the regulation of Wnt pathway altered the CSCs phenotype. For example, miR-320 can alter CSCs phenotype by downregulating Wnt pathway [89], as Wnt pathway regulates the self-renewal and symmetric cell division of PCSCs. Overexpression of PHF21b activates Wnt signaling pathway, leading to an increase in PCSCs traits. Such overexpression is associated with poor prognosis of PCa [90]. Many Wnt signaling inhibitors were tested in clinical trials as they decrease the function of CSCs. Most of these inhibitors are fusion-protein, such as the frizzled 8 receptor (FZD8) that is fused to IgG1 Fc fragment [92]. FZD8 is an important protein, which is upregulated and positively correlated with PCa progression and bone metastasis clinically [93]. Wnt signaling occurs in FZD8-mediated increase in stem cell phenotype, cell migration, and invasion in PCa cell lines [94]. Activation of Wnt signaling pathway requires the corresponding ligands, including WNT2B, WNT3, and canonical Wnt ligands to bind with Wnt receptors, leading to suppression of β-catenin, resistance to degradation by GSK protease, resulting in transcriptional activation of target genes, such as ATOH1, CCND1, CD44, LGR5 and Snail [95]. Recently, DAB2IP was identified as a new tumor suppressor gene in PCa. It regulates the stemness of PCSCs through modulation of CD117 transcription, in which the Wnt/β-catenin signaling pathway plays an important role [96]. DAB2IP also inhibits the EMT and PCa bone metastasis [97, 98]. However, SOX9 was found to regulate Wnt signaling, enhance the EMT process in PCa, resulting in PCa metastasis [99]. The miRNAs play an important role in the regulation of CSCs. For example, miR-1301-3p promotes the expansion of PCSCs via activation of Wnt/β-catenin signaling pathway. Prostate CSC-related genes, such as OCT4, SOX2, NANOG, CD44, KLF4, c-MYC, and MMP2, are upregulated by miR-1301-3p, while GSK3β and SFRP1 genes are downregulated [100].
Hedgehog pathway
Hedgehog (Hh) signaling pathway is highly conserved. It plays an important role in embryonic development, stem cells maintenance, and tissue regeneration [101]. Abnormal Hh signaling in PCa leads to enhanced progression, relapse, metastasis, and drug resistance [102,103]. Recently, several studies have shown that Hh signaling pathway can maintain CSC phenotype. This pathway includes several important components such as Hh ligands, transmembrane receptors PTCH1 and PTCH2, G-protein-coupled receptors such as Smoothened (SMO), and glioma-associated oncogene transcription factors 1 to 3 (Gli1, Gli2, and Gli3) [104,105]. When the Hh ligand binds to a PTCH receptor, the signaling pathway is activated, and a Gli transcription factor is translocated to the nucleus, where it promotes target gene expression [106-108]. In PCa, the Hh ligands are often synthesized in an autocrine or paracrine manner. For example, bone marrow stromal cells (BMSCs) express Hh ligands and maintain the stemness of PCSCs. Hedgehog signaling also induce the expression of CSC markers, such as BMI1, WNT2, and CD44.
Hh signaling pathway is critically important for CSC functions, and therefore strategies to inhibit the pathway using pharmacological or genetic methods were studied. Cyclopamine, vismodegib, saridegib, and sonidegib are potent and specific SMO inhibitors, with clinical trials indicating therapeutic benefit for patients diagnosed with cancer [109-113]. Sonidegib was found to decrease CSC markers, including NANOG, OCT4, SOX2, and c-MYC [114]. And Gli inhibitors, such as GANT61, also inhibit PCa growth and proliferation [115]. A recent study showed that inhibition of Hh signaling pathway increases the anti-tumor effects of paclitaxel in PCa [116]. Perhaps SHH signaling inhibition of PCSCs represents a therapeutic strategy in patients with prostate cancer bone metastasis.
Dynamic evolution of prostate cancer stem cells in bone metastasis
In fact, the interaction between PCSCs and bone microenvironment is a dynamic process that drives CSC evolution. Rapid division of PCa cells leads to a hypoxic environment, which affects the PCSCs function by enhance their stemness and self-renewal [117].
Disseminated PCa cells can be converted to CSCs in the bone environment, depending on the supply of cytokines and growth factors in the stem cell niches. The CSC phenotype in the bone metastasis is usually defined by the dual expression of CD133 and CD44 [118]. GAS6, expressed by osteoblast, plays an important role in converting non-CSCs into CSCs through the mTOR signaling pathway. PCSCs in the bone marrow of mice with GAS6 knockout showed a significant decrease compared with wildtype GAS6 mice [119]. The downstream signaling pathway of mTOR also plays a critical role in maintaining the CSC phenotype. This signaling is mediated by the Mer receptor on PCa cells. This GAS6/Mer/mTOR pathway reflects the interaction between osteoblast and CSCs in the bone metastatic site. Collectively, the fate of prostate cancer bone metastatic CSCs mainly depends on the bone environment.
Stemness of CSCs is not stable and changes dynamically. It is a transient state that is influenced by genetic, epigenetic, and bone microenvironment. The characteristics of CSCs are affected by their interactions with the bone microenvironment, while factors released by them affect stromal cells in the microenvironment, hijacking them to promote tumor growth. Stromal cells include fibroblast, endothelial, and immune cells, as well as other cell types. The extracellular matrix produced by the stromal cells can also influence CSC function [120,121]. Cancer-associated fibroblasts (CAFs) are an important component of cancer stromal cells and are associated with oxidative stress in the microenvironment, with potential for inducing cancer transformation. CAFs induce metabolic changes such as oxidative phosphorylation in cancer cells [122,123]. CAFs were shown to regulate the proliferative capability of CSCs in a PTEN-gene knockout PCa mouse model. They also activate Wnt and Notch signaling pathways in CSCs to maintain their stemness [124]. CAFs can induce EMT and stemness of PCSCs via production of MMPs and activation of pro-inflammatory signaling factors such as NF-κB and HIF.
Recent studies reported that the hypoxic cancer environment plays an important role in CSC evolution. Hypoxia was found to be conducive to establish an acidic tumor microenvironment and activate several proteases facilitating prostate cancer metastasis. The hypoxic environment can induce cancer cells, including CSCs, to express HIF, which enables cellular adaptation to hypoxia [125,126]. Further, several studies have shown that hypoxia and HIF induce cancer cells to express stem cells-like phenotype by upregulating the expression of OCT4, SOX2, and NANOG [127,128]. The hypoxic environment also activates Notch signaling pathway, thus increasing the population of CSCs and regulating their self-renewal and differentiation. Therefore, when the hypoxic conditions are eliminated, CSCs can be more easily depleted and is conducive for prostate cancer bone metastasis. Evolution of PCSCs is a complex process, affected by various factors that will always occur.
Dormancy of PCSCs
The dormant state of PCSCs can be induced under a foreign environment of the bone marrow. The dormant state can be maintained for prolonged durations, and might be related to their anti-apoptotic and DNA repair capability [129]. When PCSCs enter the dormant state, they become non-proliferative (G0 phase of cell cycle), and are highly conserved. In this state, they can bypass traditional therapies. Actually, cell quiescence is closely related to cell dormancy, and CSCs usually utilize these characteristics to evade harsh environment stimuli [130,131]. This is not, however, a passive state, but rather an active one that requires various signaling pathways. Factors such as p53, RB, p21, p27, and various miRNAs regulate this process. The dormancy of PCSCs is also supported by the CAFs in the tumor hypoxic environment [132,133]. Moreover, disseminated PCa cells in the bone microenvironment also exist in a dormant state for a long time. As dormancy is an important feature of PCSCs, some studies showed that mitotic quiescence rather than surface phenotype can be used to accurately identify PCSCs [134]. The PCSCs exit the dormant state in the presence of external factors so PCSCs switch between dormant and proliferative state.
The HSC niche plays an important role in inducing HSCs dormancy. When the PCSCs arrive to the bone marrow, they reside in the HSCs niche and become dormant [135]. Inactivation of oncogenes, such as Myc, or activation of p38 stress signaling can contribute to tumor dormancy [136,137]. c-MYC is a crucial cell cycle regulator, and its inactivation induce p21 and p27 protein accumulation and dormancy in the cells. Activation of Myc can maintain the stemness of PCSCs, while inactivation of Myc can inhibit PCSCs tumorigenicity. TANK binding kinase 1 (TBK1) plays an important role in maintaining the stemness of PCSCs and inducing dormancy in CSCs. Knockdown of the TBK1 using short hairpin RNA (shRNA) led to activation of mTOR signaling and a decrease in CSCs, which suggests that mTOR signaling pathway is very important for tumor dormancy [138]. The PI3K signaling pathway was also shown to be related to the dormant state of PCSCs and inhibition of the PI3K signaling can reduce the PCSCs.
Dormant PCSCs are also an important factor contributing to treatment failure. During therapeutic intervention, the PCSCs often become dormant to evade death induced by anti-proliferating agents, such as docetaxel. Therefore, new strategies should be developed to target the dormant PCSCs, which is a challenge. Strategies that redirect the dormant PCSCs to enter the G1 phase of the cell cycle might be an effective approach for future investigation.
Treatment resistance related to cancer stem cells
Traditional treatment can effectively destroy the non-stem cells cancer cells but not the dormant CSCs [139,140]. Current therapies mainly target the fast-growing, differentiated cells, but they do not affect the relatively slow-growing CSCs. The CSCs also express high levels of ATP-binding cassette (ABC) transporters that can induce active efflux of drugs and prevent drug uptake. In such cases, P-glycoprotein, and multidrug resistance associated proteins 1 and 2 (MRP1 and MRP2) are usually overexpressed in CSCs [141-144]. Moreover, PCSCs exhibit potent anti-apoptotic capacity, with anti-apoptotic genes such as Bcl-2, Bcl-xl, and survivin overexpressed in some CSCs. These cells have active DNA damage detection and repair systems. Further, PCSCs were shown to be resistant to radiation therapy, which might be related to activation of Chk1 and Chk2 [145]. Treatment of prostate cancer bone metastases with radiation induced reactive oxygen species (ROS) generation in cancer cells, leading to cancer cell death. However, when the PCSCs were treated with radiation, only limited levels ROS were induced resulting in reduced DNA damage [146-148]. Castration is an important treatment method for PCa. However, the cancer cell becomes castration-resistant following prolonged androgen deprivation. Prostate CSCs might play a role in castration-resistance, in which cancer cells show self-renewal and tumor propagation in the absence of androgen receptors [149,150]. Growing evidence shows that CSCs surface markers, including CXCR4 and EpCAM, are involved in chemotherapy resistance. Inhibition of CXCR4 by AMD3100 led to enhanced chemotherapeutic efficiency of docetaxel [151]. Knockdown of EpCAM by short interfering RNA (siRNA) in PCa cell lines increased their chemosensitivity [152].
Androgen receptor (AR) is very important for the survival of normal prostate cells and prostate cancer cells. In primary and untreated PCa, the PCSCs are AR- and therefore do not respond well to androgen deprivation therapy (ADT). Isolated CD44+/ integrin α2β1+/CD133+ CSCs from several human PCa samples expressed ABCG2, but AR expression was almost undetectable [153]. Conversely, loss of AR contributes to PCSCs generation, which might explain the substantially higher levels of PCSCs in castration-resistant PCa than in androgen-sensitive cancers. ADT induces the activation of HIFs, which then lead to PSCSs proliferation and differentiation. ADT downregulates the vascular endothelial growth factor (VEGF) expression, which in turn impairs the tumor vasculature, and thereby induces the hypoxic tumor microenvironment. Both AR+ and AR- clones exist in CRPCs. Most PCSCs do not express AR, while some do, and both clones exhibit distinct biological features [154]. Conversely, the PCSCs also contributed to ADT resistance.
It is evident that cancer cells are driven and maintained by a set of stem cells. Treatment strategies should be directed to CSCs, which are proposed to be the root cause of cancer. A study of PCSCs showed that 5-lipoxygenase inhibition induced apoptosis of PCSCs by activating c-jun N-terminal kinase and downregulating their stemness [155]. The dormant state of PCSCs is also an important factor underlying drug resistance. Strategies for identifying dormant CSCs will lead to therapies targeted at this subclone population of cancer.
Novel treatment strategies and assay systems should be adapted to effectively identify the CSCs, such as those expressing high levels of CD44. When miR-34a was used to inhibit the expression of CD44 in PCSCs, the progression and metastasis of the PCa were remarkably reduced.
Remaining questions and future direction
PCSCs have gained increasing attention in recent years and are supposed to play an important role in prostate cancer bone metastasis and drug resistance. PCSCs are believed to the precursor of bone metastasis. It is proposed that PCSCs occupy the HSCs niche and remain dormant for extended duration, followed by activation by some signals resulting in bone metastasis. The origin of PCSCs needs to be further studied. How to identify the PCSCs in the samples of bone prostate cancer bone metastasis, and how to accurately discriminate the dormant PCSCs using the proper molecular biology techniques are two important challenges that we are faced. Additionally, the gene instability of PCSCs is closely related to drug resistance. Therefore, the in-depth research for the mechanism of PCSCs gene instability will shed some light on prostate cancer bone metastasis.
Conclusion
The importance of CSCs in cancer initiation, progression, recurrence, and metastasis is gaining increasing attention. We have discussed here a number of important aspects of PCa: 1) the role of CSCs in PCa bone metastasis and their drug resistance; 2) the complex interactions between PCSCs and the bone microenvironment following bone metastasis of PCa; 3) relevant signaling pathways related to this interaction; 4) the dynamic evolution of PCSCs in the bone microenvironment, as this is a dynamic rather than static process; 5) the dormant state of PCSCs that is vital for the successful formation of bone metastasis; 6) the role of PCSCs in drug resistance, including castration resistance. We hope that this review will advance our understanding of bone metastasis and drug resistance of PCa.
Acknowledgements
The authors would like to gratefully acknowledge the financial support from the Doctoral Fund of Jining No. 1 People's Hospital (2022-BS-002), the National Natural Science Foundation of China (No. 81803097) and the Natural Science Foundation of Shandong Province (No. ZR2017QH005).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev. 2018;32(17-18):1105-1140
2. Rawla P. Epidemiology of Prostate Cancer. World J Oncol. 2019;10(2):63-89
3. Arriaga JM, Panja S, Alshalalfa M, Zhao J, Zou M, Giacobbe A, Madubata CJ, Yeji Kim J, Rodriguez A, Coleman I, Virk RK, Hibshoosh H, Ertunc O, Ozbek B, Fountain J, Karnes RJ, Luo J, Antonarakis ES, Nelson PS, Feng FY, Rubin MA, De Marzo AM, Rabadan R, Sims PA, Mitrofanova A, Abate-Shen C. A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance. Nat Cancer. 2020;1(11):1082-1096
4. Lin SC, Yu-Lee LY, Lin SH. Osteoblastic Factors in Prostate Cancer Bone Metastasis. Curr Osteoporos Rep. 2018;16(6):642-647
5. Fizazi K, Carducci M, Smith M, Damião R, Brown J, Karsh L, Milecki P, Shore N, Rader M, Wang H, Jiang Q, Tadros S, Dansey R, Goessl C. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813-22
6. Tian JY, Guo FJ, Zheng GY, Ahmad A. Prostate cancer: updates on current strategies for screening, diagnosis and clinical implications of treatment modalities. Carcinogenesis. 2018;39(3):307-317
7. Drazer MW, Stadler WM. The Role of Testosterone in the Treatment of Castration-Resistant Prostate Cancer. Cancer J. 2016;22(5):330-333
8. Jamroze A, Chatta G, Tang DG. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021;518:1-9
9. Rashid N, Javed MM, Hassan A. Fibular findings in carcinoma prostate; a challenging situation for reporting physician. J Pak Med Assoc. 2019;69(10):1572-1573
10. Wen L, Valderrama A, Costantino ME, Simmons S. Real-World Treatment Patterns in Patients with Castrate-Resistant Prostate Cancer and Bone Metastases. Am Health Drug Benefits. 2019;12(3):142-149
11. Francini E, Montagnani F, Nuzzo PV, Gonzalez-Velez M, Alimohamed NS, Rosellini P, Moreno-Candilejo I, Cigliola A, Rubio-Perez J, Crivelli F, Shaw GK, Zhang L, Petrioli R, Bengala C, Francini G, Garcia-Foncillas J, Sweeney CJ, Higano CS, Bryce AH, Harshman LC, Lee-Ying R, Heng DYC. Association of Concomitant Bone Resorption Inhibitors With Overall Survival Among Patients With Metastatic Castration-Resistant Prostate Cancer and Bone Metastases Receiving Abiraterone Acetate With Prednisone as First-Line Therapy. JAMA Netw Open. 2021;4(7):e2116536
12. Suominen MI, Fagerlund KM, Rissanen JP, Konkol YM, Morko JP, Peng Z. et al. Radium-223 Inhibits Osseous Prostate Cancer Growth by Dual Targeting of Cancer Cells and Bone Microenvironment in Mouse Models. Clin Cancer Res. 2017;23(15):4335-4346
13. Salamanna F, Borsari V, Brogini S, Giavaresi G, Parrilli A, Cepollaro S. et al. An in vitro 3D bone metastasis model by using a human bone tissue culture and human sex-related cancer cells. Oncotarget. 2016;7(47):76966-76983
14. Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L. et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med. 2019;216(2):428-449
15. Zhang X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun (Lond). 2019;39(1):76
16. Szade K, Gulati GS, Chan CKF, Kao KS, Miyanishi M, Marjon KD. et al. Where Hematopoietic Stem Cells Live: The Bone Marrow Niche. Antioxid Redox Signal. 2018;29(2):191-204
17. Koren E, Fuchs Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist Updat. 2016;28:1-12
18. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, Zhang G, Wang X, Dong Z, Chen F, Cui H. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8
19. Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124(6):1111-5
20. López-Lázaro M. The stem cell division theory of cancer. Crit Rev Oncol Hematol. 2018;123:95-113
21. Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488(7412):527-30
22. Mohammad K, Dakik P, Medkour Y, Mitrofanova D, Titorenko VI. Quiescence Entry, Maintenance, and Exit in Adult Stem Cells. Int J Mol Sci. 2019 20(9). pii: E2158
23. Bonnet D, Dick J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737
24. Archer LK, Frame FM, Maitland NJ. Stem cells and the role of ETS transcription factors in the differentiation hierarchy of normal and malignant prostate epithelium. J Steroid Biochem Mol Biol. 2017;166:68-83
25. Mei W, Lin X, Kapoor A, Gu Y, Zhao K, Tang D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers (Basel). 2019 11(4). pii: E434
26. Wang X, Xu H, Cheng C, Ji Z, Zhao H, Sheng Y, Li X, Wang J, Shu Y, He Y, Fan L, Dong B, Xue W, Wai Chua C, Wu D, Gao WQ, He Zhu H. Identification of a Zeb1 expressing basal stem cell subpopulation in the prostate. Nat Commun. 2020;11(1):706
27. Sampayo RG, Bissell MJ. Cancer stem cells in breast and prostate: Fact or fiction? Adv Cancer Res. 2019;144:315-341
28. Olson AW, Le V, Wang J, Hiroto A, Kim WK, Lee DH, Aldahl J, Wu X, Kim M, Cunha GR, You S, Sun Z. Stromal androgen and hedgehog signaling regulates stem cell niches in pubertal prostate development. Development. 2021;148(19):dev199738
29. Tolkach Y, Kristiansen G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology. 2018;85(1-2):108-116
30. Flores-Téllez TDNJ, Baena E. Experimental challenges to modeling prostate cancer heterogeneity. Cancer Lett. 2022;524:194-205
31. Talukdar S, Das SK, Pradhan AK, Emdad L, Windle JJ, Sarkar D, Fisher PB. MDA-9/Syntenin (SDCBP) Is a Critical Regulator of Chemoresistance, Survival and Stemness in Prostate Cancer Stem Cells. Cancers (Basel). 2019 12(1). pii: E53
32. Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98(4):756-65
33. Buczek ME, Reeder SP, Regad T. Identification and Isolation of Cancer Stem Cells Using NANOG-EGFP Reporter System. Methods Mol Biol. 2018;1692:139-148
34. Tang B, Raviv A, Esposito D, Flanders KC, Daniel C, Nghiem BT, Garfield S. et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Reports. 2015;4(1):155-169
35. Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene. 2011;30(11):1261-71
36. Kim D, Choi BH, Ryoo IG, Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018;9(9):896
37. Sharpe B, Beresford M, Bowen R, Mitchard J, Chalmers AD. Searching for prostate cancer stem cells: markers and methods. Stem Cell Rev Rep. 2013;9(5):721-30
38. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946-51
39. Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30(36):3833-45
40. Jia L, Wu D, Wang Y, You W, Wang Z, Xiao L. et al. Orphan nuclear receptor TLX contributes to androgen insensitivity in castration-resistant prostate cancer via its repression of androgen receptor transcription. Oncogene. 2018;37(25):3340-3355
41. Gorodetska I, Lukiyanchuk V, Peitzsch C, Kozeretska I, Dubrovska A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int J Cancer. 2019;145(11):2974-2985
42. Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K. et al. Defining a population of stem-like human prostate cancer cells that can generate and propagate castration-resistant prostate cancer. Clin Cancer Res. 2016;17:4505-16
43. Sharpe B, Beresford M, Bowen R, Mitchard J, Chalmers AD. Searching for prostate cancer stem cells: markers and methods. Stem Cell Rev Rep. 2013;9(5):721-30
44. Sánchez BG, Bort A, Vara-Ciruelos D, Díaz-Laviada I. Androgen Deprivation Induces Reprogramming of Prostate Cancer Cells to Stem-Like Cells. Cells. 2020;9(6):1441
45. Lee HC, Ou CH, Huang YC, Hou PC, Creighton CJ, Lin YS, Hu CY, Lin SC. YAP1 overexpression contributes to the development of enzalutamide resistance by induction of cancer stemness and lipid metabolism in prostate cancer. Oncogene. 2021;40(13):2407-2421
46. Vaddi PK, Stamnes MA, Cao H, Chen S. Elimination of SOX2/OCT4-Associated Prostate Cancer Stem Cells Blocks Tumor Development and Enhances Therapeutic Response. Cancers (Basel). 2019 11(9). pii: E1331
47. Lee A, Moon BI, Kim TH. BRCA1/BRCA2 Pathogenic Variant Breast Cancer: Treatment and Prevention Strategies. Ann Lab Med. 2020;40(2):114-121
48. Lecarpentier J, Silvestri V, Kuchenbaecker KB, Barrowdale D, Dennis J, McGuffog L. et al. Prediction of Breast and Prostate Cancer Risks in Male BRCA1 and BRCA2 Mutation Carriers Using Polygenic Risk Scores. J Clin Oncol. 2017;35(20):2240-2250
49. Omari A, Nastały P, Stoupiec S, Bałabas A, Dąbrowska M, Bielińska B, Huss S, Pantel K, Semjonow A, Eltze E, Brandt B, Bednarz-Knoll N. Somatic aberrations of BRCA1 gene are associated with ALDH1, EGFR, and tumor progression in prostate cancer. Int J Cancer. 2019;144(3):607-614
50. DiNatale A, Fatatis A. The Bone Microenvironment in Prostate Cancer Metastasis. Adv Exp Med Biol. 2019;1210:171-184
51. Charles JF, Aliprantis AO. Osteoclasts: more than 'bone eaters'. Trends Mol Med. 2014;20(8):449-59
52. Shi C, Wu T, He Y, Zhang Y, Fu D. Recent advances in bone-targeted therapy. Pharmacol Ther. 2020 107473
53. Zhao B. TNF and Bone Remodeling. Curr Osteoporos Rep. 2017;15(3):126-134
54. Unal M, Creecy A, Nyman JS. The Role of Matrix Composition in the Mechanical Behavior of Bone. Curr Osteoporos Rep. 2018;16(3):205-215
55. Chen X, Wang Z, Duan N, Zhu G, Schwarz EM, Xie C. Osteoblast-osteoclast interactions. Connect Tissue Res. 2018;59(2):99-107
56. Kim B, Huang G, Ho WB, Greenspan DS. Bone morphogenetic protein-1 processes insulin-like growth factor-binding protein 3. J Biol Chem. 2011;286(33):29014-25
57. Minuto F, Palermo C, Arvigo M, Barreca AM. The IGF system and bone. J Endocrinol Invest. 2005;28(8 Suppl):8-10
58. Ueland T, Lekva T, Otterdal K, Dahl TB, Olarescu NC, Jørgensen AP, Fougner KJ, Brixen K, Aukrust P, Bollerslev J. Increased serum and bone matrix levels of transforming growth factor {beta}1 in patients with GH deficiency in response to GH treatment. Eur J Endocrinol. 2011;165(3):393-400
59. Guise TA, Mohammad KS, Clines G, Stebbins EG, Wong DH, Higgins LS, Vessella R, Corey E, Padalecki S, Suva L, Chirgwin JM. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin Cancer Res. 2006;12(20 Pt 2):6213s-6216s
60. Bubendorf L, Schöpfer A, Wagner U, Sauter G, Moch H, Willi N, Gasser TC, Mihatsch MJ. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31(5):578-83
61. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704-15
62. Celià-Terrassa T, Jolly MK. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition in Cancer Metastasis. Cold Spring Harb Perspect Med. 2019 pii: a036905
63. Park M, Cho YJ, Kim B, Ko YJ, Jang Y, Moon YH, Hyun H, Lim W. RANKL immunisation inhibits prostate cancer metastasis by modulating EMT through a RANKL-dependent pathway. Sci Rep. 2021;11(1):12186
64. Plaks V, Koopman CD, Werb Z. Cancer. Circulating tumor cells. Science. 2013;341(6151):1186-8
65. Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell. 2013;24(4):410-21
66. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563-72
67. Chiang AC, Massagué J. Molecular basis of metastasis. N Engl J Med. 2008;359(26):2814-23
68. Luo G, He Y, Yu X. Bone Marrow Adipocyte: An Intimate Partner With Tumor Cells in Bone Metastasis. Front Endocrinol (Lausanne). 2018;9:339
69. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, Walenkamp AM. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49(1):219-30
70. Li JJ, Shen MM. Prostate Stem Cells and Cancer Stem Cells. Cold Spring Harb Perspect Med. 2019;9(6):a030395
71. Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S, Rhim JS. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007;67(7):3153-61
72. Rhim JS, Li H, Furusato B. Novel human prostate epithelial cell culture models for the study of carcinogenesis and of normal stem cells and cancer stem cells. Adv Exp Med Biol. 2011;720:71-80
73. Juarez J, Bendall L. SDF-1 and CXCR4 in normal and malignant hematopoiesis. Histol Histopathol. 2004;19(1):299-309
74. Karpova D, Bonig H. Concise Review: CXCR4/CXCL12 Signaling in Immature Hematopoiesis-Lessons From Pharmacological and Genetic Models. Stem Cells. 2015;33(8):2391-9
75. Liu C, Liu R, Zhang D, Deng Q, Liu B, Chao HP. et al. MicroRNA-141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro-metastasis genes. Nat Commun. 2017;8:14270
76. Chakravarthi BVSK, Chandrashekar DS, Agarwal S, Balasubramanya SAH, Pathi SS, Goswami MT. et al. miR-34a Regulates Expression of the Stathmin-1 Oncoprotein and Prostate Cancer Progression. Mol Cancer Res. 2018;16(7):1125-1137
77. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211-5
78. Lee CH, Decker AM, Cackowski FC, Taichman RS. Bone microenvironment signaling of cancer stem cells as a therapeutic target in metastatic prostate cancer. Cell Biol Toxicol. 2019 doi: 10.1007/s10565-019-09483-7. [Epub ahead of print]
79. Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB, Kuppanna G, Kallappa CG, Basalingappa KM. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018;5:5
80. Sethi S, Macoska J, Chen W, Sarkar FH. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res. 2010;3(1):90-9
81. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R. et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22(3):373-88
82. Stoyanova T1, Riedinger M2, Lin S3, Faltermeier CM4, Smith BA4, Zhang KX5. et al. Activation of Notch1 synergizes with multiple pathways in promoting castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113(42):E6457-E6466
83. Cui D, Dai J, Keller JM, Mizokami A, Xia S, Keller ET. Notch Pathway Inhibition Using PF-03084014, a γ-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Clin Cancer Res. 2015;21(20):4619-29
84. Liu X, Luo X, Wu Y, Xia D, Chen W, Fang Z. et al. MicroRNA-34a Attenuates Paclitaxel Resistance in Prostate Cancer Cells via Direct Suppression of JAG1/Notch1 Axis. Cell Physiol Biochem. 2018;50(1):261-276
85. Zhong J, Huang R, Su Z, Zhang M, Xu M, Gong J. et al. Downregulation of miR-199a-5p promotes prostate adeno-carcinoma progression through loss of its inhibition of HIF-1α. Oncotarget. 2017;8(48):83523-83538
86. Li C, Liu S, Yan R, Han N, Wong KK, Li L. CD54-NOTCH1 axis controls tumor initiation and cancer stem cell functions in human prostate cancer. Theranostics. 2017;7(1):67-80
87. Huang TB, Li YZ, Yu K, Yu Z, Wang Y, Jiang ZW, Wang HM, Yang GL. Effect of the Wnt signal-RANKL/OPG axis on the enhanced osteogenic integration of a lithium incorporated surface. Biomater Sci. 2019;7(3):1101-1116
88. Bisson I, Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009;19(6):683-97
89. Hsieh IS, Chang KC, Tsai YT, Ke JY, Lu PJ, Lee KH, Yeh SD, Hong TM, Chen YL. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/beta-catenin signaling pathway. Carcinogenesis. 2013;34(3):530-8
90. Li Q, Ye L, Guo W, Wang M, Huang S, Peng X. PHF21B overexpression promotes cancer stem cell-like traits in prostate cancer cells by activating the Wnt/β-catenin signaling pathway. J Exp Clin Cancer Res. 2017;36(1):85
91. Zhang K, Guo Y, Wang X, Zhao H, Ji Z, Cheng C. et al. WNT/β-Catenin Directs Self-Renewal Symmetric Cell Division of hTERThigh Prostate Cancer Stem Cells. Cancer Res. 2017;77(9):2534-2547
92. Chakravarthi BVSK, Chandrashekar DS, Hodigere Balasubramanya SA, Robinson AD, Carskadon S, Rao U. et al. Wnt receptor Frizzled 8 is a target of ERG in prostate cancer. Prostate. 2018;78(16):1311-1320
93. Li Q, Ye L, Zhang X, Wang M, Lin C, Huang S. et al. FZD8, a target of p53, promotes bone metastasis in prostate cancer by activating canonical Wnt/β-catenin signaling. Cancer Lett. 2017;402:166-176
94. Murillo-Garzón V, Gorroño-Etxebarria I, Åkerfelt M, Puustinen MC, Sistonen L, Nees M. et al. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat Commun. 2018;9(1):1747
95. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol. 2017;51(5):1357-1369
96. Yun EJ, Baek ST, Xie D, Tseng SF, Dobin T, Hernandez E. et al. DAB2IP regulates cancer stem cell phenotypes through modulating stem cell factor receptor and ZEB1. Oncogene. 2015;34(21):2741-52
97. Xie D, Gore C, Liu J, Pong RC, Mason R, Hao G. et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc Natl Acad Sci U S A. 2010;107(6):2485-90
98. Wang B, Huang J, Zhou J, Hui K, Xu S, Fan J. et al. DAB2IP regulates EMT and metastasis of prostate cancer through targeting PROX1 transcription and destabilizing HIF1α protein. Cell Signal. 2016;28(11):1623-30
99. Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, Ayala GE, Peterson LE, Ittmann M, Spencer DM. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12(6):559-71
100. Song XL, Huang B, Zhou BW, Wang C, Liao ZW, Yu Y, Zhao SC. miR-1301-3p promotes prostate cancer stem cell expansion by targeting SFRP1 and GSK3β. Biomed Pharmacother. 2018;99:369-374
101. Salaritabar A, Berindan-Neagoe I, Darvish B, Hadjiakhoondi F, Manayi A, Devi KP. et al. Targeting Hedgehog signaling pathway: Paving the road for cancer therapy. Pharmacol Res. 2019;141:466-480
102. Sheng T, Li C, Zhang X, Chi S, He N, Chen K, McCormick F, Gatalica Z, Xie J. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer. 2004;3:29
103. Suzman DL, Antonarakis ES. Clinical Implications of Hedgehog Pathway Signaling in Prostate Cancer. Cancers (Basel). 2015;7(4):1983-93
104. Espinosa-Bustos C, Mella J, Soto-Delgado J, Salas CO. State of the art of Smo antagonists for cancer therapy: advances in the target receptor and new ligand structures. Future Med Chem. 2019;11(6):617-638
105. Jeng KS, Chang CF, Lin SS. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int J Mol Sci. 2020 21(3). pii: E758
106. Wong ALA, Bellot GL, Hirpara JL, Pervaiz S. Understanding the cancer stem cell phenotype: A step forward in the therapeutic management of cancer. Biochem Pharmacol. 2019;162:79-88
107. Jeng KS, Chang CF, Lin SS. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int J Mol Sci. 2020 21(3). pii: E758
108. Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res. 2015;21(3):505-13
109. Liu Q, Gu J, Zhang E, He L, Yuan ZX. Targeted Delivery of Therapeutics to Urological Cancer Stem Cells. Curr Pharm Des. 2020;26(17):2038-2056
110. Ishii A, Shigemura K, Kitagawa K, Sung SY, Chen KC, Yi-Te C, Liu MC, Fujisawa M. Anti-tumor Effect of Hedgehog Signaling Inhibitor, Vismodegib, on Castration-resistant Prostate Cancer. Anticancer Res. 2020;40(9):5107-5114
111. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19(11):1410-22
112. Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD. et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366(23):2171-9
113. Ross AE, Hughes RM, Glavaris S, Ghabili K, He P, Anders NM, Harb R, Tosoian JJ, Marchionni L, Schaeffer EM, Partin AW, Allaf ME, Bivalacqua TJ, Chapman C, O'Neal T, DeMarzo AM, Hurley PJ, Rudek MA, Antonarakis ES. Pharmacodynamic and pharmacokinetic neoadjuvant study of hedgehog pathway inhibitor Sonidegib (LDE-225) in men with high-risk localized prostate cancer undergoing prostatectomy. Oncotarget. 2017;8(61):104182-104192
114. Nanta R, Kumar D, Meeker D, Rodova M, Van Veldhuizen PJ, Shankar S, Srivastava RK. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rγ null mice by regulating Bmi-1 and microRNA-128. Oncogenesis. 2013;2:e42
115. Lauth M, Bergström A, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455-60
116. Yang R, Mondal G, Wen D, Mahato RI. Combination therapy of paclitaxel and cyclopamine polymer-drug conjugates to treat advanced prostate cancer. Nanomedicine. 2017;13(2):391-401
117. Marhold M, Tomasich E, El-Gazzar A, Heller G, Spittler A, Horvat R, Krainer M, Horak P. HIF1α Regulates mTOR Signaling and Viability of Prostate Cancer Stem Cells. Mol Cancer Res. 2015;13(3):556-64
118. Shiozawa Y, Berry JE, Eber MR, Jung Y, Yumoto K, Cackowski FC. et al. The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget. 2016;7(27):41217-41232
119. Jung Y, Decker AM, Wang J, Lee E, Kana LA, Yumoto K. et al. Endogenous GAS6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget. 2016;7(18):25698-711
120. Cortini M, Massa A, Avnet S, Bonuccelli G, Baldini N. Tumor-Activated Mesenchymal Stromal Cells Promote Osteosarcoma Stemness and Migratory Potential via IL-6 Secretion. PLoS One. 2016;11(11):e0166500
121. Park TS, Donnenberg VS, Donnenberg AD, Zambidis ET, Zimmerlin L. Dynamic Interactions Between Cancer Stem Cells And Their Stromal Partners. Curr Pathobiol Rep. 2014;2(1):41-52
122. Koochekpour S, Majumdar S, Azabdaftari G, Attwood K, Scioneaux R, Subramani D. et al. Serum glutamate levels correlate with Gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin Cancer Res. 2012;18(21):5888-901
123. Mishra R, Haldar S, Placencio V, Madhav A, Rohena-Rivera K, Agarwal P. et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J Clin Invest. 2018;128(10):4472-4484
124. Kwon OJ, Zhang L, Wang J, Su Q, Feng Q, Zhang XH. et al. Notch promotes tumor metastasis in a prostate-specific Pten-null mouse model. J Clin Invest. 2016;126(7):2626-41
125. Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology. 2014;143(4):512-9
126. Parks SK, Cormerais Y, Pouysségur J. Hypoxia and cellular metabolism in tumour pathophysiology. J Physiol. 2017;595(8):2439-2450
127. Bhuria V, Xing J, Scholta T, Bui KC, Nguyen MLT, Malek NP, Bozko P, Plentz RR. Hypoxia induced Sonic Hedgehog signaling regulates cancer stemness, epithelial-to-mesenchymal transition and invasion in cholangiocarcinoma. Exp Cell Res. 2019;385(2):111671
128. Zuo J, Guo Y, Peng X, Tang Y, Zhang X, He P. et al. Inhibitory action of pristimerin on hypoxia-mediated metastasis involves stem cell characteristics and EMT in PC-3 prostate cancer cells. Oncol Rep. 2015;33(3):1388-94
129. Ruppender NS, Morrissey C, Lange PH, Vessella RL. Dormancy in solid tumors: implications for prostate cancer. Cancer Metastasis Rev. 2013;32(3-4):501-9
130. Kleffel S, Schatton T. Tumor dormancy and cancer stem cells: two sides of the same coin? Adv Exp Med Biol. 2013;734:145-79
131. Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res. 2006;66(19):9349-51
132. De Wever O, Van Bockstal M, Mareel M, Hendrix A, Bracke M. Carcinoma-associated fibroblasts provide operational flexibility in metastasis. Semin Cancer Biol. 2014;25:33-46
133. Gordon JA, Lisle JW, Alman BA, Lian JB. Disruption of crosstalk between mesenchymal stromal and tumor cells in bone marrow as a therapeutic target to prevent metastatic bone disease. J Cell Physiol. 2014;229(12):1884-6
134. Talukdar S, Bhoopathi P, Emdad L, Das S, Sarkar D, Fisher PB. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv Cancer Res. 2019;141:43-84
135. Turner CJ, Edwards CM. The Role of the Microenvironment in Prostate Cancer-Associated Bone Disease. Curr Osteoporos Rep. 2016;14(5):170-7
136. Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB. et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell. 2017;169(5):807-823.e19
137. Guo F, Yuan D, Zhang J, Zhang H, Wang C, Zhu L. et al. Silencing of ARL14 Gene Induces Lung Adenocarcinoma Cells to a Dormant State. Front Cell Dev Biol. 2019;7:238
138. Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, Krebsbach PH, Pienta KJ, Shiozawa Y, Taichman RS. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia. 2013;15(9):1064-74
139. Bighetti-Trevisan RL, Sousa LO, Castilho RM, Almeida LO. Cancer Stem Cells: Powerful Targets to Improve Current Anticancer Therapeutics. Stem Cells Int. 2019;2019:9618065
140. Garcia-Mayea Y, Mir C, Masson F, Paciucci R, LLeonart ME. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol. 2019 pii: S1044-579X(19)30164-6
141. Riganti C, Contino M, Guglielmo S, Perrone MG, Salaroglio IC, Milosevic V, Giampietro R, Leonetti F, Rolando B, Lazzarato L, Colabufo NA, Fruttero R. Design, Biological Evaluation, and Molecular Modeling of Tetrahydroisoquinoline Derivatives: Discovery of A Potent P-Glycoprotein Ligand Overcoming Multidrug Resistance in Cancer Stem Cells. J Med Chem. 2019;62(2):974-986
142. Cho Y, Kim YK. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front Oncol. 2020;10:764
143. Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, Kim KS, Lee YK, Kwon HY. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018;2018:5416923
144. Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int J Mol Sci. 2017;18(12):2574
145. Wang X, Ma Z, Xiao Z, Liu H, Dou Z, Feng X, Shi H. Chk1 knockdown confers radiosensitization in prostate cancer stem cells. Oncol Rep. 2012;28(6):2247-54
146. Yin H, Glass J. The phenotypic radiation resistance of CD44+/CD24(-or low) breast cancer cells is mediated through the enhanced activation of ATM signaling. PLoS One. 2011;6(9):e24080
147. Ogawa K, Yoshioka Y, Isohashi F, Seo Y, Yoshida K, Yamazaki H. Radiotherapy targeting cancer stem cells: current views and future perspectives. Anticancer Res. 2013;33(3):747-54
148. Wang K, Zhang T, Dong Q, Nice EC, Huang C, Wei Y. Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 2013;4:e537
149. Mei W, Lin X, Kapoor A, Gu Y, Zhao K, Tang D. The Contributions of Prostate Cancer Stem Cells in Prostate Cancer Initiation and Metastasis. Cancers (Basel). 2019 11(4). pii: E434
150. Chen X, Rycaj K, Liu X, Tang DG. New insights into prostate cancer stem cells. Cell Cycle. 2013;12(4):579-86
151. Domanska UM, Timmer-Bosscha H, Nagengast WB, Oude Munnink TH, Kruizinga RC, Ananias HJ, Kliphuis NM, Huls G, De Vries EG, de Jong IJ, Walenkamp AM. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia. 2012;14(8):709-18
152. Ni J, Cozzi P, Beretov J, Duan W, Bucci J, Graham P, Li Y. Epithelial cell adhesion molecule (EpCAM) is involved in prostate cancer chemotherapy/radiotherapy response in vivo. BMC Cancer. 2018;18(1):1092
153. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946-51
154. Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol. 2014;11(6):365-76
155. Sarveswaran S, Varma NRS, Morisetty S, Ghosh J. Inhibition of 5-lipoxygenase downregulates stemness and kills prostate cancer stem cells by triggering apoptosis via activation of c-Jun N-terminal kinase. Oncotarget. 2019;10(4):424-436
Author contact
![]() Corresponding author: Xiangyu Zhang, Department of Pathology, Jining First People's Hospital, Jining Medical University, No. 6 Jiankang Road, Jining 272000, China. Tel./Fax: +86 537 6051547; E-mail: zhangxiangyu666com.
Corresponding author: Xiangyu Zhang, Department of Pathology, Jining First People's Hospital, Jining Medical University, No. 6 Jiankang Road, Jining 272000, China. Tel./Fax: +86 537 6051547; E-mail: zhangxiangyu666com.