Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(9):2884-2892. doi:10.7150/jca.71992 This issue Cite
Research Paper
From Immunosuppression to Immunomodulation - Turning Cold Tumours into Hot
Mariangela Garofalo1, ![]() , Katarzyna Wanda Pancer2, Magdalena Wieczorek2, Monika Staniszewska3, Stefano Salmaso1, Paolo Caliceti1, Lukasz Kuryk2,
, Katarzyna Wanda Pancer2, Magdalena Wieczorek2, Monika Staniszewska3, Stefano Salmaso1, Paolo Caliceti1, Lukasz Kuryk2, ![]()
1. Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Via F. Marzolo 5, 35131 Padova, Italy
2. Department of Virology, National Institute of Public Health NIH —National Research Institute, Chocimska 24, 00-791 Warsaw, Poland
3. Centre for Advanced Materials and Technologies, Warsaw University of Technology, Poleczki 19, Warsaw 02-822, Poland
Received 2022-2-13; Accepted 2022-6-4; Published 2022-7-4
Abstract

Cancer cells employ various mechanisms to evade and suppress anti-cancer immune responses generating a “cold” immunosuppressive tumour microenvironment. Oncolytic viruses are a promising tool to convert tumour immunosuppression to immunomodulation and improve the efficacy of cancer treatment. Emerging preclinical and clinical findings confirm that oncolytic viruses act in a multimodal scheme, triggering lyses, immunogenic cell death and finally inducing anti-cancer immune responses. In this paper, we tested the local administration of a novel oncolytic adenovirus AdV-D24-ICOSL-CD40L expressing co-stimulatory molecules ICOSL and CD40L to induce the production of tumour infiltrating lymphocytes to the site of injection. Subsequently, in immunocompetent mouse models, we studied possible correlation between tumour infiltrates and anti-cancer efficacy. Described results showed that the delivery of oncolytic viruses encoding immunomodulatory transgenes in combination with anti-PD1 resulted in synergistic inhibition of both melanoma and mesothelioma tumours. Importantly anti-cancer effect positively correlated with cytotoxic CD8+ tumour-infiltrating lymphocytes exerting a central role in the tumour volume control thus generating beneficial outcomes that will undoubtedly provide new insights into possible future treatment strategies to combat cancer. Altogether our findings highlight the importance of oncolytic vectors able to modulate anti-cancer immune responses that can correlate with efficacy in solid malignancies.
Keywords: oncolytic virus, TILs, immunotherapy, mesothelioma, melanoma, drug delivery
Introduction
The tumour-infiltrating lymphocytes (TILs) is represented by a subgroup of the heterogeneous group of immune cells including mono- and poly-morphonuclear cells of the innate immunity such as NKs, macrophages, DCs, mastocytes, basophils, neutrophils, eosinophils and adaptive T and B cells [1]. Over the years it has become clear how important it is to analyse the nature of the TILs in the tumour microenvironment (TME). In fact, the ratio between anti-tumour immune cells and the immune-suppressive cells infiltrated in the TME is correlated with the prognosis of cancer [2,3]. More precisely, when an immune activator agent reaches the tumour tissue an immune response is triggered, aiming to enhance tumour-specific TILs' infiltration. Consequently, the TME becomes progressively more and more inflamed, overcoming the immune-suppressive conditions that would allow the tumour's immune surveillance escape.
There is increasing evidence supporting a role for immunotherapy in solid tumours such as melanoma and malignant pleural mesothelioma (MPM). Sobhani et al. reported that tumours with mild levels of TILs from MPM patients with epithelioid histological type markedly correlated with improved survival compared to tumours with absent or low TILs expression [4]. Further concerning melanoma, a longer survival was observed in melanoma patients presenting high density of CD8+ cytotoxic T cell infiltration, while in contrast, the infiltration of immunosuppressive tumour-associated macrophages (TAMs) and Treg cells within the tumour has proven to have a negative impact on tumour progression [5,6]. These results stressed the relevance of immunotherapy in cancer treatment. However, it's important to distinguish between T-cell inflamed (“hot”) and non-T-cell inflamed (“cold”) TME, relying on the presence or absence of TILs, which are considered as fundamental biomarkers for solid tumours [7,8]. Beside the T cell infiltration level, also the amount and variety of pro-inflammatory cytokine is a relevant factor, and as a matter of fact hot tumours are classified by molecular signatures of immune activation, differently from the cold tumours [9]. As a consequence, intensive efforts have been invested in the development of novel strategies aiming to allow the “cold- to-hot” conversion of the TMEs, to improve the outcome of the disease for patients treated with immunotherapeutic drugs [10]. A cold tumour, in fact, represents a non-immunoreactive environment, and it has been associated to a scarce or absent production of effector tumour-specific T-cells, and consequently, patients bearing these tumours don't respond to anti- immune checkpoint inhibitors, since the drug target is missing, or is not involved in the process of immune cells inactivation [8]. Collectively, tumorigenesis process is not related only to tumour cell characteristics but is deeply influenced by the TME. It is not surprising, therefore, how currently several different therapeutic strategies aim at inflaming the cold TMEs by increasing the lymphocytic infiltration in the tumour core and thus, leading to a better clinical prognosis.
A promising approach that is currently tested in various clinical studies consist of a novel class of innate immunity activators called oncolytic viruses (OVs), such vectors able to cause the infection and the lysis of cancer cells, spearing the healthy ones [11-13]. It has been shown that virotherapy can modulate anti-cancer immune responses that enhance the efficacy of check-point inhibitors (CPI). Therefore, the combination therapies of oncolytic vectors with CPIs are an encouraging regime for cancer treatment. Furthermore, the efficacy of combining OVs plus CPIs has been shown in pre-clinical studies, and there are currently many ongoing clinical trials assessing combination therapies with inspiring findings [14]. Nevertheless, despite extensive research, oncolytic viruses have shown limited efficacy against solid tumours as monotherapy [15]. Therefore, the advancement of novel and more powerful oncolytic vectors is needed. We have previously engineered an innovative AdV-D24-ICOSL-CD40L able to encode for two co-stimulatory molecules ICOSL and CD40L [16]. Upon cell lysis, both ICOSL and CD40L are released and act as soluble molecules, modulating the host anti-cancer activity by binding to their targets respectively activating the T cells and the APCs [16]. The process of cell lysis induced by the novel OVs may lead to the elimination of the primary tumour and distant metastases, thus the infiltration of TILs in the tumour core, is enhanced, leading to the inflammation of the TME. Therefore, OVs can be able to break the cancer immune tolerance, encouraging also, the development of an immune memory against tumour antigens, which prevents further tumour recurrence.
To date, many oncolytic viruses are under development, at different phases of preclinical and clinical trials [11,17-20]. A fundamental milestone in the development of oncolytic virotherapy for the management of advanced melanoma is Talimogene laherparepvec (T-VEC), under the trade name Imlygic [21]. T-VEC is the first oncolytic virus approved for melanoma metastasis, with a reported overall response rate (ORR) of 25% and complete response rate (CRR) of 10% [22]. Promising oncolytic adenovirus is called ONCOS-102, which is a chimeric 5/3 capsid expressing GM-CSF [23,24]. The treatment with the virus in Phase I clinical study was able to induce anti-tumour immunity and signals of clinical efficacy [25]. Another novel oncolytic virus called Coxsackievirus A21 (CAVATAK) exhibited synergy when administered with immune checkpoint inhibitors. A clinical trial evaluating CAVATAK in combination with ipilimumab resulted in 50% objective responses in melanoma patients [26]. Promising results have been also reported in treatment with DNX-2401, where dramatic responses with long-term survival in recurrent high-grade gliomas were reported [27]. Finally, VALO-D102 is a novel oncolytic adenovirus, expressing OX40L and CD40L, used in PeptiCRAd cancer vaccine platform. Intratumoral administration of PeptiCRAd profoundly elevated tumour-specific T cell responses, inhibited tumour growth, and activated systemic anti-cancer immunity in tested mouse models of melanoma. The combination of PeptiCRAd with anti-PD1, significantly improved anti-cancer effect [28].
Herein, the objective of this research was to assess the efficacy of administration of AdV-D24-ICOSL-CD40L in combination with anti-PD1 and their ability to generate infiltration of TILs. Moreover, we investigated possible correlation between the altitude of infiltration and anti-cancer effect- tumour volume and tumour weight- in tested immunocompetent melanoma and mesothelioma mouse models. Our results demonstrated that the local delivery of the oncolytic virus encoding immunomodulatory transgenes resulted in vivo synergistic anti-cancer effect in mice with established AB12 and B16V tumours. This study showcases the possibility of using a novel OV-formulation to induce the production of tumour-reactive TILs.
Materials and Methods
Cell lines, Virus, Anti-PD1 antibody
Juvenile Fibroblasts, isolated from human male foreskin [29-31] and B16V mouse melanoma cells were kindly provided by Prof. Rinner from the Medical University of Graz (Austria), while mouse mesothelioma cell line AB12 was obtained from Cell Bank Australia. Juvenile fribroblasts were cultured in in Dulbecco's Modified Eagle Medium (DMEM), 1% L-glutamine (Gibco Laboratories), 1% of penicillin/streptomycin (Gibco Laboratories), and 10% fetal bovine serum (FBS, Gibco Laboratories). Murine cell lines were cultured in RPMI 1640 medium (Gibco Laboratories, USA) supplemented with 1% of penicillin/streptomycin (Gibco Laboratories), 1% L-glutamine (Gibco Laboratories) and 10% FBS (Gibco Laboratories). The adenovirus vector AdV-D24-ICOSL-CD40L used in this work has a chimeric serotype 5/3 adenovirus and was generated and amplified using standard adenovirus preparation techniques as previously described [16]. For this experiment a surrogate of the vector encoding mouse CD40L and ICOSL was used. The transgenes expression was induced by a CMV promoter (a CMV-ICOSL-IRES-CD40L expression cassette inserted in place of the E3 region). Purified anti-mouse CD279 (PD1) antibody has been resuspended according to manufacturers' instructions (BioLegend).
CAR and DSG2 Expression in Cancer Cell Lines
AB12 and B16V cells were stained firstly with mouse monoclonal anti-CAR antibody (Santa Cruz Biotech, Dallas, TX, USA) and then with 1:2000 Alexa-Fluor 488 secondary antibody (Abcam, Cambridge, UK) or mouse monoclonal anti-DSG2 antibody (Abcam, Cambridge, UK) and then with 1:2000 Alexa-Fluor 488 secondary (Beckman-Coulter Cytomics FC500) (at least 104 cells/events were analyzed by flow cytometry).
Cell Viability: MTS Cytotoxicity Assay
Juvenile fibroblasts, B16V and AB12, were seeded at a density of 1 × 104 cells/well in a 96-well plate and maintained under standard growth condition (DMEM or RPMI 1640, completed with 5% FBS, 1% L-glutamine and 1% of penicillin/streptomycin (all from Gibco Laboratories). After overnight incubation, cells were treated as follows: (i) PBS, (ii) AdV-D24-ICOSL-CD40L (0.1, 1, 10, 100 VP/cell). All treatments have been diluted in growth media with 2% FBS and cells were then incubated in 5% FBS containing media. Cell viability was determined 96 hrs after treatment, by using CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) according to the manufacturer's instructions (Promega, Madison, WI, USA). The absorbance was measured with a 96-well plate spectrophotometer (Victor NivoTM, PerkinElmer, Milano, Italy) at 490 nm. The experiments were independently carried three times and each experiment contained triple replicates.
In vivo efficacy studies
All animal procedures were performed and approved by the Austrian Federal Ministry of Science and Research (BMWF) (GZ 66.010/0058-V/3b/2019) and Italian Ministry of Health (117/2020-PR). For the efficacy experiments, melanoma murine xenografts were established by subcutaneously (s.c.) injecting respectively 1 × 106 B16V cells into both flanks of 10-week-old C57BL/6 male mice (6 tumours/group) while mesothelioma murine xenografts were established by subcutaneously (s.c.) injecting respectively 1.5 × 107 AB12 cells into both flanks of 10-week-old Balb/c male mice (6-10 tumours/group). Tumours (two tumours per mouse, ~5 × 5 mm in diameter) were randomized prior the treatment initiation as follows: mock (100 μL of PBS) administered intratumorally (i.t.), AdV-D24-ICOSL-CD40L administered i.t. at a concentration of 1.75 × 1010 VP/tumour (3.5 × 1010 VP/mouse), murine anti-PD1 (purified anti-mouse CD279 (PD1) antibody BioLegend) administered intravenously (i.v.) (200 μg/mouse), AdV-D24-ICOSL-CD40L administered i.t. at a concentration of 1.75 × 1010 VP/tumour (3.5 × 1010 VP/mouse) followed by i.v. treatment with anti-PD1 (200 μg/mouse) (Table 1). Tumour size was recorded using calliper on two dimensions every three days. The longest and shortest diameter of tumour at each timepoint were recorded and the tumour volume was calculated using a formula of 0.52 × length x (width)2. All animals were observed for clinical signs, morbidity, or mortality daily during the acclimatization and administration period and additionally 30 min after each treatment (Table S1). Characteristics of clinical signs in animal health scoring.
Treatment characteristics in immunocompetent mouse models.
| # | Treatment group | Cell injection, both flanks | Dose | Schedule (days) |
|---|---|---|---|---|
| Immunocompetent BALB/c AB12 xenograft mesothelioma mouse model | ||||
| 1 | Mock (PBS) | 1.5 × 107 AB12 cells into both flanks of 10-week-old BALB/c male mice (6-10 tumours/group). | PBS i.t. and i.v. | 1-6 |
| 2 | AdV-D24-ICOSL-CD40L | 1.75 × 1010 VP/tumour i.t. | ||
| 3 | Anti-PD1 | 200 μg anti-PD1 i.v. | ||
| 4 | AdV-D24-ICOSL-CD40L + Anti-PD1 | 1.75 × 1010 VP/tumour i.t. + 200 μg anti-PD1 i.v. | ||
| Immunocompetent C57BL/6 B16Vxenograft melanoma mouse model | ||||
| 1 | Mock (PBS) | 1 × 106 B16V cells into both flanks of 10-week-old C57BL/6 male mice (6 tumours/group) | PBS i.t. and i.v. | 1-6 |
| 2 | AdV-D24-ICOSL-CD40L | 1.75 × 1010 VP/tumour i.t. | ||
| 3 | Anti-PD1 | 200 μg anti-PD1 i.v. | ||
| 4 | AdV-D24-ICOSL-CD40L + Anti-PD1 | 1.75 × 1010 VP/tumour i.t. + 200 μg anti-PD1 i.v. | ||
Immune cell infiltrates
The percentage number of mouse immune cell populations were monitored by flow cytometry: mouse CD45+ (cat. number: 550994, BD) lymphocytes: whole T cells (mCD3+ (cat. number: 561798, BD), CD4+ T cells (mCD3+ hCD4+ (cat. number: 552775, BD), CD8+ T cells (mCD3+ mCD8+ (cat. number: 560182, BD). Tumours were harvested and subsequently dissociated with cell strainer. Immune cells were isolated by following the protocol described earlier [32]. After dissociation, cells were washed and stained with antibodies 30 min at 4ºC in the dark and then suspended in PBS. Samples were acquired using BD FACSAriaTM III instrument. The populations were gated with forward and side scattering (FSC-A/SSC-A dot plot) in leukocytic regions. Flow cytometry analysis was performed on FlowJo v10 software.
Histopathological studies
Tissues from murine tumour, spleen and liver underwent routine paraffin processing followed by sectioning at 4 μm and staining with Hematoxylin and Eosin (HE). The histopathological evaluation was performed as a blind using 40×magnification using the microscope Zeiss Axio Imager A2, Carl Zeiss Microscopy GmbH, Jena, Germany.
Statistical Analysis
GraphPad Prism software (Version 9) was used to analyse in vivo variables. Statistical analysis was comprised of a repeated measures one way ANOVA test. The FTV calculation method was used to assess therapeutic synergism [17,18,33]. Briefly, the observed FTV equated the mean tumour volume for each experimental group divided by the mean tumour volume of the PBS control group. The expected FTV for a combination (AdV-D24-ICOSL-CD40L plus anti-PD1) equals the product of the observed FTV for the individual groups (expected combinatory therapy = FTVAdV-D24-ICOSL-CD40L* FTVanti-PD1). The ratio of the expected FTV divided by the observed FTV indicated the nature of the interaction: >1 indicated synergism, 1 indicated additive, and < 1 indicated less than additive (antagonism). Pearson correlation coefficient was used to identify potential relationships between tumour volume and percentage of CD4+ and CD8+ T cells in the tumour infiltrating lymphocytes in tumour tissue.
Results and Discussion
Oncolytic virotherapy is a promising and game-changing cancer treatment, getting increasing interest among researchers worldwide. In the field of cancer immunotherapy, the immunological profile of TME is a key determinant of disease prognosis and therapeutic outcome. Based on reported results it has been shown that OVs are able to modulate and alter TME landscape, resulting in enhanced anti-cancer activity alone or in a combination with other agents. Therefore, OVs are potent immune activators able to promotes profound, long-lasting anti-cancer immune responses and clinical efficacy [33-36].
In this paper, we investigated the ability of novel oncolytic adenovirus AdV-D24-ICOSL-CD40L expressing co-stimulatory molecules ICOSL and CD40L [16] to induce infiltration of TILs to the site of injection. Subsequently, in tested animal models, we evaluated possible correlation between tumour infiltrates and anti-cancer efficacy. Therefore, a central hypothesis of our study was that the intratumoral treatment with the novel oncolytic adenovirus AdV-D24-ICOSL-CD40L combined with CPIs can reshape TME by enhancing infiltration of TILs and inducing immune response against tumour, which will then correlate with clinical efficacy.
Firstly, the oncolytic properties of AdV-D24-ICOSL-CD40L have been evaluated through the MTS cell viability assay (Figure S1). We assessed the overall tumour cell selectivity of the novel oncolytic adenovirus on non-tumour cells juvenile fibroblasts. The results show that none of the tested conditions displayed an impact on the growth of non-tumour cells (cell availability > 90%), confirming the tumour-cell selectivity of the virus. In contrast, cell viability experiments carried out on B16V and AB12 demonstrated that tested virus was able to infect and induce cell death in those cell lines, especially at a concentration of 100 VP/cell (cell viability was 40.4% and 44.7% for B16V and AB12, respectively) (Figure S1). This is not surprising as it is known that genetically modified adenovirus, delta-24, which has a 24-base pair deletion in the Rb-binding region of the E1A gene, shows selective replication and oncolysis in various malignant cells [37,38]. Expression level of adenovirus cell entry receptors on the surface of cancer cells show that the B16V and AB12 cell line expressed DSG2 receptors (approximately 21% and 98% of cells positive for the marker respectively). The expression of CAR receptors low in B16V (negative for CAR expression) compared to AB12 (approx. 90% (Figure S2), thus, suggesting that both cancer cell lines can be targeted with oncolytic adenoviruses exhibiting affinity to DSG2 receptors for therapeutic treatment [22]. Subsequently, to evaluate the ability of AdV-D24-ICOSL-CD40L alone or in combination with anti-PD1 to induce infiltration of TILs we established immunocompetent melanoma and mesothelioma mouse models in syngeneic tumour models: C57BL/6 and BALB/c mice. In our previous preclinical study, we tested anti-cancer efficacy of oncolytic adenovirus expressing GM-CSF in humanized mice engrafted with A2058 melanoma cells. Our previous results reveal that the combination of anti-PD1 with the virus significantly reduced tumour volume and exhibited synergistic anti-cancer effect [39]. Herein, in vivo research was carried out to investigate the possible anti-tumour effects triggered by the combination therapy according to the scheme listed down in Table 1. We showed that the therapy with the virus in combination with anti-PD1 was the most effective regimen in both tested animal models. At the end point (day 33), mesothelioma tumour volumes of mice treated with the combinatory therapy were markedly smaller compared to the other ones treated virus alone or in mock group (9,9 mm3, 72 mm3, 219 mm3, p≤ 0.001, respectively for the combination therapy, virus, and mock group) (Figure 1A). The therapy with anti-PD1 did not significantly reduce the tumour volume (vs mock). Similar observations have been reported in melanoma B16V model, where the combination treatment of AdV-D24-ICOSL-CD40L + anti-PD1was superior over other treatments (102 mm3, 137 mm3, 484 mm3, p≤ 0.001, respectively for the combination therapy, virus, and mock group) (Figure 1B). In line with previous findings also in this model, the therapy with anti-PD1 did not exhibit anti-cancer efficacy.
Anti-cancer properties of tested agents in immunocompetent mouse models. A BALB/c AB12 xenograft mesothelioma mouse model. Mice were engrafted with 1x106 cells/flank. The virus was administered intratumorally on days 1-6 i.t, anti-PD1 was given i.p. on days 1-6. The tumour volumes and weights, clinical health scores were monitored 2-3 times per week. At the end of the study mice were euthanized and tumour collected for immunological analyses. The average tumour volumes and weights are presented as mm3 ± SEM. B C57BL/6 B16V xenograft melanoma mouse model. Mice were engrafted with 5x106 cells/flank cells/flank. The virus was administered intratumorally on days 1-6 i.t, anti-PD1 was given i.p. on days 1-6. The tumour volumes and weights, clinical health scores were monitored 2-3 times per week. At the end of the study mice were euthanized and tumour collected for immunological analyses. The average tumour volumes and weights are presented as mm3 ± SEM. #1 mice in mock group, #2 mice in anti-PD1 treated group (C57BL/6), have been euthanized due to ethical reason before end of the study (tumour volume exceeded 1000 mm3). Therefore, the latest available tumour volume/weight measurements have been considered. Results are expressed as mean ± SEM. ANOVA was used. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
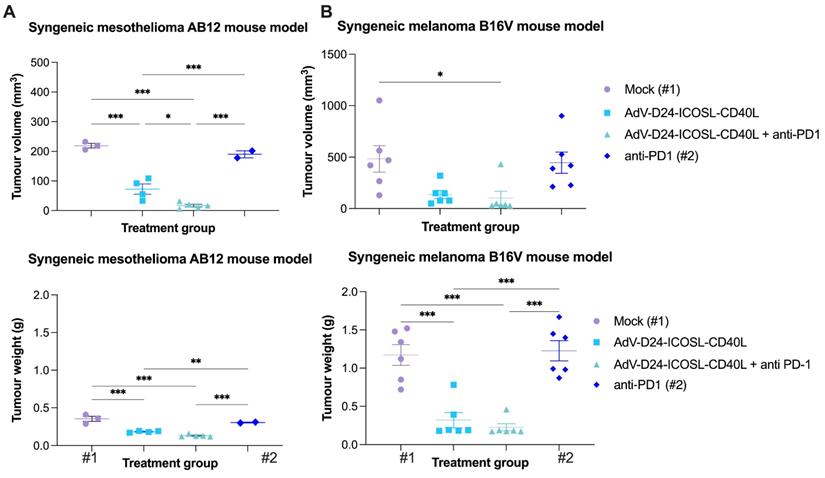
No significant changes in body weight have been reported thorough the study (Figure S3). Interestingly, mice bearing B16V melanoma tumours and treated with combination scheme (Table 1), exhibited 100% survival thus suggesting its safe profile (data not shown). In contrast mice in mock and anti-PD1 treated group in C57BL/6 mice have been euthanized due to ethical reason before end of the study (tumour volume exceeded 1000 mm3). In fact, the B16 is one of the most aggressive melanoma cell lines for C57BL/6 [40]. This cell line is highly aggressive and can metastasize from a primary subcutaneous site to the lungs [41]. Therefore, this model allowed us to test the anti-cancer efficacy of the combinatory approach in highly aggressive skin cancer.
It has been previously reported that the use of OVs in combination with chemotherapy has resulted in synergistic anti-cancer interactions in mesothelioma BALB/c nude mouse model [17]. Further research also demonstrated that OVs given together with doxorubicin or cisplatin plus carboplatin resulted in synergistic anti-tumour activity against soft-tissue sarcoma in Syrian hamster [42]. Moreover, Zhang et al. described that oncolytic viruses synergistically enhanced anti-PD-L1 and anti-CTLA-4 immunotherapy by reshaping the TME [43]. Therefore, to check this possibility, we performed fractional tumour volume analyses which revealed that the combination therapy resulted in synergistic anti-cancer effect in both cancer models (3.7 and 1.2 FTV ratio for mesothelioma and melanoma models respectively) (Table 2), thus suggesting the potential of the proposed approach for improved anti-cancer efficacy and induction of antineoplastic immunity.
Assessment of therapeutic anti-cancer synergism in the AB12 and B16V models in immunocompetent mice (BALB/c and C57BL/6 respectively) with the fractional tumour volume (FTV) calculation method. The latest available tumour volume measurements have been taken for the purpose of the FTV calculation.
| AB12 BALB/c mesothelioma immunocompetent mouse model | |||||
|---|---|---|---|---|---|
| Tumour growth | FTV | AdV-D24-ICOSL-CD40L + Anti-PD1 | |||
| AdV-D24-ICOSL-CD40L | Anti-PD1 | Expa | Obsb | Ratio | |
| FTV | FTV | Exp/Obs | |||
| EoS | 0.331 | 0.868 | 0.287 | 0.077 | 3.718 |
| B16V C57BL/6 melanoma immunocompetent mouse model | |||||
| Tumour growth | FTV | AdV-D24-ICOSL-CD40L + Anti-PD1 | |||
| AdV-D24-ICOSL-CD40L | Anti-PD1 | Expa | Obsb | Ratio | |
| FTV | FTV | Exp/Obs | |||
| EoS | 0.284 | 0.922 | 0.261 | 0.212 | 1.235 |
aExp, expected; bObs, observed; EoS, end of study/last measurements.
Considering that advanced carcinomas are often associated with impaired immune recognition and a highly immunosuppressive TME, the presence of TILs prior a treatment relates to a good clinical prognosis, while the opposite is linked to TME immunosuppression [44]. Despite the importance of both CD4+ and CD8+ T cells subsets, the different effector function of these adaptive immunity cells makes fundamental the characterization of the TILs' composition within the tumour mass, which might represent an important prognostic factor. Focusing on the results obtained on B16V and AB12 model, concerning the infiltration of CD4+ helper and cytotoxic CD8+ T cells it's possible to see how the tested treatment induced an infiltration. Phenotyping analyses of TILs isolated from collected tutors in both animal models showed that the virus alone or combined with anti-PD1 was able to increase the level of immune cell infiltrates, especially CD8+ T cells (AB12: mock: 1.36%, virus: 6%, anti-PD1: 2.4%, combination therapy: 7.38%; B16V: mock: 0.33%, virus: 1.94%, anti-PD1: 0.31%, combination therapy: 2.33%) (Figure 2, Figure S4). Collectively, looking at these results it is possible to speculate that this novel chimeric adenovirus is effectively able to enhance the infiltration of CD8+ cytotoxic T lymphocytes, and therefore exerting a central role in the tumour volume control. Our results show that CD8+ TILs (but not CD4+) count significantly correlated with the tumour volume (p=0.001, p=0.007 respectively for AB12 and B16V models) (Figure 2B) and the tumour weight (p=0.001, p=0.012 respectively for AB12 and B16V models) (Figure 2C). These findings suggest that AdV-D24-ICOSL-CD40L therapy sensitizes tumours to other immunotherapies (e.g. CPIs).
Anti-cancer and immunomodulatory properties of tested agents in immunocompetent mouse models. At the end of the study mice were euthanized and tumour collected for immunological analyses (the latest available tumour samples have been collected for TILs isolation). A Tumour infiltrating lymphocytes CD4+ and CD8+ expression has been assessed in collected tumours. The populations were gated with forward and side scattering (FSC-A/SSC-A dot plot) in leukocytic regions (analysed by flow cytometry, 6-10 tumours/experimental group). B-E Pearson correlation coefficient was used to identify potential relationships between tumour volume/tumour weight and percentage of CD4+ and CD8+ T cells in the tumour infiltrating lymphocytes in tumour tissue. Individual data and mean +/- SEM are presented for each group. Results are expressed as mean ± SEM. ANOVA was used. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
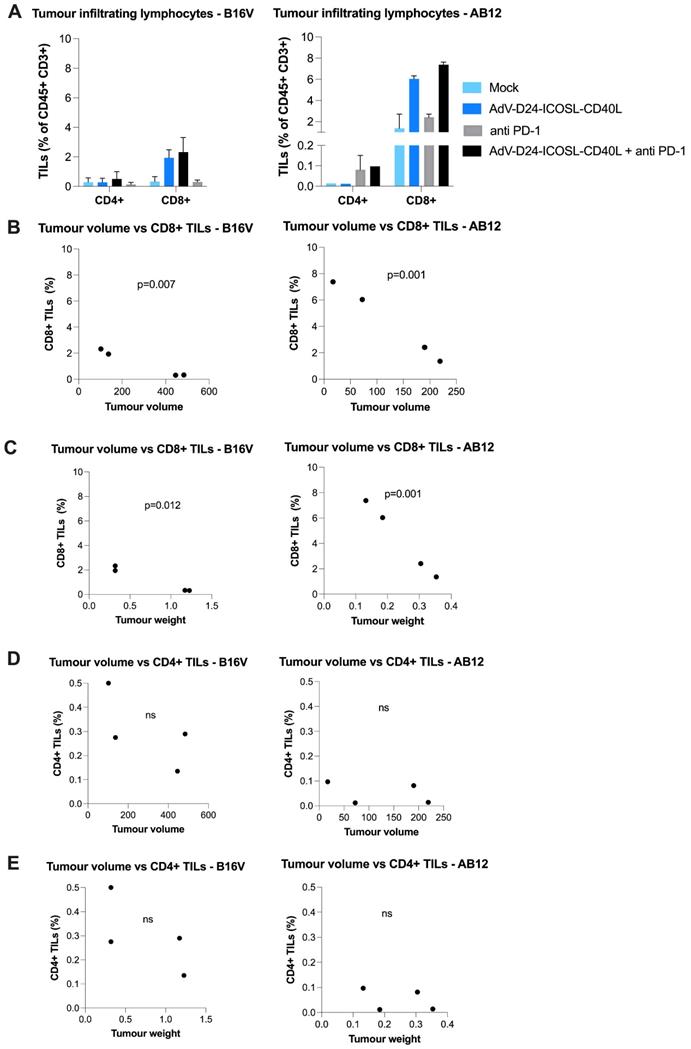
In point of fact, in Phase I clinical study (NCT01598129) the therapy with oncolytic virus ONCOS-102 (AdV5/3-D24-GM-CSF), in patients resistant to available treatments resulted in 40% of disease control rate in evaluable patients (4/10) at 3 months and median overall survival (mOS) was over 9 months. A sound infiltration of TILs to tumour lesions was reported post-treatment in 11 out of 12 patients. Similarly, correlation between cytotoxic CD8+ TILs and macrophages CD68+ in tumours and overall survival was reported, suggesting that the virus was able to reshape local immunological TME at tumours and recruit activated cytotoxic CD8+ T cells able to control disease progression [25,45]. In line with this observation other study evaluated the prognostic role of CD8+ TILs in cancer patients treated with checkpoint inhibitors. Obtained results suggested that high intertumoral infiltration but not circulating cytotoxic T cells, can foresee treatment outcomes in a population with checkpoint inhibitor therapy across different cancer indications [46].
Furthermore, histological analysis showed that samples treated with the combination therapy exhibited no significant histopathological findings: normal liver lobule and normal spleen (Figure S5). Indeed, tumour samples from mice treated with AdV-D24-ICOSL-CD40L plus anti-PD1 show necrotic remnants of cells in a small necrotic area and apoptotic cell remnants in degenerative area, with profound presence of immune cell infiltrates, while tumours treated with PBS showed less frequent presence of immune cell infiltrates with lower number of apoptotic/necrosis cells.
Nevertheless, despite these findings, further research aiming at more detailed phenotyping analyses of TILs, including activation and exhaustion markers is required to better understand how anti-tumour immune responses induced locally, contributes to clinical systemic efficacy. Investigation of tumour antigen specific T cells from the peripheral blood can also provide supportive findings. Although there are still challenges to overcome in regard to oncolytic virotherapy, the combination regime analysed throughout this research has shown potentialities to induce immune response against the tumour, offering a glimmer of light to both melanoma and mesothelioma patients.
Conclusions
Overall, our study reveal that combined treatment with AdV-D24-ICOSL-CD40L and anti-PD1 showed higher anti-tumour activity and a synergistic anti-cancer effect against both melanoma and mesothelioma animal models. Interestingly, as demonstrated in the presented research, anti-cancer effects positively correlated with CD8+cytotoxic T cell infiltrates, thus suggesting possible favourable therapeutic outcomes for cancer patients. Moreover, the development of novel oncolytic vectors armed with potent co-stimulatory molecules could induce durable anti-cancer immune responses and have important implications to advance further clinical therapeutic strategies for the treatment of solid malignancies such as melanoma and mesothelioma. The benefits of compounding various ICIs with oncolytic viruses-encoding many immunomodulatory molecules for patients with solid tumours may be a greater area of focus in the next years.
Supplementary Material
Supplementary figures and table.
Acknowledgements
Ethics Committee Approval and Patient Consent
All animal procedures were performed and approved by the Austrian Federal Ministry of Science and Research (BMWF) (GZ 66.010/0058-V/3b/2019) and Italian Ministry of Health (117/2020-PR).
Source of Funding
L.K. was supported by the National Science Centre, Poland SONATINA (2019/32/C/NZ7/00156) and the National Institute of Public Health NIH - National Research Institute, Poland (1BWBW/2021, BW-3/2022). M.G. acknowledges PRID-J (Grant Number: GARO_SID19_02) funded by University of Padua. M.S. was supported by the Centre for Advanced Materials and Technologies, WUT, Poland and grant POB BioTechMed-1 no. 504/04496/1020/45.010406 (Excellence Initiative Research University).
Competing Interests
L.K. is a shareholder in Targovax.
References
1. Antohe M, Nedelcu RI, Nichita L, Popp CG, Cioplea M, Brinzea A. et al. Tumor infiltrating lymphocytes: The regulator of melanoma evolution (Review). Oncol Lett. 2019;17:4155-4161
2. Mardanpour K, Rahbar M, Mardanpour S, Mardanpour N, Rezaei M. CD8+ T-cell lymphocytes infiltration predict clinical outcomes in Wilms' tumor. Tumor Biol. 2020;42:1010428320975976
3. Pagès F, Galon J, Dieu-Nosjean M-C, Tartour E, Sautès-Fridman C, Fridman W-H. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29:1093-1102
4. Sobhani N, Roviello G, Pivetta T, Ianza A, Bonazza D, Zanconati F. et al. Tumour infiltrating lymphocytes and PD-L1 expression as potential predictors of outcome in patients with malignant pleural mesothelioma. Mol Biol Rep. 2019;46:2713-2720
5. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A. et al. CD103 þ Tumor-Resident CD8 þ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Signi fi cantly During Anti - PD-1 Treatment. 2018; 24:3036-3046.
6. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Emma J, Taylor M. et al. immune resistance. 2015; 515:568-571.
7. Linette GP, Carreno BM. Tumor-Infiltrating Lymphocytes in the Checkpoint Inhibitor Era. Curr Hematol Malig Rep. 2019;14:286-291
8. Ochoa de Olza M, Navarro Rodrigo B, Zimmermann S, Coukos G. Turning up the heat on non-immunoreactive tumours: opportunities for clinical development. The Lancet Oncology. 2020;21:e419-e30
9. Gajewski TF. The Next Hurdle in Cancer Immunotherapy_ Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin Oncol. 2015;42:663-671
10. Liu Z, Han C, Fu Y-X. Targeting innate sensing in the tumor microenvironment to improve immunotherapy. Cell Mol Immunol. 2020;17:13-26
11. Capasso C, Magarkar A, Cervera-Carrascon V, Fusciello M, Feola S, Muller M. et al. A novel in silico framework to improve MHC-I epitopes and break the tolerance to melanoma. Oncoimmunology. 2017;6:e1319028
12. Kuryk L, Møller A-S, Vuolanto A, Pesonen S, Garofalo M, Cerullo V. et al. Optimization of Early Steps in Oncolytic Adenovirus ONCOS-401 Production in T-175 and HYPERFlasks. Int J Mol Sci. 2019;20:621
13. Kuryk L, Bertinato L, Staniszewska M, Pancer K, Wieczorek M, Salmaso S. et al. From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review. Cancers. 2020;12:3057
14. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O. et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109-1119.e10
15. Hwang JK, Hong J, Yun C-O. Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int J Mol Sci. 2020;21:8627
16. Garofalo M, Bertinato L, Staniszewska M, Wieczorek M, Salmaso S, Schrom S. et al. Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model. Pharmaceutics. 2021;13:547
17. Kuryk L, Haavisto E, Garofalo M, Capasso C, Hirvinen M, Pesonen S. et al. Synergistic anti-tumor efficacy of immunogenic adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and standard of care chemotherapy in preclinical mesothelioma model. Int J Cancer. 2016;139:1883-1893
18. Garofalo M, Iovine B, Kuryk L, Capasso C, Hirvinen M, Vitale A. et al. Oncolytic Adenovirus Loaded with L-carnosine as Novel Strategy to Enhance the Antitumor Activity. Mol Cancer Ther. 2016;15:651-660
19. Hirvinen M, Capasso C, Guse K, Garofalo M, Vitale A, Ahonen M. et al. Expression of DAI by an oncolytic vaccinia virus boosts the immunogenicity of the virus and enhances antitumor immunity. Mol Ther - Oncolytics. 2016;3:1-9
20. Garofalo M, Saari H, Somersalo P, Crescenti D, Kuryk L, Aksela L. et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J Control Release. 2018;283:223-234
21. Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum Vaccin Immunother. 2018;14:839-846
22. Kuryk L, Møller AW. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J Med Virol. 2020;92:1309-1315
23. Kuryk L, Møller A-SW, Garofalo M, Cerullo V, Pesonen S, Alemany R. et al. Antitumor-specific T-cell responses induced by oncolytic adenovirus ONCOS-102 (AdV5/3-D24-GM-CSF) in peritoneal mesothelioma mouse model. J Med Virol. 2018;90:1669-1673
24. Kuryk L, Vassilev L, Ranki T, Hemminki A, Karioja-Kallio A, Levälampi O. et al. Toxicological and bio-distribution profile of a GM-CSF-expressing, double-targeted, chimeric oncolytic adenovirus ONCOS-102 - Support for clinical studies on advanced cancer treatment. PLoS One. 2017;12:1-15
25. Ranki T, Pesonen S, Hemminki A, Partanen K, Kairemo K, Alanko T. et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer. 2016;4:1-18
26. Cook M, Chauhan A. Clinical Application of Oncolytic Viruses: A Systematic Review. Int J Mol Sci. 2020;21:7505
27. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS. et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol. 2018;36:1419-1427
28. Ylösmäki E, Ylösmäki L, Fusciello M, Martins B, Ahokas P, Cojoc H. et al. Characterization of a novel OX40 ligand and CD40 ligand-expressing oncolytic adenovirus used in the PeptiCRAd cancer vaccine platform. Mol Ther - Oncolytics. 2021;20:459-469
29. Kretschmer N, Deutsch A, Durchschein C, Rinner B, Stallinger A, Higareda-Almaraz JC. et al. Comparative Gene Expression Analysis in WM164 Melanoma Cells Revealed That β-β-Dimethylacrylshikonin Leads to ROS Generation, Loss of Mitochondrial Membrane Potential, and Autophagy Induction. Mol. 2018;23:2823
30. Durchschein C, Hufner A, Rinner B, Stallinger A, Deutsch A, Lohberger B. et al. Synthesis of Novel Shikonin Derivatives and Pharmacological Effects of Cyclopropylacetylshikonin on Melanoma Cells. Mol. 2018;23:2820
31. Garofalo M, Bellato F, Magliocca S, Malfanti A, Kuryk L, Rinner B. et al. Polymer Coated Oncolytic Adenovirus to Selectively Target Hepatocellular Carcinoma Cells. 2021; 13:949.
32. Tan YS, Lei YL. Isolation of Tumor-Infiltrating Lymphocytes by Ficoll-Paque Density Gradient Centrifugation. Methods in molecular biology. 2019;1960:93-9
33. Kuryk L, Moller AW, Jaderberg M. Quantification and functional evaluation of CD40L production from the adenovirus vector ONCOS-401. Cancer Gene Ther. 2019;26:26-31
34. Kessler T, Wick W. Oncolytic virotherapy: Potentially a game-changing tumor treatment. Cancer Cell. 2021;39:753-755
35. Yin J, Markert JM, Leavenworth JW. Modulation of the Intratumoral Immune Landscape by Oncolytic Herpes Simplex Virus Virotherapy. Frontiers in oncology. 2017;7:136
36. Hemminki O, dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. 2020;13:84
37. Stolarek R, Gomez-Manzano C, Jiang H, Suttle G, Lemoine MG, Fueyo J. Robust infectivity and replication of Delta-24 adenovirus induce cell death in human medulloblastoma. Cancer Gene Ther. 2004;11:713-720
38. Mantwill K, Klein FG, Wang D, Hindupur SV, Ehrenfeld M, Holm PS. et al. Concepts in Oncolytic Adenovirus Therapy. Int J Mol Sci. 2021;22:10522
39. Kuryk L, Møller ASW, Jaderberg M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology. 2019;8:1-11
40. Danciu C, Oprean C, Coricovac DE, Andreea C, Cimpean A, Radeke H. et al. Behaviour of four different B16 murine melanoma cell sublines: C57BL/6J skin. Int J Exp Pathol. 2015;96:73-80
41. Overwijk WW, Restifo NP. B16 as a Mouse Model for Human Melanoma. Curr Protoc Immunol. 2000;39:20.1.1-20.1.29
42. Siurala M, Bramante S, Vassilev L, Hirvinen M, Parviainen S, Tähtinen S. et al. Oncolytic adenovirus and doxorubicin-based chemotherapy results in synergistic antitumor activity against soft-tissue sarcoma. Int J Cancer. 2015;136:945-954
43. Zhang H, Xie W, Zhang Y, Dong X, Liu C, Yi J. et al. Oncolytic adenoviruses synergistically enhance anti-PD-L1 and anti-CTLA-4 immunotherapy by modulating the tumour microenvironment in a 4T1 orthotopic mouse model. Cancer Gene Ther. 2021;29:456-465
44. Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F. et al. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45-53
45. Ranki T, Joensuu T, Jäger E, Karbach J, Wahle C, Kairemo K. et al. Local treatment of a pleural mesothelioma tumor with ONCOS-102 induces a systemic antitumor CD8+ T-cell response, prominent infiltration of CD8+ lymphocytes and Th1 type polarization. Oncoimmunology. 2014;3:e958937-1 -e958937-4
46. Li F, Li C, Cai X, Xie Z, Zhou L, Cheng B. et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine. 2021;41:101134
Author contact
![]() Corresponding authors: mariangela.garofaloit (M.G.) & lkurykgov.pl (L.K.)
Corresponding authors: mariangela.garofaloit (M.G.) & lkurykgov.pl (L.K.)