Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(13):3415-3426. doi:10.7150/jca.63514 This issue Cite
Research Paper
Silibinin inhibits the migration, invasion and epithelial-mesenchymal transition of prostate cancer by activating the autophagic degradation of YAP
Weichao Dan, Yizeng Fan, Tao Hou, Yi Wei, Bo Liu, Taotao Que, Bixin Yu, Jin Zeng ![]() , Lei Li
, Lei Li ![]()
Department of Urology, the First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710061, P.R. China.
Received 2021-6-3; Accepted 2022-4-16; Published 2022-9-25
Abstract

Silibinin (SB), a flavonoid extracted from milk thistle seeds, has been found to exert antitumor effects in numerous tumor types. Our previous study reported that SB had anti-metastatic effects in prostate cancer (PCa). However, the exact underlying molecular mechanisms remain to be determined. The present study aimed to investigate the effects of SB on the migration, invasion and epithelial-mesenchymal transition (EMT) of castration-resistant PCa (CRPC) cells using wound healing, Transwell assays, and western blotting. The results revealed that SB treatment significantly inhibited the migration and invasion of CRPC cell lines. Moreover, SB was confirmed to activate autophagy, as determined using LC3 conversion, LC3 turnover and LC3 puncta assays. Further mechanistic studies indicated that the expression levels of Yes-associated protein (YAP) were downregulated in an autophagy-dependent manner after SB treatment. In addition, the SB-induced autophagic degradation of YAP was associated with the anti-metastatic effects of SB in CRPC. In conclusion, the findings of the present study suggested that SB might inhibit the migration, invasion and EMT of PCa cells by regulating the autophagic degradation of YAP, thus representing a potential novel treatment strategy for metastatic CRPC.
Keywords: silibinin, migration, invasion, epithelial-mesenchymal transition, autophagy, Yes-associated protein
Introduction
Prostate cancer (PCa) is one of the most common types of malignant tumor in men worldwide, accounting for 21% of all new male adult cancer diagnoses in 2020 [1]. According to the statistics, there are currently ~1 million new cases of PCa diagnosed worldwide each year [1]. In the past few decades, the incidence of PCa in China has rapidly increased from 3.52/100,000 individuals in 1998 to 10.23 /100,000 individuals in 2015, with an average annual growth rate of 11.17% [2]. Therefore, PCa poses a serious public health risk among Chinese men. In total, ~1/3 of patients with PCa in China are diagnosed with advanced metastatic PCa, and androgen deprivation therapy is currently an effective treatment option. However, almost all patients will eventually develop into castration-resistant PCa (CRPC) after 12-36 months of treatment [3]. Although combination therapy with chemotherapy and novel endocrine treatments are effective in some cases of CRPC, the majority of patients do not respond well; thus, novel effective treatments are urgently required [3]. In recent years, scientists have paid particular attention to the effects of traditional Chinese medicine on the treatment of CRPC.
Silibinin (SB), a flavonoid extracted from milk thistle seeds, has been proven in our and other previous studies to exert antitumor effects in a wide variety of cancer types, including bladder [4], renal [5], breast [6], gastric [7], lung [8] and colon cancer [9]. Mechanistically, the inhibition of metastasis is considered to be one of the key molecular mechanisms underlying the effects of SB against cancer [10]. Our previous study revealed that SB treatment decreased the invasion and migration of renal cell carcinoma both in vitro and in vivo. In addition, SB attenuated metastasis and epithelial-mesenchymal transition (EMT) in bladder carcinoma [4]. Moreover, our research group also discovered that SB inhibited PCa cell invasion and migration by downregulating the expression levels of Vimentin and matrix metalloproteinase-2 (MMP2), which led to the morphological reversal of the EMT phenotype [11]. However, the exact underlying mechanisms responsible for the anti-metastatic effects of SB in PCa remain to be determined.
Yes-associated protein (YAP) is the core module of the Hippo signaling pathway and has been found to regulate organ size, stem cell functions, tissue regeneration and tumorigenesis [13]. As a transcription factor, YAP translocates to the nucleus to exert its biological functions. Previous studies have reported that YAP was aberrantly upregulated in the majority of patients with PCa, where it served as an oncogene [14,15]. Moreover, YAP was found to promote tumor progression and metastasis through its transcriptional enhanced associate domain (TEAD) [16]. Therefore, developing targeted biological agents against YAP may represent a promising cancer treatment strategy. Notably, a previous study reported that SB could inhibit the YAP signaling pathway, which subsequently promoted apoptosis in human glioblastoma cells; however, the underlying molecular mechanism was not investigated [17]. The present study aimed to investigate the effects of SB on the migration, invasion and EMT of CRPC cells and to determine the underlying molecular mechanisms, with a specific focus on the autophagy-regulated Hippo/YAP signaling pathway. The results revealed that SB could inhibit the migration, invasion and EMT of CRPC cells, which were associated with regulating the autophagic degradation of YAP.
Materials and methods
Reagents and antibodies
SB was purchased from Sigma-Aldrich; Merck KGaA and dissolved in DMSO (50 mM). Hydroxychloroquine sulfate (cat. no. H0915) and 3-methyladenine (3-MA; cat. no. M9281) were also purchased from Sigma-Aldrich; Merck KGaA. Rabbit primary antibodies against YAP (cat. no. 14074), Vimentin (cat. no. 5741) and β-actin (cat. no. 4970) were all purchased from Cell Signaling Technology, Inc. Antibodies against LC3B (cat. no. ab48394), E-cadherin (cat. no. ab15148), N-cadherin (cat. no. ab76057) and GAPDH (cat. no. ab181602) were purchased from Abcam.
Cell lines and culture
The human PCa cell lines, C4-2 and PC-3, were obtained from the American Type Culture Collection and cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 0.1 mg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at a temperature of 37°C. The intracellular vacuoles of PC-3 and C4-2 cells were captured by an inverted light microscope.
MTT assay
Briefly, PCa cells were plated into 96-well plates at a density of 5.0×104 cells/ml and treated with different concentrations (0, 25, 50, 75, 100, 125, 150, 175, 200 µM) of SB for 24 or 48 h. DMSO was used as a negative control. Following the incubation, MTT (150 μl) was added/well for a further 4-h incubation at a temperature of 37°C. DMSO was subsequently added/well to dissolve the purple formazan crystals, and cell proliferation was measured using a 96-well microplate reader (Bio-Rad Laboratories, Inc.) at a wavelength of 490 nm. The equation of MTT assay was:
% Cytotoxicity=(100×(Control-Sample))/Control
Preparation of cytoplasmic and nuclear extracts
Cells at a density of 20.0×104 cells/well were seeded into 6-well plates and treated with SB (50 µM) for 48 h at a temperature of 37°C. DMSO was used as a negative control, the concentration of DMSO was 1‰. The cytoplasmic and nuclear proteins were separated and extracted using a Nuclei EZ Prep Nuclei Isolation kit (Sigma-Aldrich; Merck KGaA) according to the manufacturer's protocol. The expression levels of the different proteins were analyzed using western blotting.
Western blotting
Cells were treated with SB (0, 25, 50 µM) for 48 h at a temperature of 37°C, then total protein was extracted using RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% NP40 and 0.5% sodium deoxycholate; pH 7.4) supplemented with proteinase inhibitors (cat. no. 04693132001; Sigma-Aldrich; Merck KGaA) and phosphatase inhibitors (cat. no. 04906837001; Sigma-Aldrich; Merck KGaA). DMSO was used as a negative control. Total protein was quantified using a Bradford assay and equal amounts (20 µg) of denatured proteins were separated via 10% SDS-PAGE. The separated proteins were subsequently transferred onto polyvinylidene fluoride membranes and blocked with 5% skimmed milk for 1 h at room temperature. The membranes were then incubated with the following primary antibodies overnight at 4°C: Anti-YAP (1:1,000), anti-Vimentin (1:2,000), anti-β-actin (1:5,000), anti-LC3B (1:1,000), anti-E-cadherin (1:2,000), anti-N-cadherin (1:1,000) and anti-GAPDH (1:5,000). Following the primary antibody incubation, the membranes were washed with TBS with 0.1%Tween 20 thrice and incubated with a goat anti-rabbit IgG HRP-conjugated antibody (1:5,000; cat. no. A9169; Sigma-Aldrich; Merck KGaA) for 1 h at a room temperature. Protein bands were visualized using the Clarity Max Western ECL substrate (cat. no. 1705062; Bio-Rad Laboratories, Inc.) and exposure to Bio-Rad's ChemiDoc XRS+ system.
Immunofluorescence
Cells at a density of 5.0×104 cells/well were plated onto round cover slips and treated with DMSO or SB (50 µM) for 48 h at a temperature of 37°C. Following treatment, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized with 0.5% Triton X- 100 solution. Then cells were blocked with 5% serum for 1 h at room temperature. The cells were subsequently incubated with an anti-YAP primary antibody (1:100) overnight at 4°C. Following the primary antibody incubation, the cells were incubated with a Cy3-conjugated goat anti-rabbit IgG secondary antibody (1:200; cat. no. P0183; Beyotime Institute of Biotechnology) for 1 h at room temperature. Cells were then counterstained with 1 µg/ml DAPI for 5 min at a room temperature. YAP expression was captured with a fluorescent microscope (Olympus, Inc., magnification, ×400).
Dual-luciferase reporter assay
Cells at a density of 20.0×104 cells/well were seeded into 6-well plates and transfected with monomeric red fluorescent protein (mRFP)-enhanced green fluorescent protein (EGFP)-LC3 reporter plasmids which were mixed with Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and diluted in Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.). After 6 h, medium was changed and maintained for 18 h. Then cells were treated with DMSO or SB (50 µM) for 48 h at a temperature of 37°C. The expression of EGFP and mRFP was detected with a fluorescent microscope (Olympus, Inc., magnification, ×400).
Wound healing assay
PCa cells were seeded into 6-well plates and cultured with RPMI-1640 medium supplemented with 10% FBS. Upon cells reaching 80-90% confluence, artificial wounds were made by scratching the cell monolayer with a 200‐μl pipette tip. After scratching, the wells were washed twice with PBS to remove the detached cells and cultured with serum‐free RPMI-1640 medium with or without SB (50 µM). DMSO was used as a negative control. The cells were visualized and photographed at 0 and 48 h using an inverted light microscope. The gap distances were semi-quantitatively analyzed using ImageJ 1.48v software (National Institutes of Health).
Transwell migration and invasion assays
Cells were resuspended in serum-free RPMI-1640 medium and adjusted to a density of 5×105 cells/ml. For the invasion assay, a mixture of RPMI-1640/Matrigel (Sigma-Aldrich; Merck KgaA; duration of precoating, 3 h) containing PCa cells was plated into the upper chamber of Transwell plates. RPMI-1640 medium supplemented with 10% FBS was added to the lower chamber. Following incubation at 37°C for 24 or 48 h, the Transwell plates were washed with PBS, then cells were fixed with 4% paraformaldehyde for 15 min at room temperature and stained with 0.1% aniline violet (dissolved in ethanol) for 15 min at room temperature. Stained cells were visualized using an inverted light microscope (magnification, ×100).
Small interfering RNA (siRNA/si) and plasmid transfections
siRNAs targeting autophagy related gene 5 (ATG5) and YAP were designed and purchased from Shanghai Gene Pharma Co., Ltd. A negative control (NC) siRNA (si-NC) (Shanghai Gene Pharma Co., Ltd) was also used. The sequences for the siRNAs were as follows: si-ATG5 sequence, 5'-GAAGTTTGTCCTTCTGCTA-3'; si-NC, 5'-UUCUCCGAACGUGUCACGUTT-3'; si-YAP sequence, 5'-GGUGAUACUAUCAACCAAATT-3'. Briefly, 100 pmol siRNAs (si-NC and siATG5) and Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) were diluted in Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.) and incubated with the cells at 37°C. Medium was changed after 4-6 hours and then subsequent experiments were conducted. YAP overexpression plasmid (the sequence was inserted into the pcDNA3.1 vector) (Shanghai Gene Pharma Co., Ltd.), and their negative controls (Shanghai Gene Pharma Co., Ltd.) were mixed with Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) in Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.). Then the mixture was transfected into cultured PC-3 cells according to standard procedures. The transfection efficiency was analyzed using western blotting following 48 h of transfection.
Quantitative real-time PCR (qRT-PCR)
After SB treatment (50 µM) for 48 h, total cell RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). DMSO was used as a negative control. Then total RNA was reverse-transcribed using the Primer Script RT reagent kit (Takara Bio, Inc), according to tech manufacturer's protocol. The mRNA levels of YAP, E-cadherin and N-cadherin were detected using qRT-PCR. The thermocycling conditions of the qPT-PCR: an activation stage of 50 °C for 2min and 95 °C for 2min; an amplification stage of 95 °C for 15 s, 55 °C for 30 s, and 72 °C for 30 s for 40 cycles. The sequences of the primers PCR amplification were: 5'-CGAGAGCTACACGTTCACGG-3' (forward) and 5'-GGGTGTCGAGGGAAAAATAGG-3' (reverse) (E-cadherin), 5'-TCAGGCGTCTGTAGAGGCTT-3' (forward) and 5'-ATGCACATCCTTCGATAAGACTG-3' (reverse) (N-cadherin), 5'-TAGCCCTGCGTAGCCAGTTA-3' (forward) and 5'- TCATGCTTAGTCCACTGTCTGT-3' (reverse) (YAP), 5'-GGAGCGAGATCCCTCCAAAAT-3' (forward) and 5'-GGCTGTTGTCATACTTCTCATGG-3' (reverse) (GAPDH). Quantification was calculated using the 2-∆∆CT method [18].
Immunohistochemistry assay
Tumors were embedded with paraffin and sectioned into 5-μm slices. After deparaffinized, the sections were hydrated through a graded ethanol series. Then in methanol supplemented with 3% H2O2 was used to block endogenous peroxidase activity. The sections were subsequently washed twice with 0.01M PBS and blocked with FBS (Gibco; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. After rinsing, samples were incubated with primary anti-rabbit YAP (1:200, cat. no. 14074, Cell Signaling Technology, Inc.), Vimentin (1:200, cat. no. 5741, Cell Signaling Technology, Inc.) and E-cadherin (1:50, cat. no. ab15148, Abcam) at 4°C overnight, and incubated with the appropriate secondary antibodies (1-3 drops; cat. no. 8114; Cell Signaling Technology, Inc.) at room temperature for 1h. After stained with diaminobenzidine (DAB), the sections were photographed with inverted light microscope (magnification, ×200). Quantitative analysis was performed using ImageJ v1.47 software (National Institute of Health).
Xenograft animal model
A total of 20 male BALB/c nude mice (weight, 15-20 g; age, 4 weeks) were obtained from the Laboratory Animal Center of Xi'an Jiaotong University (Xi'an, China). All animal experiments were approved by the Biomedical Ethics Committee, Health Science Center of Xi'an Jiaotong University. The health and behavior of the nude mice were monitored daily, high ethical and welfare standards were maintained in all operation involving interactions with animals. The nude mice were housed in a specific pathogen-free environment at a temperature of 22-25°C, with a 12-h light/dark cycle and free access to water and food. PC-3 cells and PC-3 YAP overexpressing cells were resuspended in serum-free RPMI-1640 medium containing Matrigel (Sigma-Aldrich; Merck KGaA) at the density of 2.0×107 cells/ml. In total, 4.0×106 cells in suspension solution were subcutaneously injected into the right flank of nude mice (age, 4 weeks), and upon the tumor volume reaching ~150 mm3 in size, the mice were separated into the following four groups: PC-3 control (n=5), PC-3 SB treatment (n=5), PC-3(YAP overexpressing) control (n=5) and PC-3(YAP overexpressing) SB treatment (n=5) groups. When tumor diameter reached 0.3-0.5mm, the mice were received intraperitoneal injection with the DMSO (control groups) and SB 150 mg/kg (SB-treated groups) every 3 days. The tumor volume was calculated using the following formula: Volume (mm3) =0.5×length×width2. After 30 days, mice were sacrificed by CO2 (30% of the chamber volume/min) and tumors were harvested; the animals were exposed to CO2 until complete cessation of breathing was observed for 10 min. Tumors were embedded with paraffin for immunohistochemistry staining and western blotting analysis.
Statistical analysis
Data are presented as the mean ± SD of three independent experiments. Statistical analyses were performed using GraphPad Prism 5.2 software (GraphPad Software, Inc.). Statistical differences between two groups were determined using unpaired Student's t-test and one-way ANOVA. P<0.05 was considered to indicate a statistically significant difference.
Results
SB inhibits migration, invasion and EMT of CRPC cells
To determine the inhibitory effects of SB, CRPC cell lines PC-3 and C4-2 were treated with different concentrations of SB. As shown in Fig. 1A, SB treatment inhibited the proliferation of CRPC cells in a concentration- and time-dependent manner, with an IC50 value of 30 µM in PC-3 cells and 42 µM in C4-2 cells at 48 h. Furthermore, we detected the effects of SB on cell viability of human benign prostatic hyperplasia cell line BPH-1. The results showed that silibinin didn't have significant inhibitory effects on cell viability of BPH-1. (Fig. S1). Wound healing and Transwell assays were subsequently performed to determine the effect of SB on the migratory and invasive abilities of CRPC cells. The results revealed that SB inhibited the migration and invasion of CRPC cells in a concentration-dependent manner (Fig. 1B-D). It is well known that EMT plays important roles in cancer metastasis [19]. To investigate the effect of SB on the EMT of CRPC, western blotting was used to analyze the expression levels of EMT-related markers, E-cadherin, N-cadherin and Vimentin, in PC-3 cells. The results demonstrated that the expression level of the epithelial marker, E-cadherin, was gradually upregulated, while the expression levels of the mesenchymal markers, N-cadherin and Vimentin, were gradually downregulated following the treatment with SB in both a dose- and time-dependent manner (Fig. 1E and F). To further determine if SB could affect EMT through transcriptional regulation, we performed qRT-PCR to detect message RNA (mRNA) level of EMT-related markers (Fig. S2). The results showed that SB significantly upregulated E-cadherin transcript levels and downregulated N-cadherin transcript levels. These results indicate that SB may inhibit the migration, invasion and EMT of CRPC cells.
SB inhibits the migration, invasion and EMT of prostate cancer cells. (A) PC-3 and C4-2 cells were treated with different concentrations of SB for 24 or 48 h, and the cell viability was analyzed using a MTT assay. (B) Wound healing assay was performed using PC-3 and C4-2 cells following the treatment with 50 µM SB for 48 h. (C) PC-3 and C4-2 cells were treated with 0, 25 or 50 µM SB and Transwell (C) migration and (D) invasion assays were performed. Magnification, x100; scale bar, 200-µm. **P<0.01. (E) PC-3 cells were treated with 0, 25 or 50 µM SB for 48 h. The protein expression levels of E-cadherin, N-cadherin and Vimentin (typically running as a doublet) were analyzed using western blotting. (F) PC-3 cells were treated with 50 µM SB for 0, 12, 24 or 48 h. The protein expression levels of E-cadherin, N-cadherin and Vimentin (typically running as a doublet) were analyzed using western blotting. SB: silibinin.
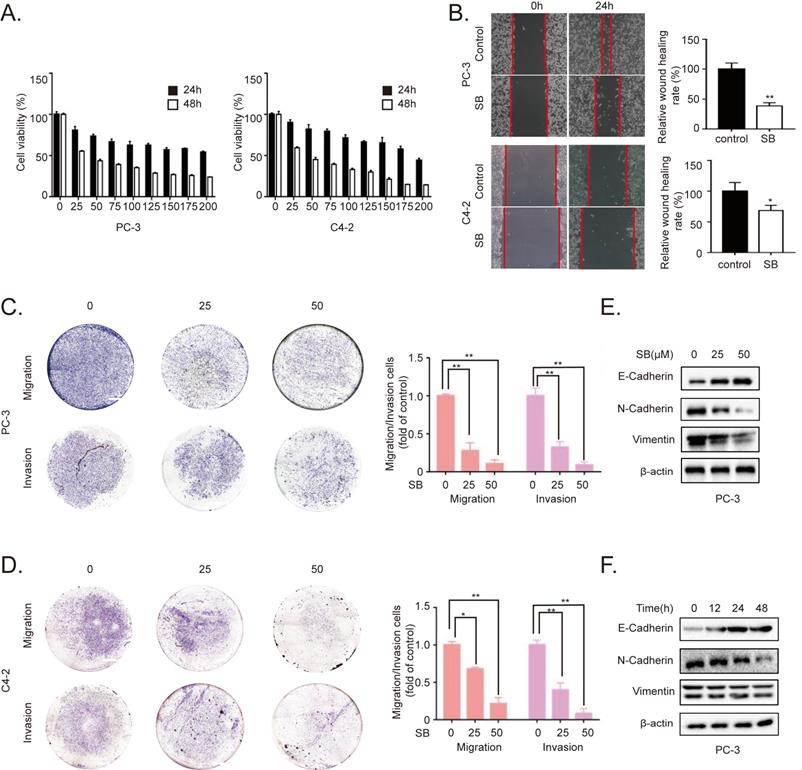
SB induces autophagy in CRPC cells
To determine the antitumor mechanism of SB in CRPC, PC-3 and C4-2 cells were incubated with 50 µM SB for 48 h and morphological changes were subsequently visualized. The number of intracellular vacuoles was significantly increased in PC-3 and C4-2 cells following SB treatment compared with the control group (Fig. 2A). To further determine the association between the formation of intracellular vacuoles and cell autophagy, monomeric red fluorescent protein (mRFP)-enhanced green fluorescent protein (EGFP)-LC3 reporter plasmids were transfected into PC-3 and C4-2 cells. As shown in Fig. 2B, the number of yellow LC3 and red LC3 puncta was increased in CRPC cells treated with SB, indicating that SB could activate autophagy. The expression levels of LC3 were further analyzed using western blotting, and the results showed that LC3-II expression levels were upregulated following SB treatment (Fig. 2C and D). In addition, inhibition of autophagy by 3-MA (which had little impact on CRPC cells proliferation, shown in Fig. S3) significantly attenuated SB-induced inhibition of migration (Fig. 2E) and EMT (Fig. 2F), suggesting an important role for autophagy in the anticancer effects of SB. These results indicate that SB may activate autophagy, and the inhibition of autophagy may attenuate SB-induced antitumor effects in CRPC cells.
Effect of SB on autophagy in prostate cancer cells. (A) PC-3 and C4-2 cells were treated with 50 µM SB for 48 h and then observed under an inverted light microscope. The red box corresponds to the image with a higher magnification ratio. Upper scale bar, 200-µm; lower scale bar, 50-µm. (B) PC-3 and C4-2 cells were transfected with mRFP-EGFP-LC3 reporter plasmids and treated with 50 µM SB for 48 h. Visualization of LC3 puncta was performed using a fluorescence microscope. Scale bar, 50-µm. (C) PC-3 and (D) C4-2 cells were treated with 0, 25 or 50 µM SB for 48 h. The protein expression levels of LC3 were analyzed using western blotting. (E) Transwell migration assays were performed in PC-3 cells following the treatment with 50 µM SB and/or 5 mM 3-MA. Magnification, x100; scale bar, 200-µm. **P<0.01. (F) PC-3 cells were treated with 50 µM SB and/or 5 mM 3-MA. The protein expression levels of E-cadherin, N-cadherin and LC3 were analyzed using western blotting. SB: silibinin; mRFP-EGFP-LC3: monomeric red fluorescent protein-enhanced green fluorescent protein-LC3.
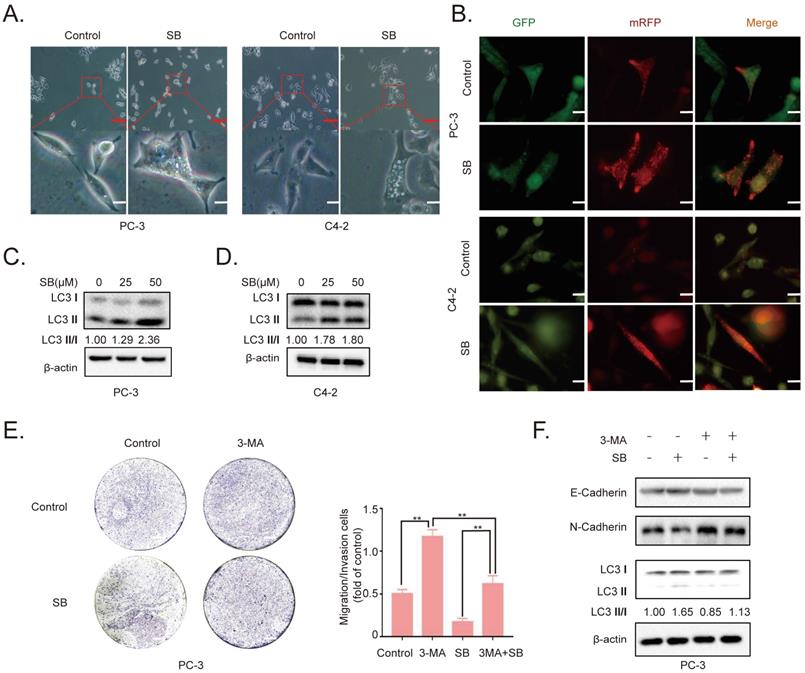
SB promotes the autophagic degradation of YAP
To further determine the role of the YAP signaling pathway in the SB-mediated antitumor effects on CRPC cells, the expression of YAP was determined. The results revealed that SB treatment downregulated YAP expression levels in a concentration- and time-dependent manner in PC-3 cells (Fig. 3A). Similar results were obtained from the immunofluorescence assay (Fig. 3B, Fig. S4). A previous study reported that YAP exerted its functions by entering the nucleus [20]. As shown in Fig. 3C, SB treatment decreased both the cytoplasmic and nuclear expression of YAP. The decrease YAP level could be caused by either decreased synthesis or enhanced degradation by the tumor cells. By examining the YAP transcript level in PC-3 cells cultured with or without SB, we found that SB increased YAP mRNA expression, suggesting that SB might promote YAP protein degradation (Fig. S5). To further explore the mechanism of SB-induced downregulation of YAP, cycloheximide (CHX) was used to inhibit the translation of proteins. PC-3 cells were treated with 50 µM SB for 40 h, and then incubated with 100 µg/ml CHX for 0, 1, 2, 4, 6, and 8 h. The results revealed that YAP expression levels were downregulated to a greater extent in combined treatment group, comparing with CHX treatment alone (Fig. 3D and E). To determine the autophagic mechanism of YAP downregulation, chloroquine (CQ) and si-ATG5 were used to inhibit autophagy-dependent protein degradation. As shown in Fig. 3F and G, inhibition of autophagy attenuated the SB-mediated downregulation of YAP expression. These results indicate that SB may induce the autophagic degradation of YAP in CRPC cells.
SB promotes the autophagic degradation of YAP. (A) PC-3 cells were treated with 0, 25 or 50 µM SB for 48 h or with 50 µM SB for 0, 24 or 48 h. The protein expression levels of YAP were analyzed using western blotting. (B) PC-3 cells were treated with 50 µM SB for 48 h. Immunofluorescence was used to analyze the expression of YAP in PC-3 cells. Scale bar, 50-µm. (C) PC-3 cells were treated with 50 µM SB for 48 h. The cytosolic and nuclear protein expression levels of YAP were detected using western blotting. (D) PC-3 cells were treated with 50 µM SB and/or 100 µg/ml CHX. The protein expression levels of YAP were analyzed using western blotting. (E) Semi-quantification of the band intensities from part (D). YAP protein bands were normalized to β-actin, then normalized to the 0 h time point. (F) PC-3 cells were treated with 50 µM SB and/or 50 µM CQ. The protein expression levels of YAP and LC3 were analyzed using western blotting. (G) PC-3 cells were treated with 50 µM SB and/or transfected with si-ATG5. The protein expression levels of YAP and ATG5 were analyzed using western blotting. SB: silibinin; YAP: Yes-associated protein; CHX: cycloheximide; CQ, chloroquine; ATG5: autophagy related gene 5; si: small interfering RNA.
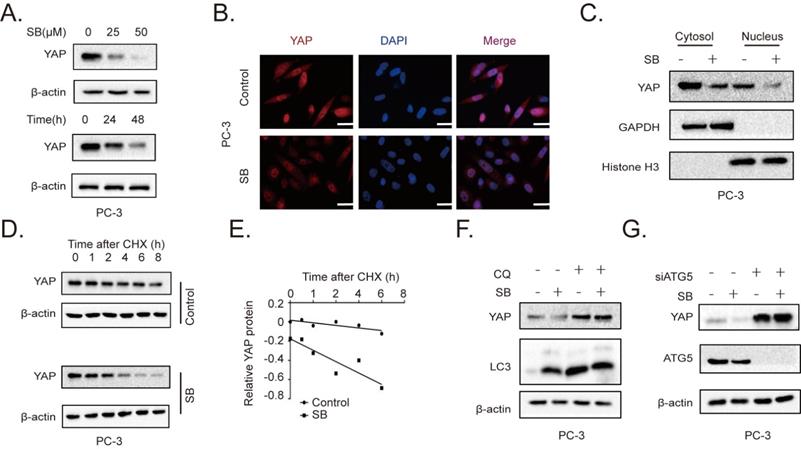
SB inhibits the migration, invasion and EMT of CRPC cells by downregulating YAP expression
To further determine the association between SB-induced YAP degradation and the migration, invasion of CRPC cells, we established YAP-overexpressing PC-3 cells. The results of the Transwell assay demonstrated that the overexpression of YAP attenuated the effects of SB (Fig. 4A and B). Next, siRNA was used to silence YAP expression in C4-2 cells. As expected, the knockdown of YAP further enhanced the inhibitory effects of SB treatment (Fig. 4C and D). The underlying inhibitory effects of SB on EMT and YAP were subsequently explored. As shown in Fig. 4E and F, the results found that YAP overexpression attenuated the suppressive effects of SB, while YAP knockdown promoted the suppressive effects of SB.
SB inhibits the migration, invasion and epithelial-mesenchymal transition of prostate cancer cells by downregulating YAP expression. Transwell (A, B) migration and invasion assays were performed with YAP-overexpressing PC-3 cells. Cells were treated with 50 µM SB for 48 h. Magnification, x100; scale bar, 200-µm. *P<0.05, **P<0.01. Transwell (C, D) migration and invasion assays were performed with YAP-knockdown C4-2 cells. Cells were treated with 50 µM SB for 48 h. Magnification, x100; scale bar, 200-µm. **P<0.01. (E) YAP-overexpressing PC-3 cells were treated with 50 µM SB for 48 h. The protein expression levels of E-cadherin, N-cadherin and YAP were analyzed using western blotting. (F) C4-2 cells were treated with siYAP and SB (50 µM) for 48 h. The protein level of E-cadherin, N-cadherin and YAP were detected by western blotting. SB: silibinin; YAP: Yes-associated protein; si, small interfering RNA.
Antitumor effects of SB on CRPC cells in vivo
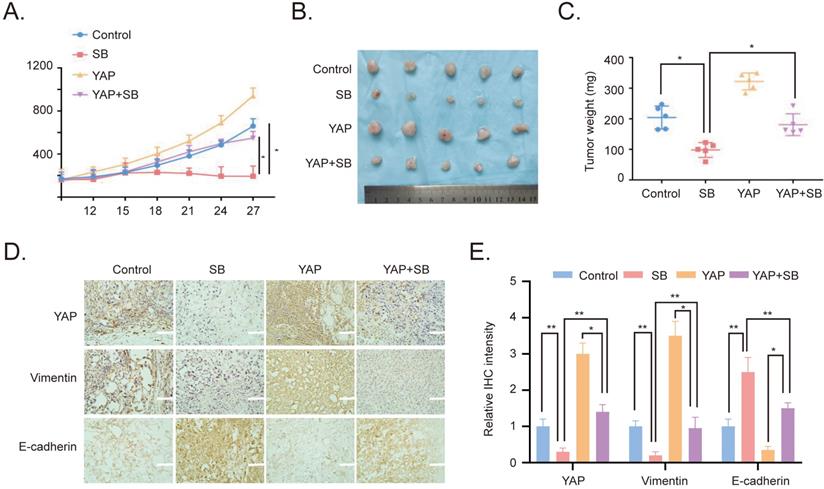
To verify the effects of SB in vitro, YAP-overexpressing PC-3 cells were used to establish a xenograft tumor model in male BALB/c nude mice. The results revealed that the overexpression of YAP increased the growth of the xenograft tumors, while SB treatment had a negative effect on tumor growth (Fig. 5A). Similarly, the weight of the tumors in the YAP-overexpressing group was the largest, while the tumor weight in the SB treatment group was decreased compared with the other groups (Fig. 5B and C). In addition, the expression levels of E-cadherin, Vimentin and YAP in the tumors were analyzed using immunohistochemistry staining. The results demonstrated that the expression levels of YAP and Vimentin were downregulated following SB treatment, while SB treatment upregulated E-cadherin expression levels (Fig. 5D and E). These results suggested that, at least to some extent, SB may be able to reverse the promoting effects of YAP on EMT in vivo.
Antitumor effects of SB on prostate cancer cells in vivo. (A) Tumor volumes from mice were measured every 3 days. (B) Images of the subcutaneous xenograft tumors formed from the different groups following treatment with SB for 30 days. (C) Weight of the dissected xenografts tumors. n=5 mice/experimental group. The data are presented as the mean ± SD. *P<0.05. (D) Expression levels of E-cadherin, Vimentin and YAP in the tumors were analyzed using immunohistochemistry. Scale bar, 100-µm. (E) Quantification of the protein expression from part (D). SB: silibinin; YAP: Yes-associated protein.
Discussion
In the past few decades, scientists have been focused on exploring effective strategies to treat human cancers and searching for novel antitumor drugs[21]. A large number of drugs have been proven to exert a wide range of antitumor effects and are currently used in the clinic, including plant-derived drugs, targeted therapies and chemotherapy drugs. Among them, SB, a flavonoid compound extracted from Silybum marianum, has been suggested as a promising novel treatment for numerous cancer types [22-24]. SB has been reported to exert antitumor effects in breast carcinoma [25], hepatocellular carcinoma [26], bladder cancer [27], PCa [28] and renal cell carcinoma [29].
CRPC, the main reason for PCa metastasis, is inevitable to occur for most PCa patients after initial androgen-deprivation therapy. Different from untreated (hormone-naive) prostate adenocarcinoma, CRPC is characterized by a peculiar genetics molecular landscape. CRPC patients exhibit the main characteristics of hormone-independent, phenotypic plasticity, a degree of stemness signatures, chemotherapy resistance and high rates of metastasis [30]. In the past several years, many novel agents have been designed and applied to treat CRPC, but a considerable number of patients still face the problem of primary resistance to new generation agents [31]. As novel natural compounds, SB has been confirmed to have potential antitumor effects on multiple cancers, including PCa [28]. Despite increasing evidence demonstrating a solid molecular and mechanistic level of positive activity of SB on prostate cancer, its precise effects and underlying mechanism in CRPC are not completely elucidated. The results of the present study revealed that SB inhibited the migration and invasion of CRPC in vitro. Furthermore, the present data demonstrated that SB treatment upregulated E-cadherin expression levels, which is an epithelial marker [32], and downregulated N-cadherin and Vimentin expression levels, which are mesenchymal markers [32]. These results indicated the important inhibitory role of SB in CRPC.
Autophagy is a cellular process that involves transporting cellular materials to lysosomes for degradation [33]. As a self-digestion mechanism, autophagy has been discovered to play important roles in numerous diseases types, particularly in cancer [34]. Previous studies have reported that autophagy suppressed tumorigenesis via multiple possible mechanisms [35,36]. In prostate and breast cancer, the expression of several essential autophagy genes was found to be partially downregulated, such as Beclin-1 (BECN1) [37,38]. Further studies have also demonstrated that the suppression of autophagy promoted cancer cell proliferation [39-41]. Previous studies showed that BECN1 heterozygous mutant mice were more susceptible to tumor development [42-44]. Moreover, accumulating evidence verified that autophagy could inhibit EMT. EMT regulators Snail and Slug were upregulated in glioblastoma cells after autophagy inhibition by silencing Beclin-1, leading to enhancement of migration and invasion [45]. Vice versa, nutrient deprivation-mediated autophagy induction could suppress the mesenchymal phenotype and tumor metastasis [46]. Additionally, it was reported that death effector domain-containing DNA-binding protein (DEDD) physically interacted with the class III PtdIns 3-kinase complex, which controlled the initiation of autophagy. This interaction activated autophagy and induced the autophagy-mediated lysosomal degradation of Snail and Twist, two master inducers of the EMT process [47,48]. Therefore, autophagy is generally considered as a tumor-suppressive mechanism and targeting autophagy has been hypothesized to represent a promising cancer treatment strategy. The findings of the present study revealed that SB treatment induced autophagy in PCa cells, which was verified using inverted microscopy, a LC3 turnover assay and western blotting. In addition, SB-mediated inhibition of migration and invasion was found to be closely associated with autophagy. Pretreatment with 3-MA, an inhibitor of autophagy, partially rescued SB-induced anti-metastatic effects in PCa cells.
As a transcriptional co-activator that mediates multiple biological functions, YAP has been discovered to be essential for cancer initiation, progression and metastasis in the majority of tumor types [49]. In PCa, previous studies have identified that YAP was hyperactivated and drove tumorigenesis and development [30,50,51]. Another study found that the expression levels of the YAP inhibitors, LATS1/2, were significantly downregulated in metastatic PCa compared with non-metastatic PCa, which indicated a role for YAP in PCa metastasis [52]. Several treatments targeting YAP signaling pathways have shown therapeutic potential in CRPC and other types of urological cancer [53]. Verteporfin, an inhibitor of the YAP/TEAD interaction, was found to inhibit the proliferation of CRPC [54]. Sphingomab, a sphingosine-1-phosphate antagonist that inhibited YAP transcriptional activity, was discovered to attenuate the metastasis of bladder cancer cells [55]. In the present study, SB treatment downregulated the protein expression levels of YAP, which resulted in the inhibition of the invasion, migration and EMT of CRPC cells. Moreover, the current data found that post-translational regulation was the main underlying mechanism of the SB-induced downregulation of YAP expression. Previous studies have reported that YAP was degraded through the autophagic-lysosomal [56-58] and ubiquitin system [59-61]. The present study used CQ and si-ATG5 to inhibit protein degradation via the autophagy-lysosomal pathway. The expression levels of YAP were significantly upregulated when autophagy was blocked by CQ or si-ATG5. Furthermore, blocking autophagy also attenuated the inhibitory effects of SB on the migration, invasion and EMT of CRPC. Therefore, the present research discovered a potential novel mechanism of autophagy-dependent degradation of YAP that was mediated by SB in CRPC. Basically, SB is reported to modulate invasion, migration and EMT phenotype in cancers through multiple mechanisms, such as AR signaling [62] and Wnt/β-catenin pathway [63]. Our present study proposed a novel viewpoint which partially contributed to SB-mediated cancer inhibition. However, whether the SB-mediated downregulation of YAP expression is also dependent on other mechanisms, and the underlying association between YAP and metastatic features requires further investigations.
In conclusion, the findings of the present study suggested that SB might promote the degradation of YAP by activating autophagy, which highlighted a potential novel mechanism for the antitumor effects of SB in CRPC. These findings indicate that SB may represent an effective agent for CRPC treatment.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NO. 82073304 and 81925028).
Ethics Committee Approval
The study was approved by Biomedical Ethics Committee, Health Science Center of Xi'an Jiaotong University (NO: 2021-1537-2).
Ethic statement
All of the experimental procedures involving animals were conducted in according with the Institutional Animal Care Guideline of Xi'an Jiaotong University and approved by Biomedical Ethics Committee, Health Science Center of Xi'an Jiaotong University.
Author contributions
L.L. and J.Z. designed and supervised all experiments and contributed to the preparation of the manuscript. W.-C.D., Y.-Z.F., J.Z., T.H., Y.W, B.L, T.-T.Q, and B.-X.Y. performed the experiments, analyzed the data and contributed to the preparation of the manuscript. W.-C.D., Y.-Z.F and T.H. collected animal samples and helped conduct the animal experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7-30
2. Zeng H, Chen W, Zheng R, Zhang S, Ji JS, Zou X. et al. Changing cancer survival in China during 2003-15: a pooled analysis of 17 population-based cancer registries. Lancet Glob Health. 2018;6:e555-67
3. Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I. et al. Prostate cancer. Lancet. 2016;387:70-82
4. Wu K, Ning Z, Zeng J, Fan J, Zhou J, Zhang T. et al. Silibinin inhibits beta-catenin/ZEB1 signaling and suppresses bladder cancer metastasis via dual-blocking epithelial-mesenchymal transition and stemness. Cell Signal. 2013;25:2625-33
5. Ma Z, Liu W, Zeng J, Zhou J, Guo P, Xie H. et al. Silibinin induces apoptosis through inhibition of the mTOR-GLI1-BCL2 pathway in renal cell carcinoma. Oncol Rep. 2015;34:2461-8
6. Si L, Fu J, Liu W, Hayashi T, Nie Y, Mizuno K. et al. Silibinin inhibits migration and invasion of breast cancer MDA-MB-231 cells through induction of mitochondrial fusion. Mol Cell Biochem. 2020;463:189-201
7. Wang YX, Cai H, Jiang G, Zhou TB, Wu H. Silibinin inhibits proliferation, induces apoptosis and causes cell cycle arrest in human gastric cancer MGC803 cells via STAT3 pathway inhibition. Asian Pac J Cancer Prev. 2014;15:6791-8
8. Hou X, Du H, Quan X, Shi L, Zhang Q, Wu Y. et al. Silibinin Inhibits NSCLC Metastasis by Targeting the EGFR/LOX Pathway. Front Pharmacol. 2018;9:21
9. Zheng R, Ma J, Wang D, Dong W, Wang S, Liu T. et al. Chemopreventive Effects of Silibinin on Colitis-Associated Tumorigenesis by Inhibiting IL-6/STAT3 Signaling Pathway. Mediators Inflamm. 2018;2018:1562010
10. Deep G, Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer Metastasis Rev. 2010;29:447-63
11. Wu KJ, Zeng J, Zhu GD, Zhang LL, Zhang D, Li L. et al. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol Sin. 2009;30:1162-8
12. Wu K, Zeng J, Li L, Fan J, Zhang D, Xue Y. et al. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol Rep. 2010;23:1545-52
13. Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811-28
14. Collak FK, Demir U, Ozkanli S, Kurum E, Zerk PE. Increased expression of YAP1 in prostate cancer correlates with extraprostatic extension. Cancer Biol Med. 2017;14:405-13
15. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747-61
16. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci U S A. 2012;109:E2441-50
17. Bai ZL, Tay V, Guo SZ, Ren J, Shu MG. Silibinin Induced Human Glioblastoma Cell Apoptosis Concomitant with Autophagy through Simultaneous Inhibition of mTOR and YAP. Biomed Res Int. 2018;2018:6165192
18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8
19. Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. J Clin Med. 2016;5:1-17
20. Misra JR, Irvine KD. The Hippo Signaling Network and Its Biological Functions. Annu Rev Genet. 2018;52:65-87
21. Gupta SC, Sung B, Prasad S, Webb LJ, Aggarwal BB. Cancer drug discovery by repurposing: teaching new tricks to old dogs. Trends Pharmacol Sci. 2013;34:508-17
22. Cheung CW, Gibbons N, Johnson DW, Nicol DL. Silibinin-a promising new treatment for cancer. Anticancer Agents Med Chem. 2010;10:186-95
23. Jahanafrooz Z, Motamed N, Rinner B, Mokhtarzadeh A, Baradaran B. Silibinin to improve cancer therapeutic, as an apoptotic inducer, autophagy modulator, cell cycle inhibitor, and microRNAs regulator. Life Sci. 2018;213:236-47
24. Li L, Zeng J, Gao Y, He D. Targeting silibinin in the antiproliferative pathway. Expert Opin Investig Drugs. 2010;19:243-55
25. Si L, Fu J, Liu W, Hayashi T, Nie Y, Mizuno K. et al. Silibinin inhibits migration and invasion of breast cancer MDA-MB-231 cells through induction of mitochondrial fusion. Mol Cell Biochem. 2020;463:189-201
26. Varghese L, Agarwal C, Tyagi A, Singh RP, Agarwal R. Silibinin efficacy against human hepatocellular carcinoma. Clin Cancer Res. 2005;11:8441-8
27. Wu K, Ning Z, Zeng J, Fan J, Zhou J, Zhang T. et al. Silibinin inhibits beta-catenin/ZEB1 signaling and suppresses bladder cancer metastasis via dual-blocking epithelial-mesenchymal transition and stemness. Cell Signal. 2013;25:2625-33
28. Wu K, Zeng J, Li L, Fan J, Zhang D, Xue Y. et al. Silibinin reverses epithelial-to-mesenchymal transition in metastatic prostate cancer cells by targeting transcription factors. Oncol Rep. 2010;23:1545-52
29. Ma Z, Liu W, Zeng J, Zhou J, Guo P, Xie H. et al. Silibinin induces apoptosis through inhibition of the mTOR-GLI1-BCL2 pathway in renal cell carcinoma. Oncol Rep. 2015;34:2461-8
30. Salem O, Hansen CG. The Hippo Pathway in Prostate Cancer. Cells-Basel. 2019;8:370
31. Snipaitiene K, Bakavicius A, Lazutka JR, Ulys A, Jankevicius F, Jarmalaite S. Urinary microRNAs can predict response to abiraterone acetate in castration resistant prostate cancer: A pilot study. Prostate. 2021;2021:1-8
32. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
33. Levy J, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528-42
34. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726-34
35. Amaravadi RK, Kimmelman AC, Debnath J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019;9:1167-81
36. Levy J, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528-42
37. Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E. et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59-65
38. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651-62
39. Strohecker AM, White E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014;4:766-72
40. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
41. Yang A, Kimmelman AC. Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status. Autophagy. 2014;10:1683-4
42. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077-82
43. Wei Y, An Z, Zou Z, Sumpter R, Su M, Zang X. et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife. 2015;4:e05289
44. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809-20
45. Catalano M, D'Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C. et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9:1612-25
46. Chen HT, Liu H, Mao MJ, Tan Y, Mo XQ, Meng XJ. et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Cancer. 2019;18:101
47. Lv Q, Hua F, Hu ZW. DEDD, a novel tumor repressor, reverses epithelial-mesenchymal transition by activating selective autophagy. Autophagy. 2012;8:1675-6
48. Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H. et al. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012;72:3238-50
49. Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811-28
50. Zhang L, Yang S, Chen X, Stauffer S, Yu F, Lele SM. et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol Cell Biol. 2015;35:1350-62
51. Holden JK, Cunningham CN. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers (Basel). 2018;10:81
52. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26:54-68
53. Lin KC, Park HW, Guan KL. Deregulation and Therapeutic Potential of the Hippo Pathway in Cancer. Annual Review of Cancer Biology. 2018;2:30617-50202
54. Kuser-Abali G, Alptekin A, Lewis M, Garraway IP, Cinar B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat Commun. 2015;6:8126
55. Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM. et al. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. Embo Mol Med. 2012;4:761-75
56. Liang N, Zhang C, Dill P, Panasyuk G, Pion D, Koka V. et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med. 2014;211:2249-63
57. Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML. et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun. 2018;9:4962
58. Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T. et al. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKɛ-mediated phosphorylation. Nat Immunol. 2017;18:733-43
59. Neufeld TP. Hippo Signaling: Autophagy Waits in the Wings. Dev Cell. 2020;52:544-5
60. Toloczko A, Guo F, Yuen HF, Wen Q, Wood SA, Ong YS. et al. Deubiquitinating Enzyme USP9X Suppresses Tumor Growth via LATS Kinase and Core Components of the Hippo Pathway. Cancer Res. 2017;77:4921-33
61. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF (beta-TRCP). Genes Dev. 2010;24:72-85
62. Zhu W, Zhang JS, Young CY. Silymarin inhibits function of the androgen receptor by reducing nuclear localization of the receptor in the human prostate cancer cell line LNCaP. Carcinogenesis. 2001;22:1399-403
63. Lu W, Lin C, King TD, Chen H, Reynolds RC, Li Y. Silibinin inhibits Wnt/β-catenin signaling by suppressing Wnt co-receptor LRP6 expression in human prostate and breast cancer cells. Cell Signal. 2012;24:2291-6
Author contact
![]() Corresponding authors: Professor Lei Li or Professor Jin Zeng; Department of Urology, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an, Shaanxi 710061, P.R. China. E-mail: lilydrcom (Lei Li); E-mail: zengjin1984com (Jin Zeng)
Corresponding authors: Professor Lei Li or Professor Jin Zeng; Department of Urology, The First Affiliated Hospital of Xi'an Jiaotong University, 277 Yanta West Road, Xi'an, Shaanxi 710061, P.R. China. E-mail: lilydrcom (Lei Li); E-mail: zengjin1984com (Jin Zeng)