Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2023; 14(3):393-402. doi:10.7150/jca.77322 This issue Cite
Research Paper
Platycodin D confers oxaliplatin Resistance in Colorectal Cancer by activating the LATS2/YAP1 axis of the hippo signaling pathway
Chien-Hao Wang1,2, Rathinasamy Baskaran3, Shawn Shang-Chuan Ng4,5, Tso-Fu Wang6,7, Chi-Cheng Li6,8, Tsung-Jung Ho9,10, Dennis Jine-Yuan Hsieh11,12, Chia-Hua Kuo13,14, Ming-Cheng Chen15,16 ![]() , Chih-Yang Huang17,18,19,20,21
, Chih-Yang Huang17,18,19,20,21 ![]()
1. Department of Chinese Medicine, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Tzu Chi University, Hualien, Taiwan
2. Integration Center of Traditional Chinese and Modern Medicine, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien, Taiwan
3. Department of Bioinformatics and Medical Engineering, Asia University, Taichung, Taiwan
4. Department of Biological Science and Technology, College of Life Sciences, China Medical University, Taichung, Taiwan
5. Ph.D. Program for Biotechnology Industry, China Medical University, Taichung 406, Taiwan
6. Department of Hematology and Oncology, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien, Taiwan
7. School of Medicine Tzu Chi University, 701, Section 3, Chung-Yang Road, Hualien 97004, Taiwan
8. Center of Stem Cell & Precision Medicine, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien, Taiwan
9. Chinese Medicine, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Tzu Chi University, Hualien, Taiwan
10. Department of Medical Laboratory Science and Biotechnology, China Medical University, Taichung, Taiwan
11. School of Medical Laboratory and Biotechnology, Chung Shan Medical University, Taichung, Taiwan
12. Clinical Laboratory, Chung Shan Medical University Hospital, Taichung 402, Taiwan
13. Laboratory of Exercise Biochemistry, University of Taipei, Taipei, Taiwan
14. Department of Kinesiology and Health Science, College of William and Mary, Williamsburg, VA, USA
15. Department of Surgery, Division of Colorectal Surgery, Taichung Veterans General Hospital, Taichung, Taiwan
16. Faculty of Medicine, National Yang-Ming University, Taipei, Taiwan.
17. Cardiovascular and Mitochondria related diseases research center, Hualien Tzu Chi Hospital, Hualien 970, Taiwan
18. Graduate Institute of Biomedicine, China Medical University, Taichung, Taiwan
19. Department of Biotechnology, Asia University, Taichung 413, Taiwan
20. Center of General Education, Buddhist Tzu Chi Medical Foundation, Tzu Chi University of Science and Technology, Hualien 970, Taiwan
21. Department of Medical Research, China Medical University Hospital, China Medical University, Taichung 404, Taiwan
Received 2022-7-21; Accepted 2022-9-6; Published 2023-1-22
Abstract

Oxaliplatin-based therapy is used as a first-line drug to treat metastatic colorectal cancer. However, long-term and repeated drug treatment resulted in drug resistance and the failure of chemotherapy. Various natural compounds were previously reported to act as chemosensitizers to reverse drug resistance. In this study, we found that platycodin D (PD), a saponin found in Platycodon grandiflorum, inhibited LoVo and OR-LoVo cells proliferation, invasion, and migration ability. Our results indicated that combined treatment of oxaliplatin with PD dramatically reduced the cellular proliferation in both LoVo and OR-LoVo cells. Furthermore, treatment with PD dose-dependently decreased LATS2/YAP1 hippo signaling and survival marker p-AKT expression, as well as increased cyclin-dependent kinase inhibitor proteins such as p21 and p27 expression. Importantly, PD activates and promotes YAP1 degradation through the ubiquitination and proteasome pathway. The nuclear transactivation of YAP was significantly reduced under PD treatment, leading to transcriptional inhibition of the downstream genes regulating cell proliferation, pro-survival, and metastasis. In conclusion, our results showed that PD is suitable as a promising agent for overcoming oxaliplatin-resistant colorectal cancer.
Keywords: Oxaliplatin, Drug resistance, Platycodin, Cell cycle, metastasis
Introduction
Colorectal cancer (CRC) is the third prevalent malignant tumor, causing malignancies. In spite of substantial advancements in CRC therapy, it remains one of the primary causes of cancer-related mortality [1]. Oxaliplatin-based chemotherapy is employed in the front lines of CRC treatment all over the world. Nearly half of CRC patients receiving oxaliplatin-based chemotherapy are cured of CRC [2, 3]. Oxaliplatin is a platinum based third generation chemotherapeutic agent, which acts against the cancer cells by interacting with DNA and forms cross-links between the two strands in the S phase of cell division [4]. On the other hand, chemotherapy could also develop resistance to oxaliplatin, which may allow cancer cells to survive or quiescence, contributing to the cancer reappearance. Resistance to oxaliplatin is likely related to cellular transport, detoxification, DNA repair, cell death, and epigenetic alternation [5, 6]. Understanding these molecular mechanisms of oxaliplatin resistance and utilizing them for developing novel therapeutic strategies for cancer therapies have been investigated [7-9].
The key serine/threonine kinase of the hippo tumor-suppressive signaling pathway is Large tumor suppressor 2 (LATS2), which is present in chromosome 13Q1.11 in humans. LATS2 controls the cell cycle by regulating Yes-associated protein 1 (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ) (orthologues of Yorkie in Drosophila) phosphorylation, which are key downstream regulators in the hippo signaling pathway [10]. LATS2 is a key regulator of mitotic progression and activates its downstream proteins such as YAP, retinoblastoma protein (pRB), and p53, which altogether contribute to cell cycle arrest and cancer cell growth inhibition [11]. Further, LATS2 is also known to interact with other signaling pathways like estrogen signaling, and the Ras and Akt network which plays role in regulating cell proliferation, apoptosis, and metastasis of different cancer types [12, 13]. Recent studies have shown the association of hippo pathway in the development of CRC [14-16].
The remarkable oncogenic characteristics of the Hippo signaling pathway proteins, such YAP and TAZ, as well as their druggability, are gaining attention in recent researches in cancer drug resistance [17]. Paclitaxel and cisplatin resistance is conferred by overexpressing YAP-S127A in ovarian cancer cells with low baseline YAP activity, but YAP knockdown in ovarian cancer cells with higher YAP activity improves sensitivity to paclitaxel and cisplatin. This is due to YAP-S127A lacks a significant LATS1/2 phosphorylation site and accumulates in the nucleus [18]. In nasopharyngeal cancer, epithelial-mesenchymal transition (EMT) and overexpression of TAZ have been found to be positively correlated [19]. While TAZ stimulates interleukin-8 (IL-8) transcription to develop resistance to doxorubicin, YAP induces doxorubicin resistance by triggering the mitogen-activated protein kinase (MAPK) pathway [20].
Natural products have been extensively studied in the realm of drug discovery because they are a rich source of molecules with a wide structural variety. In vitro and in vivo anticancer properties have been observed in a wide range of natural compounds [21-23]. Platycodon grandifloras common Chinese medicinal plant belongs to the family Campanulaceae which was used in traditional folk medicines in China, Japan, and Korea. The root of Platycodon grandifloras is used to cure a variety of ailments heavy cough, sore throat, bronchitis, and asthma [24, 25]. Platycodin D is one of the major saponins presented in Platycodon grandifloras which possess various pharmacological properties such as anti-oxidant, anti-inflammatory, anti-obesity, anti-atherogenic, and immunomodulatory effects [26-30]. PD has remarkable antitumor effects on several cancer cell lines, reducing the proliferation of cancer cell growth by inhibiting the cell cycle and inducing apoptosis [31, 32].
On the basis of these studies, we hypothesize that LoVo colorectal cancer cell line develops resistance to chemotherapeutic drugs by activating the hippo signaling pathway and activates LATS2, and YAP by phosphorylation and PD treatment in the parental and resistance LoVo cells could effectively modulate the hippo signaling pathway by inhibiting the phosphorylation of LATS2, and YAP.
Materials and Methods
Cell culture
Food Industry Research and Development Institute, Hsinchu, Taiwan, provided the LoVo colon cell line, which is human colon cell line. LoVo cells were cultured in Dulbecco's Minimum Essential Medium (DMEM, Sigma, USA) containing 10% fetal bovine serum (FBS) (HyCloneTM, USA), streptomycin (100 g/mL) and penicillin (100 U/mL) at 37 °C in a humidified environment of 5% CO2. To establish stable colon cancer cell lines resistant to oxaliplatin, LoVo cell lines were exposed to oxaliplatin in dose-dependent manner from 0 to 25 µM for 24 hr. This was MTT-1. LoVo cell lines exposed to 21.5 µM oxaliplatin resulted around 50% cell death (IC50 of oxaliplatin). Cells treated with 21.5 µM oxaliplatin (24 hr) allowed to reach 80% confluence and passaged twice in this same concentration (21.5 µM) of oxaliplatin. The same procedure was repeated at increased doses of oxaliplatin (30 and 40 µM) until a cell population was selected that demonstrated at least a threefold greater IC50 (75 µM) to oxaliplatin than the parental cell lines. OXA-R LoVo cells were developed by the previous report [3].
MTT assay
To assess the cell viability of LoVo cells, the MTT [3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium-bromide] assay was performed. A 96-well plate was seeded with 1 x 104 parental LoVo cells and 1 x 104 OXA-R LoVo cells, and the cells were exposed to various drug doses. Oxaliplatin was treated to parental LoVo and OXA-R LoVo cells at 1, 5, 10, 15, 20, 25, 30, 40, 50, 60, 70, and 80 µM for 24 hours, After the treatment time, DMEM was removed from the cells and washed with PBS. Each well received 20 μl (5 mg/mL) of MTT, which was then left to incubate for 4 hours. A microplate reader was used to detect the absorbance at 570 nm after dissolving MTT formazan crystals in 200 μl of DMSO. Platycodin D (purity ≥98%, Sigma, USA) was dissolved in DMSO.
Western blot
An equal amount (30-40 µg) of protein was separated by using an 8-12% SDS-PAGE electrophoresis gel at 90V for 45 minutes. Protein from the gel was transferred to the polyvinylidene fluoride membrane at 4 °C using blotting apparatus (Bio-Rad Laboratories, Hercules, CA, USA). PVDF membrane was then submerged in 5% non-fat milk powder in TBST at room temperature for 1 hr. The membrane was then washed thrice with TBS 3 times, for 5 minutes each. The membrane was then incubated overnight with primary antibody (1:1000 dilution in TBST) at 4 °C in a mechanical rocker. Then, the membrane was washed thrice with TBS 3 times, for 5 minutes each, and incubated with HRP-conjugated secondary antibody (1:5000 dilution in TBST) at room temperature for 1 h in a mechanical rocker. After washing with TBS, the membrane was submerged in chemiluminescence ECL solution (Merck Millipore, Burlington, MA, USA), the protein bands were visualized using chemi-doc apparatus (Fuji-film LAS-3000, GE Healthcare), and densitometric analysis was performed using ImageJ software (version 1.4.3.67) (NIH, Bethesda, MD, USA). Antibodies details: LATS2 (#5888, Cell Signaling, Baltimore, MD, USA), p-LATS2 (ab111344, Abcam, Cambridge, UK), YAP (sc-101199, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-YAP (#13008, Cell Signaling, Baltimore, MD, USA), TAZ (sc-48805, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p21 (sc-6246, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p27 (sc-1641, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-AKT (#9275s, Cell Signaling, Baltimore, MD, USA), Ki67 (ab15580, Abcam, Cambridge, UK), β-Actin (sc-47778, Santa Cruz Biotechnology, Santa Cruz, CA, USA), HDAC1 (sc-6298, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Ubiquitin (sc-8017, Santa Cruz Biotechnology, Santa Cruz, CA, USA), GAPDH (sc-25778, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Terminal Deoxynucleotidyl Transferase-mediated Nick-End Labeling (TUNEL) assay
DNA breaks due to apoptosis was determined using Terminal Deoxynucleotidyl Transferase-mediated Nick-End Labeling (TUNEL) assay. In Situ Cell Death Detection Kit (Roche) was used to assay. After the cells were treated with appropriate dose and time DMEM was removed from the cells and washed with PBS thrice. Then the cells were fixed in 4% formalin for 1 hr. After fixation the cells were made permeable with permeation solution (0.1% Triton X‐100 in 0.1% sodium citrate) for 10 mins. Cells were washed with PBS and incubated in TUNEL solution for 1 hr at room temperature. Cells were stained with nuclear stain DAPI for 5 mins and observed under fluorescent microscope for TUNEL positive cells.
Transwell migration and invasion assays
Serum-free medium was applied to dilute the Matrigel, and 50 μl of diluted Matrigel was inoculated into each chamber. After being treated with DMSO and PD, both parental LoVo and OXA-R LoVo cells were resuspended at a density of 1 × 105 cells/mL with DMEM medium without FBS. Then, we added 0.2 mL of the cell suspension to each upper chamber of the 24-well plate, while 0.6 mL DMEM containing 20% FBS was added to the lower chambers. After 24 hr, the upper chambers were washed, fixed with 4% paraformaldehyde for 20 min, stained with 0.25% crystal violet for 30 min, and imaged by a microscope.
Immunofluorescence microscopy
1 × 105 parental or OXA‐resistant LoVo colon cancer cells were seeded 24 well plates and treated with PD for 24 hrs. At the end of treatment cells were washed with PBS and fixed with 4% paraformaldehyde for 1 hr in room temperature. After PBS was cells were permeabilized with permeabilization solution for 10 mins. To prevent non-specific binding, cells were incubated with 10% FBS for 1 hr at room temperature. 250 μl of YAP primary antibody (1:200 dilution) was added to the cells and incubated at 37°C for 3 hrs. Cells were then washed with PBS thrice and incubated with 300 μl FITC conjugated secondary antibody (1:1000 dilution) for 1 hr at room temperature. Cells were then washed and stained with DAPI for 5 mins and washed again with PBS thrice. Cells were then visualized under fluorescent microscope.
Statistical analysis
The results shown are the means ±SD of three independent experiments. Statistical analysis was performed by one-way analysis of variants followed by a Tukey's post-hoc test SPSS 16 software (SPSS, Chicago, IL, USA).
Results
Characterization of chemoresistance in OXP-LoVo colorectal cancer cells
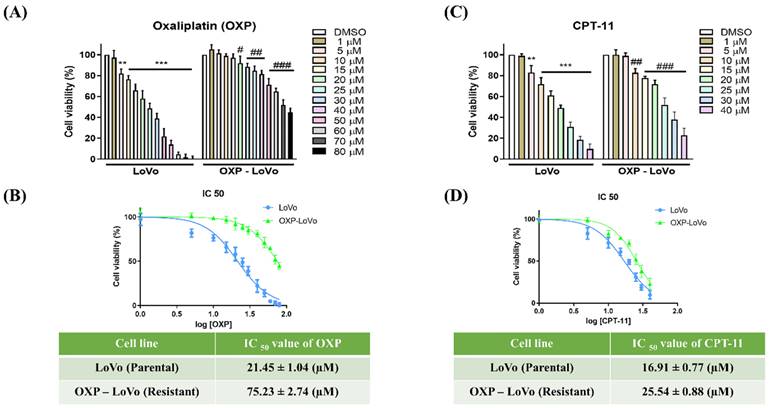
OXP-LoVo colorectal cancer cells were developed based on our previous study [3]. Both parental and OXP-LoVo cells were treated with various concentrations of oxaliplatin (1-80 μM) for 24 hrs. and cell viability was determined by MTT assay. Oxaliplatin from 5 μM induces significant cell death in LoVo cells, however in OXP-LoVo cells cell significant cell death was observed after 20 μM of oxaliplatin (Figure 1A). IC50 values of oxaliplatin in LoVo and OXP-LoVo was found to be 21.45 μM and 75.23 μM respectively (Figure 1B). In order to establish multidrug resistance, parental LoVo and OXP-LoVo cells were treated with different concentrations of irinotecan (CPT‐11) (1-40 μM) for 24 hrs and cell viability was quantified. 5 μM of CPT-11 induces cell death significantly in parental LoVo cells but in OXP-LoVo cells 10 μM of CPT-11 induced significant cell death. IC50 of CPT-11 parental LoVo was found to be 16.91 μM, whereas in OXP-LoVo cells it was around 25.5 μM (Figure 1C&D). This result demonstrated that OXP-LoVo cells were resistant to both OXA and CPT-11.
Characterization of chemoresistance in OXP-LoVo colon-rectal cancer cells. (A&C) Parental and OXP-LoVo cells were treated with various concentrations of oxaliplatin (1-80 μM) and CPT-11 (1-40 μM) for 24 hrs. and cell viability was determined by MTT assay. (B&D) IC50 values of oxaliplatin (OXP) and CPT-11 in LoVo and OXP-LoVo colon-rectal cancer cells. *** P<0.001, and ** P<0.01 compared to LoVo control group and ### P<0.001, ## P<0.01 and # P<0.05 compared to OXP-LoVo control.
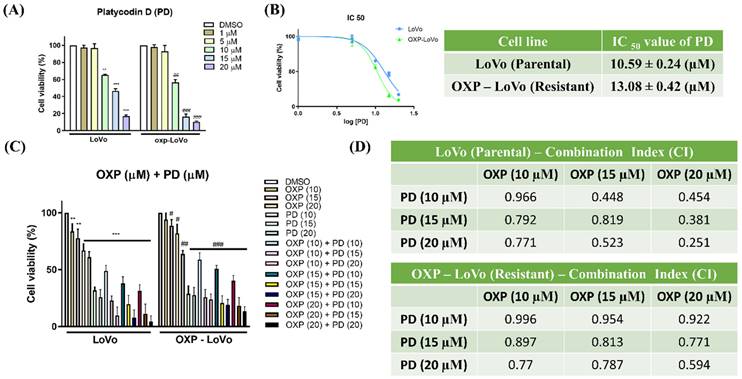
PD alone and combined with oxaliplatin inhibits CRC cell proliferation in parental and OXA-resistance cells
Several reports have established the anticancer effect of PD [31]. In the present study, we evaluated the effect of PD on parental LoVo and OXP-LoVo cell viability. Both parental LoVo and OXP-LoVo cells were treated with different concentrations of PD (1-20 μM) for 24 hrs. In both cells, PD from 10 μM induces cell death significantly and the IC50 of PD in parental LoVo and OXP-LoVo cells was found to be 10.59 μM and 13.08 μM respectively (Figure 2A&B). In order to find the combinatorial synergistic effect both the cells were treated combined with OXA (10, 15 & 20 μM) and PD (10, 15 & 20 μM) for 24 hrs, and cell viability was estimated (Figure 2C). Combined treatment of OXA and PD induces cell death in a dose-dependent manner. A combination index (CI) was calculated for the drug pair OXA and PD. CI < 1 is believed to be better compatibility with high synergistic effects. In our present study, CI was found to be below 1 for OXA (10, 15 & 20 μM) and PD (10, 15 & 20 μM) in parental LoVo and OXP-LoVo cells (Figure 2D).
PD alone and combined with oxaliplatin inhibits CRC cell proliferation in parental and OXA-resistance cells. (A) Both parental LoVo and OXP-LoVo cells were treated with different concentrations of PD (1-20 μM) for 24 hrs. (B) IC50 of PD in parental LoVo and OXP-LoVo cells. (C) Combinatorial synergistic effect of OXA (10, 15 & 20 μM) and PD (10, 15 & 20 μM) on LoVo and OXP-LoVo cells. (D) A combination index (CI) was calculated for the drug pair OXA and PD. *** P<0.001, and ** P<0.01 compared to LoVo control group and ### P<0.001, and ## P<0.01 compared to OXP-LoVo control.
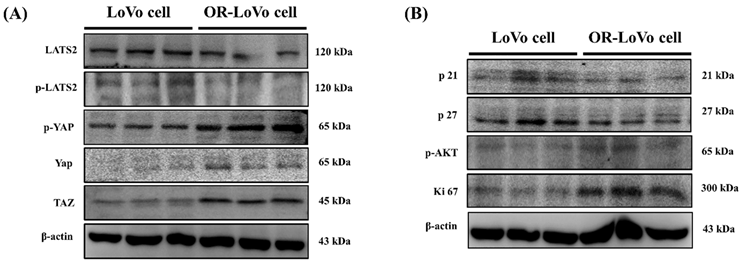
LATS2 and YAP/TAZ hippo signaling is highly activated in OXP-LoVo cells
We evaluated the hippo signaling pathway proteins and cell cycle regulating protein expression in LoVo and OXP-LoVo cells. p-LAST2 p-YAP and TAZ were upregulated in OXP-LoVo cells when compared to LoVo cells (Figure 3A). On the other hand, cy-clin-dependent kinase inhibitor proteins such as p21 and p27 and cell survival protein p-Akt and Ki 67 were increased in OXP-LoVo cells than parental LoVo cells (Figure 3B).
LATS2 and YAP/TAZ hippo signaling is highly activated in OXP-LoVo cells. (A) Immunoblot showing the expression levels of p-LAST2, p-YAP, and TAZ. (B) Immunoblot showing the expression levels of cell cycle and survival proteins p21, p27, p-AKT and Ki67.
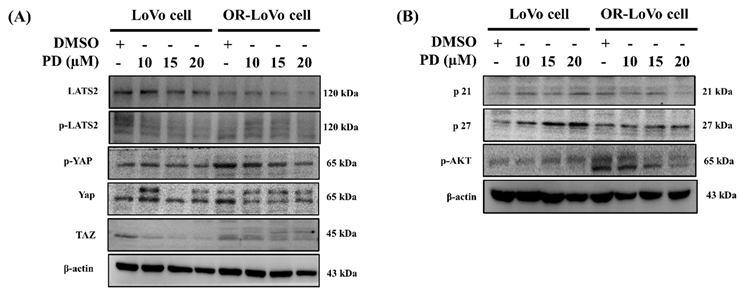
PD significantly downregulated LATS2 and YAP/TAZ hippo signaling in LoVo and OXP-LoVo cells
Then we evaluated the effect of PD on hippo signaling, cell cycle, and cell survival protein expression. PD dose-dependently decreased LATS2/YAP1 and Taz in hippo signaling and survival marker p-AKT expression, as well as increased cyclin-dependent kinase inhibitor proteins such as p21 and p27 expression in parental and OXP-LoVo cells (Figure 4).
PD significantly downregulated LATS2 and YAP/TAZ hippo signaling in LoVo and OXP-LoVo cells. Western blot analysis showing expression of p-LAST2, p-YAP, and TAZ proteins. (B) Western blot analysis for the protein expressions p21, p27, and p-AKT.
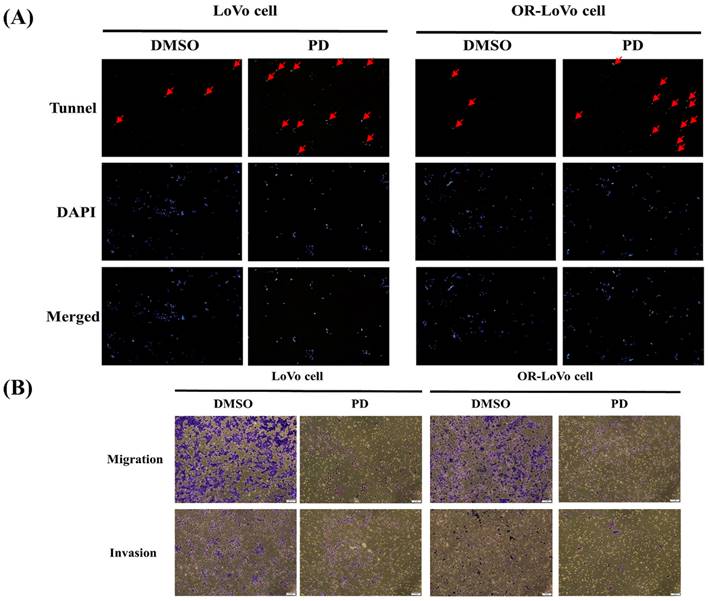
PD induces apoptosis, invasion, and migration in LoVo and OXP-LoVo cells
The effect of PD on apoptosis and metastasis was analyzed in the CRC cells. The number of TUNEL positive cells was significantly higher in the PD treated parental and OXP-LoVo cells when compared to their respective control groups (Figure 5A). Effect PD on metastasis was estimated by invasion, and migration assay. PD treatment in both type of cells effectively prevents the invasion, and migration of the CRCs (Figure 5B), the anti-metastasis effect of PD.
PD induces apoptosis, invasion, and migration in LoVo and OXP-LoVo cells. (A) TUNEL positive cells were increased in LoVo and OXP-LoVo cells after PD treatment. (B) PD treatment effectively reduced the metastasis in LoVo and OXP-LoVo CRCs by inhibiting invasion, and migration.
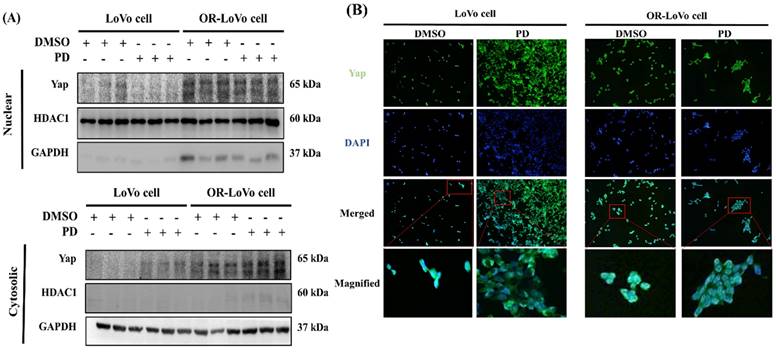
PD inhibited Yap nuclear translocation and activation in LoVo and OXP-LoVo cells
Inhibiting YAP activation and nuclear translocation might be an approach to reduce cancer progression. Then we test the effect of PD on YAP activation and nuclear translocation. We quantified the YAP expression in the nuclear and cytoplasmic extract of parental LoVo cells and OXP-LoVo cells. YAP expression in the nuclear extract was increased in OXP-LoVo cells compared to LoVo cells, however, PD treatment reduced the YAP levels in the nuclear extract. In the cytoplasmic extract, PD treatment increased YAP levels in OXP-LoVo cells (Figure 6A). Immunofluorescence assay also confirms that PD treatment in the LoVo cells and OXP-LoVo cells effectively prevents the nuclear translocation of Yap (Figure 6B).
PD inhibited Yap nuclear translocation and activation in LoVo and OXP-LoVo cells. (A) Western blot results showing YAP1 expression in nuclear and cytosolic fractions of PD treated LoVo and OXP-LoVo cells. (B) Immunofluorescence image showing the effect of PD in inhibiting YAP1 nuclear translocation.
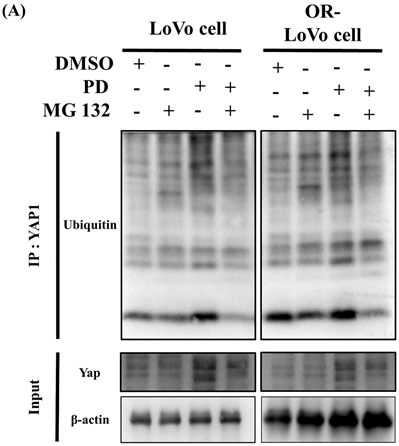
PD activates and promotes YAP1 degradation through the ubiquitination and proteasome pathway
Immunoprecipitation assay followed by immunoblot was performed for the YAP1 to study the effect of PD on ubiquitination and proteasome pathway. MG132, a proteasome inhibitor was used. PD treatment in LoVo cells and OXP-LoVo cells degrade YAP1 through activating ubiquitination mediated proteasome degradation (Figure 7).
PD activates and promotes YAP1 degradation through the ubiquitination and proteasome pathway.
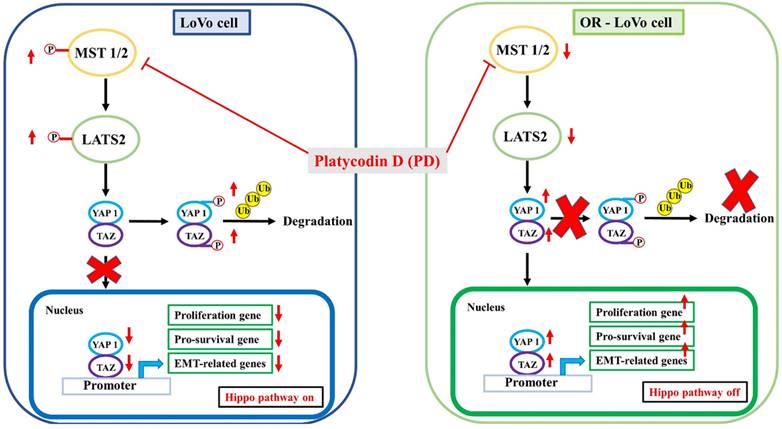
Schematic representation of the molecular mechanism of PD in inhibiting hippo pathway in parental and OXA-resistance LoVo colorectal cancer cells.
Discussion
PD has been reported to have an anti-cancer effect in different in vitro and in vivo [31]. PD has been reported to reduce cell viability and invasion while greatly increasing apoptosis in OSCC cells in a dose-dependent manner [33]. Further, PD demonstrates the anti-cancer properties in breast cancer cells in vitro and in vivo, as evidenced by decreasing cell proliferation and survival of MDA-MB-231 cells and inhibited the tumor cell volume in MDA-MB-231 xenograft tumors in BALB/c nude mice [34]. In liver cancer cells, PD exerts an anti-cancer effect by inducing apoptosis and inhibiting metastasis [35]. PD increased the expression of Fas and FasL at the mRNA and protein levels in HaCaT cells, causing proteolytic cleavage of caspase-8 and -3 in a time-dependent manner, and elicits extrinsic apoptosis [36]. In HepG2 and MCF-7 cancer cells, PD dramatically decreased the Bcl-2/Bax ratio and elevated the expression of cleaved caspase-9, caspase-3, and Poly (ADP-ribose) polymerase (PARP) [35, 37]. Furthermore, it has been discovered that platycodin D could cause G2/M phase arrest in PC3 cells [38]. It has been demonstrated that platycodin D inhibits the migration and invasion of MDA-MB-231 cells in a dose-dependent manner. While MMP-2 activity was only marginally lowered, PD significantly suppressed MMP-9 activity. MMP-9 mRNA expression shown to be down-regulated by PD [39].
Dysfunction of the hippo pathway has been linked to the development and metastasis of CRC [40, 41]. Upregulation of hippo pathway proteins such as YAP and TAZ along with other proteins Transcriptional enhancer factor TEF-1 also known as TEA domain family member 1 (TEAD), and Octamer-binding transcription factor 4 (OCT4) have a correlation of developing adenoma to CRC [42]. YAP and TAZ upregulations were shown to be substantially linked with lymph node metastases in CRC patients [43]. TAZ was sued as a prognostic marker for CRC which is also linked to downstream target genes AXL and Connective tissue growth factor (CTGF) [44].
Extracellular matrix (ECM), cell adhesion, serine/threonine kinase receptor, G protein-coupled receptor (GPCR), and cellular metabolism dysfunction are the direct upstream regulators of YAP/TAZ activity [45]. In a clinical study, YAP expression in a CRC patient have shown to regulate epithelial-mesenchymal transition (EMT) by activating Slug and inhibiting E-cadherin [46]. Higher YAP expression is linked to CRC recurrence in human CRC hepatic metastases [40]. Recently, we have shown that Irinotecan (CPT-11) resistant CRC cells have shown to increase the expression GPCR such as Gαi‐2, Gαq/11, and Gαs and metastasis [47]. Nuclear translocation of YAP and upregulation of β-catenin expression could decrease the overall and progression-free survival in CRC patients [43]. Another study correlates the YAP/TAZ expression with the chemoresistance CRC with liver metastases and suppressing/inhibiting YAP/TAZ expression in the CRC patients sensitize the CRC to the chemotherapeutic drug cetuximab and increase progression-free survival [48]. In our present study, PD treatment dose-dependently decreased LATS2/YAP1 hippo signaling and survival marker p-AKT expression, as well as increased cyclin-dependent kinase inhibitor proteins such as p21 and p27 expression.
Hippo signaling is a piece of epigenetic machinery that regulates the development and progression of cancer. According to the recent in silico analysis, promotor sites of the YAP/TAZ genes are greatly enhanced with the binding sites of many transcriptional factors including P300 which acts as a histone acetyltransferase (HAT) at the YAP1 promoter and procured chromatin remodelers of other DNA or histone-modifying components such as CTCF and HAT [16].
In recent years, hippo signaling in many human cancer types has been studied which provides a better understanding of their role in cancer progression. Targeting the YAP/TAZ transcriptional network chemical inhibitors might be a significant step forward in the combat against cancer therapy resistance [49]. The nuclear-cytoplasmic shuttling of YAP/TAZ has been given considerable attention for its regulatory function [50]. YAP/TAZ must be translocated into the nucleus to act as a transcriptional activation factor. Thus, preventing/inhibiting YAP/TAZ activation and nuclear translocation might be an approach to reduce cancer progression. In our present study, we have shown that YAP levels in the nucleus were increased OXP-LoVo cells, whereas PD significantly reduced the nuclear translocation of YAP suggesting PD sensitize OXP-LoVo cells by inhibiting nuclear translocation of YAP.
A combinatorial treatment regime is the main element of chemotherapy-resistant cancer treatment that focus to sensitize chemotherapy-resistant cancer through the sequential regime of an adjuvant drug [51]. Results of our present study demonstrate that combined treatment of oxaliplatin and PD reduce the cell viability significantly in OXA-LoVo cells in a dose-dependent manner. PD dose-dependently decreased LATS2/YAP1 hippo signaling and survival marker p-AKT expression, as well as increased cyclin-dependent kinase inhibitor proteins such as p21 and p27 expression. Importantly, PD activates and promotes YAP1 degradation through the ubiquitination and proteasome pathway. The nuclear transactivation of YAP was significantly reduced under PD treatment, leading to transcriptional inhibition of the downstream gene, including proliferation gene, pro-survival gene, and EMT-related gene. In conclusion, our results showed that PD is suitable as a promising agent for overcoming oxaliplatin-resistant colorectal cancer.
Acknowledgements
This work was supported by the Hualien Tzu Chi Hospital (TCRD110-80).
Data availability statement
The data that support the findings of this study are available from the corresponding author, CYH, upon reasonable request.
Author contributions
Chien-Hao Wang - Methodology, writing-original draft, writing-review and editing. Rathinasamy Baskaran - writing-original draft, writing-review and editing. Shawn Shang-Chuan Ng - resources, formal analysis. Tso-Fu Wang - investigation, formal analysis. Chi-Cheng Li - formal analysis, data curation. Tsung-Jung Ho - validation, investigation. Dennis Jine-Yuan Hsieh - project administration. Chia-Hua Kuo - data curation, project administration. Ming-Cheng Chen - Conceptualization, funding acquisition. Chih-Yang Huang - Conceptualization, validation, resources, supervision, project administration, funding acquisition.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Alshareef SH, Alsobaie NA, Aldeheshi SA, Alturki ST, Zevallos JC, Barengo NC. Association between race and cancer-related mortality among patients with colorectal cancer in the United States: a retrospective cohort study. International journal of environmental research and public health. 2019;16:240
2. Bahrami A, Amerizadeh F, Hassanian SM, ShahidSales S, Khazaei M, Maftouh M. et al. Genetic variants as potential predictive biomarkers in advanced colorectal cancer patients treated with oxaliplatin-based chemotherapy. Journal of cellular physiology. 2018;233:2193-201
3. Hsu HH, Chen MC, Baskaran R, Lin YM, Day CH, Lin YJ. et al. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. Journal of cellular physiology. 2018;233:5458-67
4. Ide H, Nakano T, Salem AM, Shoulkamy MI. DNA-protein cross-links: formidable challenges to maintaining genome integrity. DNA repair. 2018;71:190-7
5. Hu W, Lei L, Xie X, Huang L, Cui Q, Dang T. et al. Heterogeneous nuclear ribonucleoprotein L facilitates recruitment of 53BP1 and BRCA1 at the DNA break sites induced by oxaliplatin in colorectal cancer. Cell death & disease. 2019;10:1-16
6. Causse SZ, Marcion G, Chanteloup G, Uyanik B, Boudesco C, Grigorash BB. et al. HSP110 translocates to the nucleus upon genotoxic chemotherapy and promotes DNA repair in colorectal cancer cells. Oncogene. 2019;38:2767-77
7. Ashrafizadeh M, Mirzaei S, Hashemi F, Zarrabi A, Zabolian A, Saleki H. et al. New insight towards development of paclitaxel and docetaxel resistance in cancer cells: EMT as a novel molecular mechanism and therapeutic possibilities. Biomedicine & Pharmacotherapy. 2021;141:111824
8. Martinez-Balibrea E, Martínez-Cardús A, Ginés A, de Porras VR, Moutinho C, Layos L. et al. Tumor-related molecular mechanisms of oxaliplatin resistance. Molecular cancer therapeutics. 2015;14:1767-76
9. Liu YP, Zheng CC, Huang YN, He ML, Xu WW, Li B. Molecular mechanisms of chemo-and radiotherapy resistance and the potential implications for cancer treatment. MedComm. 2021;2:315-40
10. Sebio A, Lenz H-J. Molecular pathways: hippo signaling, a critical tumor suppressor. Clinical Cancer Research. 2015;21:5002-7
11. He C, Lv X, Huang C, Hua G, Ma B, Chen X. et al. YAP 1-LATS 2 feedback loop dictates senescent or malignant cell fate to maintain tissue homeostasis. EMBO reports. 2019;20:e44948
12. Thompson BJ. YAP/TAZ: drivers of tumor growth, metastasis, and resistance to therapy. Bioessays. 2020;42:1900162
13. Zhang S, Wang J, Wang H, Fan L, Fan B, Zeng B. et al. Hippo cascade controls lineage commitment of liver tumors in mice and humans. The American journal of pathology. 2018;188:995-1006
14. Pan Y, Tong JHM, Lung RWM, Kang W, Kwan JSH, Chak WP. et al. RASAL2 promotes tumor progression through LATS2/YAP1 axis of hippo signaling pathway in colorectal cancer. Molecular cancer. 2018;17:1-14
15. Veschi V, Stassi G. The Hippo Show Must Go On: YAP Activation as a Therapeutic Strategy in Colorectal Cancer. Cell Stem Cell. 2020;27:501-2
16. Mohajan S, Jaiswal PK, Vatanmakarian M, Yousefi H, Sankaralingam S, Alahari SK. et al. Hippo pathway: Regulation, deregulation and potential therapeutic targets in cancer. Cancer Letters. 2021;507:112-23
17. Zeng R, Dong J. The Hippo signaling pathway in drug resistance in cancer. Cancers. 2021;13:318
18. Zhang X, George J, Deb S, Degoutin J, Takano E, Fox S. et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene. 2011;30:2810-22
19. Li S, Zhang X, Zhang R, Liang Z, Liao W, Du Z. et al. Hippo pathway contributes to cisplatin resistant-induced EMT in nasopharyngeal carcinoma cells. Cell Cycle. 2017;16:1601-10
20. Basu D, Lettan R, Damodaran K, Strellec S, Reyes-Mugica M, Rebbaa A. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of hippo, TGF-β, and Wnt signaling pathways. Molecular cancer therapeutics. 2014;13:1457-67
21. Bharathi Priya L, Huang CY, Hu RM, Balasubramanian B, Baskaran R. An updated review on pharmacological properties of neferine—A bisbenzylisoquinoline alkaloid from Nelumbo nucifera. Journal of Food Biochemistry. 2021;45:e13986
22. Rejhová A, Opattová A, Čumová A, Slíva D, Vodička P. Natural compounds and combination therapy in colorectal cancer treatment. European journal of medicinal chemistry. 2018;144:582-94
23. Sauter ER. Cancer prevention and treatment using combination therapy with natural compounds. Expert review of clinical pharmacology. 2020;13:265-85
24. Ji M-Y, Bo A, Yang M, Xu J-F, Jiang L-L, Zhou B-C. et al. The pharmacological effects and health benefits of Platycodon grandiflorus—a medicine food homology species. Foods. 2020;9:142
25. Park DI, Lee JH, Moon S-K, Kim C-H, Lee YT, Cheong J. et al. Induction of apoptosis and inhibition of telomerase activity by aqueous extract from Platycodon grandiflorum in human lung carcinoma cells. Pharmacological research. 2005;51:437-43
26. Wang Y, Che J, Zhao H, Tang J, Shi G. Platycodin D inhibits oxidative stress and apoptosis in H9c2 cardiomyocytes following hypoxia/reoxygenation injury. Biochemical and biophysical research communications. 2018;503:3219-24
27. Kang S-H, Kim T-H, Shin K-C, Ko Y-J, Oh D-K. Biotransformation of food-derived saponins, platycosides, into deglucosylated saponins including deglucosylated platycodin D and their anti-inflammatory activities. Journal of agricultural and food chemistry. 2019;67:1470-7
28. Kim H-L, Park J, Jung Y, Ahn KS, Um J-Y. Platycodin D, a novel activator of AMP-activated protein kinase, attenuates obesity in db/db mice via regulation of adipogenesis and thermogenesis. Phytomedicine. 2019;52:254-63
29. Zhao HL, Cho K-H, Ha YW, Jeong T-S, Lee WS, Kim YS. Cholesterol-lowering effect of platycodin D in hypercholesterolemic ICR mice. European journal of pharmacology. 2006;537:166-73
30. Xie Y, Sun H-X, Li D. Platycodin D is a potent adjuvant of specific cellular and humoral immune responses against recombinant hepatitis B antigen. Vaccine. 2009;27:757-64
31. Khan M, Maryam A, Zhang H, Mehmood T, Ma T. Killing cancer with platycodin D through multiple mechanisms. Journal of cellular and molecular medicine. 2016;20:389-402
32. Hsu W-C, Ramesh S, Shibu MA, Chen M-C, Wang T-F, Day CH. et al. Platycodin D reverses histone deacetylase inhibitor resistance in hepatocellular carcinoma cells by repressing ERK1/2-mediated cofilin-1 phosphorylation. Phytomedicine. 2021;82:153442
33. Zhang Z, Zhao M, Zheng W, Liu Y. Platycodin D, a triterpenoid saponin from Platycodon grandiflorum, suppresses the growth and invasion of human oral squamous cell carcinoma cells via the NF-κB pathway. Journal of Biochemical and Molecular Toxicology. 2017;31:e21934
34. Kong Y, Lu Z-L, Wang J-J, Zhou R, Guo J, Liu J. et al. Platycodin D, a metabolite of Platycodin grandiflorum, inhibits highly metastatic MDA-MB-231 breast cancer growth in vitro and in vivo by targeting the MDM2 oncogene. Oncology Reports. 2016;36:1447-56
35. Li T, Xu W-S, Wu G-S, Chen X-P, Wang Y-T, Lu J-J. Platycodin D induces apoptosis, and inhibits adhesion, migration and invasion in HepG2 hepatocellular carcinoma cells. Asian Pacific Journal of Cancer Prevention. 2014;15:1745-9
36. Ahn KS, Hahn B-S, Kwack K, Lee EB, Kim YS. Platycodin D-induced apoptosis through nuclear factor-κB activation in immortalized keratinocytes. European journal of pharmacology. 2006;537:1-11
37. Yu JS, Kim AK. Platycodin D induces apoptosis in MCF-7 human breast cancer cells. Journal of medicinal food. 2010;13:298-305
38. Zhou R, Lu Z, Liu K, Guo J, Liu J, Zhou Y. et al. Platycodin D induces tumor growth arrest by activating FOXO3a expression in prostate cancer in vitro and in vivo. Current Cancer Drug Targets. 2014;14:860-71
39. Chun J, Kim YS. Platycodin D inhibits migration, invasion, and growth of MDA-MB-231 human breast cancer cells via suppression of EGFR-mediated Akt and MAPK pathways. Chemico-biological interactions. 2013;205:212-21
40. Wierzbicki PM, Rybarczyk A. The Hippo pathway in colorectal cancer. Folia histochemica et cytobiologica. 2015;53:105-19
41. Mouillet-Richard S, Laurent-Puig P. YAP/TAZ signalling in colorectal cancer: Lessons from consensus molecular subtypes. Cancers. 2020;12:3160
42. Liang K, Zhou G, Zhang Q, Li J, Zhang C. Expression of hippo pathway in colorectal cancer. Saudi journal of gastroenterology: official journal of the Saudi Gastroenterology Association. 2014;20:188
43. Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A. et al. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PloS one. 2013;8:e65539
44. Yuen H-F, McCrudden CM, Huang Y-H, Tham JM, Zhang X, Zeng Q. et al. TAZ expression as a prognostic indicator in colorectal cancer. PloS one. 2013;8:e54211
45. Yu F-X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780-91
46. Cheng D, Jin L, Chen Y, Xi X, Guo Y. YAP promotes epithelial mesenchymal transition by upregulating Slug expression in human colorectal cancer cells. International Journal of Clinical and Experimental Pathology. 2020;13:701
47. Chen MC, Baskaran R, Lee NH, Hsu HH, Ho TJ, Tu CC. et al. CXCL2/CXCR2 axis induces cancer stem cell characteristics in CPT-11-resistant LoVo colon cancer cells via Gαi-2 and Gαq/11. Journal of cellular physiology. 2019;234:11822-34
48. Lee K-W, Lee SS, Kim S-B, Sohn BH, Lee H-S, Jang H-J. et al. Significant association of oncogene YAP1 with poor prognosis and cetuximab resistance in colorectal cancer patients. Clinical Cancer Research. 2015;21:357-64
49. Battilana G, Zanconato F, Piccolo S. Mechanisms of YAP/TAZ transcriptional control. Cell Stress. 2021;5:167
50. Shreberk-Shaked M, Oren M. New insights into YAP/TAZ nucleo-cytoplasmic shuttling: new cancer therapeutic opportunities? Molecular oncology. 2019;13:1335-41
51. Yap TA, Omlin A, De Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. Journal of Clinical Oncology. 2013;31:1592-605
Author contact
![]() Corresponding author: Chih-Yang Huang Ph.D., Chair Professor, Cardiovascular and Mitochondria related diseases research center, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Tzu Chi University of Science and Technology, Hualien, Taiwan; Tel: +886-4-22053366 ext 3313. Fax: +886-4-22032295. E-mail address: cyhuangcmu.edu.tw
Corresponding author: Chih-Yang Huang Ph.D., Chair Professor, Cardiovascular and Mitochondria related diseases research center, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Tzu Chi University of Science and Technology, Hualien, Taiwan; Tel: +886-4-22053366 ext 3313. Fax: +886-4-22032295. E-mail address: cyhuangcmu.edu.tw