Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2023; 14(4):573-590. doi:10.7150/jca.82291 This issue Cite
Editorial Commentary
The most likely but largely ignored triggering factor for breast (or all) cancer invasion
Yan-gao Man1 ![]() , Ciaran Mannion1
, Ciaran Mannion1 ![]() , Alexander Stojadinovic2, George E Peoples2, William CS Cho3, Sidney W. Fu4, Xiaohui Tan4, Yi-Hsuan Hsiao5, Aijun Liu6, Andrzej Semczuk7, Paul Zarogoulidis8, Andrei B. Gapeev9, Xiyun Deng10, Xiaoning Peng10, Boris A. Reva11, Tatiana Omelchenko12, Jialian Wang13, Guohong Song14, Tingtao Chen15
, Alexander Stojadinovic2, George E Peoples2, William CS Cho3, Sidney W. Fu4, Xiaohui Tan4, Yi-Hsuan Hsiao5, Aijun Liu6, Andrzej Semczuk7, Paul Zarogoulidis8, Andrei B. Gapeev9, Xiyun Deng10, Xiaoning Peng10, Boris A. Reva11, Tatiana Omelchenko12, Jialian Wang13, Guohong Song14, Tingtao Chen15
1. Department of Pathology, Hackensack Meridian School of Medicine, Nutley, NJ, USA
2. LumaBridge, San Antonio, TX, USA
3. Queen Elizabeth Hospital, Department of Clinical Oncology, Hong Kong, China
4. Division of Genomic Medicine, Department of Medicine, and of Microbiology, Immunology & Tropical Medicine, George Washington University Medical Center, Washington DC, USA
5. Department of Obstetrics and Gynecology, Changhua Christian Hospital, Changhua, Taiwan
6. Department of Pathology, Chinese PLA General Hospital 7 th Medical Center, Beijing, China
7. IIND Department of Gynecology, Lublin Medical University, Lublin, Poland
8. Pulmonary-Oncology Department, "Theageneio" Cancer Hospital, Thessaloniki, Greece
9. Laboratory of Biological Effects of Non-Ionizing Radiation, Institute of Cell Biophysics, Russian Academy of Sciences, Russian Federation
10. Department of Pathology, Hunan Normal University School of Medicine, Changsha, Hunan, China
11. Department of Genetics and Genomics Sciences, Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
12. Laboratory of Mammalian Cell Biology and Development, The Rockefeller University, New York, NY, USA
13. Department of Sema4 Health Informatics, Stamford, CT, USA
14. Department of Medical Oncology, Peking University Cancer Hospital and Institute, China
15. Department of Gastrointestinal Surgery, The Second Affiliated Hospital of Nanchang University and National Engineering Research Center for Bioengineering Drugs and the Technologies, Institute of Translational Medicine, Nanchang University, Nanchang, China
Received 2022-12-31; Accepted 2023-1-27; Published 2023-2-27
Abstract

Breast cancer development and progression are believed to be a sequential process, from normal to hyperplastic, to in situ, and to invasive and metastatic stages. Given that over 90% of cancer deaths are caused by invasive and metastatic lesions, countless factors and multiple theories have been proposed as the triggering factor for the cascade of actions of cancer invasion. However, those factors and theories are largely based on the studies of cell lines or animal models. In addition, corresponding interventions based on these factors and theories have failed to reduce the incidence rate of invasive and metastatic lesions, suggesting that previous efforts may have failed to arm at the right target. Considering these facts and observations, we are proposing “A focal aberrant degeneration in the myoepithelial cell layer (MECL) as the most likely triggering factor for breast cancer invasion”. Our hypothesis is based on our recent studies of breast and multiple other cancers. Our commentary provides the rationale, morphologic, immunohistochemical, and molecular data to support our hypotheses. As all epithelium-derived cancers share a very similar architecture, our hypothesis is likely to be applicable to invasion of all cancer types. We believe that human tissue-derived data may provide a more realistic roadmap to guide the clinic practice.
Keywords: Breast myoepithelial cell layer, Tumor capsule, Tumor invasion, Cell interactions.
Editorial Commentary
All epithelium-derived cancers are architecturally similar with the epithelium (EP), capsule, and stroma [1,2]. The EP is the origin of a majority of the human malignances [3,4]. The breast capsule is made of the myoepithelial cell layer (MECL, a single cell layer embracing the EP) and basement membrane (BM, a thin layer of fibers and smooth muscle cells attached to the MECL) [5,6]. The prostate and salivary tissues share similar capsule constitutes with the breast, whereas other cancers have only the BM constituting the capsule [7, 8]. The stroma contains lymphocytic ducts, blood vessels, different immune cells, and EP cell metabolism-needed materials [9, 10].
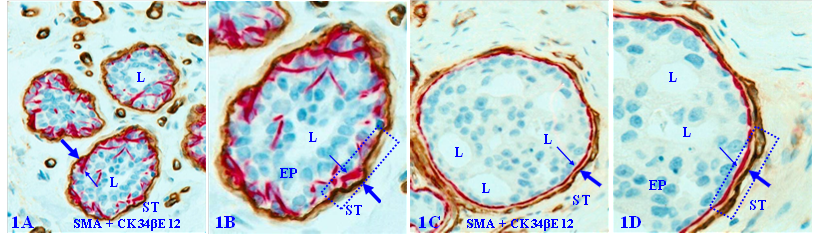
The capsule physically segregates the EP from the stroma and actively mediates the diffusion of EP growth- and metabolism-needed materials and nutrient from the stroma. In addition, as all types of EP cells belong to a self-renewal population and stem cells are normally located near the capsule, the capsule also functions as a physical confiner to force proliferating cells to migrate to the lumen and surface direction to replace aged or injured EP cells [11, 12]. Figure 1 uses breast tissues as an example to elucidate the structural relationship of different EP tissue components.
Structural relationships of breast tissue components. Formalin-fixed and paraffin-embedded human breast tissue sections are double immune-stained with a BM marker smooth muscle actin (SMA, brown) and ME cell marker CK34βE12 (red). B and D are a higher magnification of A and C. ST = stroma. EP = epithelium. L = Lumen. Thick arrows identify the BM. Thin arrows identify MECL. Squares identify capsules. Due to the conferment of the capsule, stem cell-derived proliferating cells are normally moving to the acinar or ductal lumen direction.
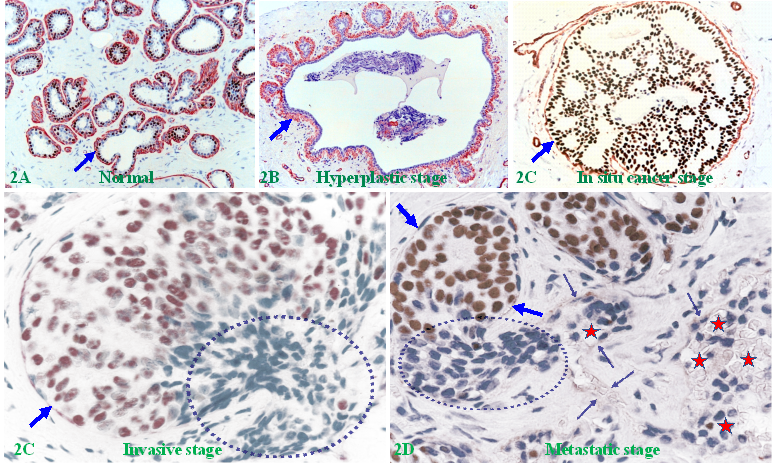
Human breast carcinogenesis is believed to follow the principle and steps proposed by “The clonal evolution theory", progressing sequentially from the normal to hyperplastic, to in situ, and finally to invasive and metastatic stages [13-15] (Figure 2).
The sequence of human breast cancer development and progression. Formalin-fixed and paraffin-embedded human breast tissue sections were double immune-stained for ME cell marker smooth muscle actin (SMA, red) and estrogen receptor (ER, brown). Arrows identify the MECL. Circles identify EP cells overlying focally disrupted MECL and within the stroma (D). Red stars identify tumor and red blood cells within a blood vessel.
With over 90% of cancer deaths resulting from invasion and metastasis-related illnesses [16-19], countless factors and theories have been proposed for triggering cancer progression:
1. Estrogen, progesterone, and their corresponding receptors, which have a dual role in regulating tumor cell proliferation and invasion [20-22].
2. Tumor suppressor genes, in which the reduction or lose can directly or indirectly lead to elevated migration and invasion of cancer cells [23-26].
3. Oncogenes, which cause aberrant expression of their oncoprotein products that facilitate tumor cell proliferation and migration [27-29].
4. Tumor dedifferentiation and dissociation, which mobilize the tumor cells out of the main tumor bulk and enables them to invade adjacent tissues by active locomotion [30-32].
5. Inflammatory signaling cascades, which are intimately involved in neoplastic processes fostering tumor cell proliferation, survival, invasion, and migration [33-35].
6. Capillary vessels derived from the periductal necklace of vessels, which breach the basement membrane, providing an escape hatch for cancer cell invasion [36-38].
7. Aberrant expression of P-Cadherin, which has been suggested as a stem cell marker associated with epithelial mesenchymal transition (EMT) and cancer invasion [39-41].
8. Tumor derived exosomes, which activate cancer-associated fibroblasts (CAFs) through miRNAs and Wnt pathway that in turn enhances invasion and metastasis [42-44].
9. Aberrant integrin expression, which supports oncogenic growth factor receptor (GFR) signaling and GFR-dependent cancer cell migration and invasion [45-47].
10. Mutational drivers, which promote chromosomal instability and genetic mutations that trigger cancer invasion [48-50].
11. Progressive changes in the structure and composition of tumor stroma, which is believed 14. to be a required transition to invasive breast cancer [51-53].
Unfortunately, these factors and theories are largely based on clinical testing results or results from in vitro studies on cancer cell lines or animal models. In addition, none of above factors or theories has elucidated the specific pathways for cancer cells to overcome the following physical and functional barriers for invading:
1. The ME or basal cells in breast, salivary and prostate gland, which embrace the entire EP system [54-56]. How do the cancer cells physically cross over ME or basal cell layers?
2. The tumor capsule in other cancer types, which is composed of smooth muscle cells and dense fibers [57-59]. How do the cancer cells physically breach these structures?
3. Intercellular junctions and adhesion molecules, which intercalate all EP cells into a single sheet [60-62]. How do the cancer cells physically disassociate into individual cells?
4. The stromal and immune surveillance system, which harbors a variety of self-defensive cells [63-65]. How do the invading cancer cells escape from this surveillance system?
5. The cancer stem cells, which are universally regarded as the direct precursor of invasive lesions [66-68]. How do the stem cells physically enter the invasion cascade?
In addition to the factors and theories alluded above, the matrix metalloproteinase (MMP) family and the associated proteolytic enzyme theory were once universally considered to be the most likely factor and mechanism for triggering the invasion of all in situ cancer types [69-74]. According to the proteolytic enzyme theory, aberrantly altered EP cells increasingly produce a wide variety of MMPs during their evolution, which reach the highest concentration at the in situ cancer stage. The elevated enzymes could selectively degrade the surrounding tumor capsule and intercellular adhesion molecules, resulting in disruptions or a total loss of the associated tumor capsule, which permits the cancer cells at the disrupted sites to freely migrate into their adjacent stroma or to invade lymphatic ducts or blood vessels [69-74] (Figure 2E).
The proteolytic theory was strongly supported by in vitro studies and animal models, which consistently showed that all matrix metalloproteinase family members could specifically degrade the tumor capsule and cause invasion, while the corresponding antagonists or neutralizing agents could partially or completely stop cancer invasion [75-81]. Those findings inspired international efforts to develop such therapeutics for clinic trials in the late 1990's. However, thousands of the world-wide clinic trials with the corresponding antagonists or neutralizing agents of MMPs have failed to show any reduction of the cancer invasion rate [82-87]. Those disappointing results of the world-wide clinic trials have led the world's top experts in the field to unanimously advocate the search for new directions and strategies to combat cancer invasion [83,88, 89].
Collectively, these facts have casted strong doubts on the validity of all proposed factors and theories on cancer invasion. Thus, we would like to propose that “A focal aberrant degeneration of the breast MECL (basal cells or capsule in other cancers) is the most likely but largely ignored triggering factor for cancer invasion” for following reasons:
A. The MECL is closely associated with both carcinogenesis and cancer progression
1. The ME cell population possess a unique anti-cancer system
ME cells have a low rate of malignancies [90-93]. Based on a 2013 article, “Myoepithelial carcinoma of the breast is extremely rare and only 33 cases have been reported in the English literature” [94]. A 2020 article reported that “Breast mucoepidermoid carcinoma (MEC) is clinically rare, with an estimated incidence of 0.2-0.3% of all primary breast tumors” [95]. It is likely that ME cells may possess a unique system that is resistant to carcinogenesis.
2. The MECL is the source of several types of tumor suppressors
A number of studies have shown that the MECL produces a number of tumor suppressors, including p63, p73, Wilms' tumor 1 (WT-1), maspin, 14-3-3 sigma, and stefin A, which exert significant inhibition on the growth of tumor cells [96-101]. The MECL also produces multiple proteinase- and angiogenic-inhibitors, which suppress cell migration [102-104]. Figure 3 shows a set of 3-consecutive sections from a biopsy sample immune-stained for 3-different suppressors. It is apparent that they are co-expressed in the MECL of all normal and hyperplastic structures.
Tumor suppressors in the MECL of normal and hyperplastic structures. A set of three consecutive sections from a female patient with morphologically normal and hyperplastic appearing acinar and ductal structures were immune-stained for maspin, p63, and WT-1. Arrows identify the MECL. Please note that the MECL in all these acinar and ductal structures are non-disrupted and express high levels of all three tumor suppressors.
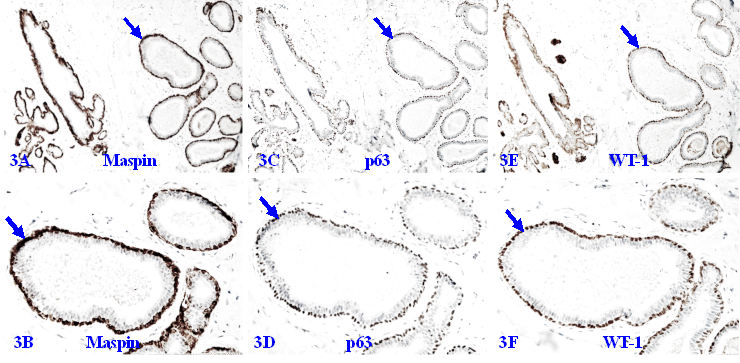
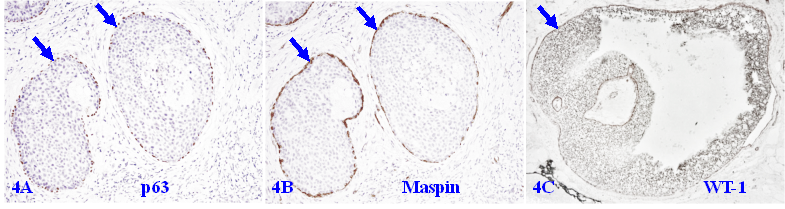
The similar co-expression of different tumor suppressors is also seen in the MECL of a vast majority of DCIS cases, provided it is morphologically distinct and intact as shown in Figure 4.
Tumor suppressors in the MECL of ductal carcinoma in situ (DCIS). Formalin-fixed and paraffin-embedded human breast tissue sections from two patients with ductal carcinoma in situ (DCIS) were immune-stained for p63, maspin, and WT-1. Please note that the MECL in all cases are non-disrupted and express high levels of all three tumor suppressors, even at the case with a large size of DCIS (4C).
3. Focal MECL disruptions confer epithelial cell invasive growth pattern
Breast EP cells are a self-renew population with stem cells normally located at the basal layer resting on the MECL [11,12]. Due to the physical confinement imposed by the surrounding MECL and BM, stem cells normally undergo an orchestrated, progressive series of proliferation and differentiation steps at the basal layer, and then move unidirectionally upward towards the acinar or ductal luminal direction to replace aged or damaged cells. However, if the surrounding capsule is disrupted, the confinement force will be lost and proliferating cells will be more easily migrating to invade into the stroma. As the EP is normally devoid of lymphatic ducts and blood vessels, whereas the stroma is very rich in both, and thus, invading cells are prone to metastasize.
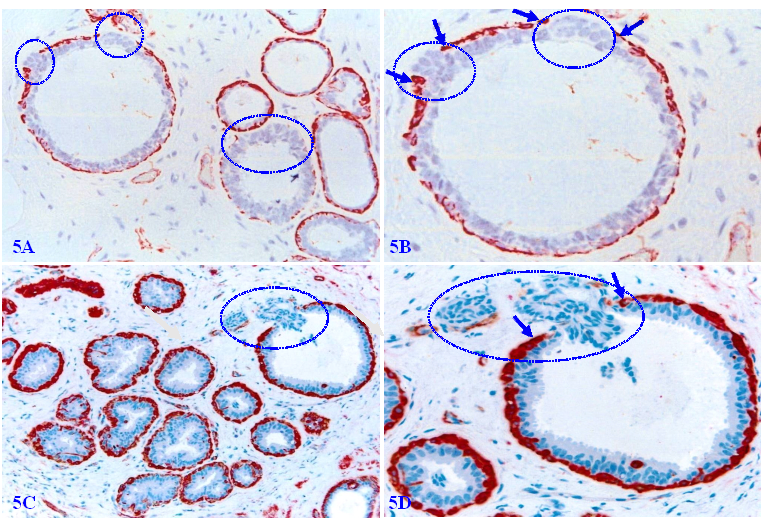
Figure 5 shows a normal (A-B) and hyperplastic (C-D) appearing duct, which harbors three focal disruptions in the MECL (the absence of ME cells resulting in a gap larger than a combined size of at least 3 ME cells in at least 2 or more consecutive sections). EP cells overlying each of these focal MECL disruptions form a tongue-like protrusion “invading” towards the stroma.
Invasive growth pattern in normal and hyperplastic appearing breast ducts. Formalin-fixed and paraffin-embedded human normal and hyperplastic appearing breast tissue sections were immune-stained for smooth muscle actin (SMA). Arrows identify residual ME cell layers. Circles identify cell clusters overlying focally disrupted MECL, which are arranged as tongue-like protrusions “invading” into the adjacent stroma.
4. Focal MECL disruptions selectively favor formation of ER (-) and Her-2 (+) cell clusters
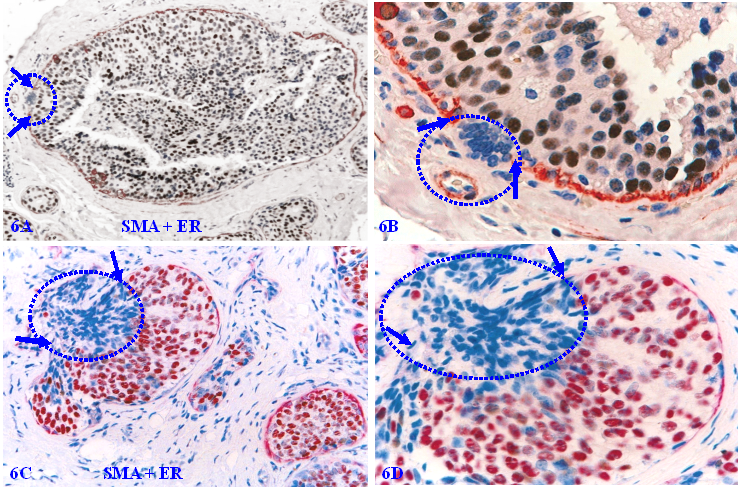
Our previous studies have consistently shown that the size of focal MECL disruptions along with the overlying tumor cell clusters in the pure cases of DCIS is very small and hard to find in H & E-stained sections [105-114]. As shown in Figure 6A - 6B, a large DCIS harbors a small disruption with only about 10-overlying ER-negative cells. In contrast, the size of focal MECL disruptions and overlying tumor cells clusters are significantly larger in cases of invasive ductal carcinoma (IDC; Figure 6C-6D).
ER-negative cell clusters overlying focally disrupted MECLs. Formalin-fixed and paraffin-embedded human breast cancer tissue sections are double immune-stained for SMA (red) and ER (brown or pink). Arrows identify residual MECL. Circles identify ER-negative cell clusters overlying focal MECL disruptions. Please note that cells overlying focal MECL are ER-negative, whereas cells within the tumor core are ER-positive.
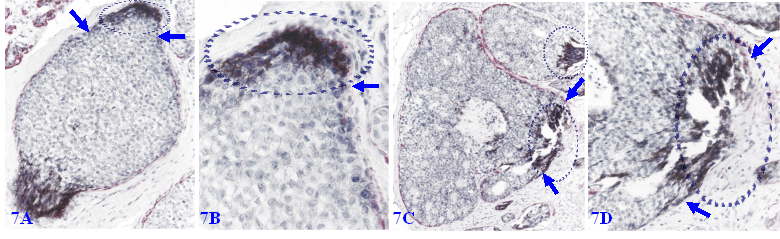
Focal MECL disruptions also facilitate the formation of Her-2 (+) cell clusters overlying focal MECL disruptions. As showed in Figure 7, EP cells surrounded by the residual MECLs are largely devoid of Her-2 expression, whereas all ductal EP cells localized at the focal MECL disruptions show high levels of cytoplasmic Her-2. Cells overlying focal MECL disruptions are arranged as tongue-like protrusions “invading” into the adjacent stroma.
Her-2 positive cell clusters overlying focally disrupted MECLs. Formalin-fixed and paraffin-embedded human breast cancer tissue sections are double immune-stained for SMA (red) and Her-2 (black). Arrows identify residual MECLs. Circles identify Her-2-positive cell clusters overlying focal MECL disruptions. Please note that the EP cells within the tumor core surrounded by the residual MECL are largely devoid of Her-2 expression.
5. Tumor cells overlying focal MECL disruption show a unique gene expression pattern
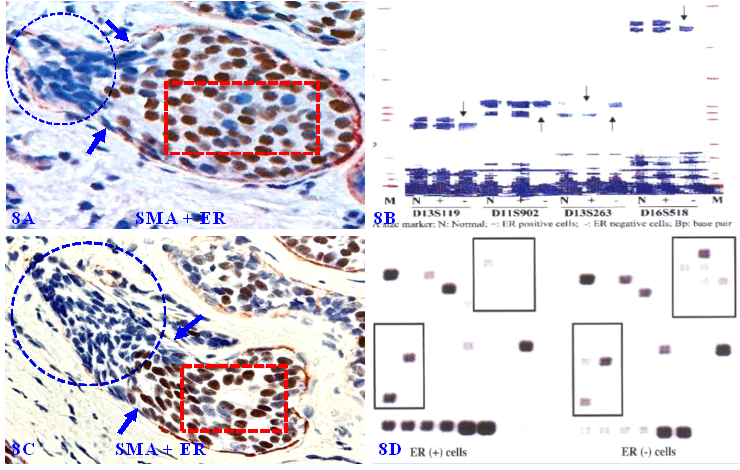
Our previous studies have consistently shown that micro-dissected ER-negative cell clusters overlying focally disrupted MECL have a different rate and pattern of the loss of heterozygosity (LOH), and expression of cell proliferation and stem cell markers compared to their comparable counterparts within the same tumor core surrounded by the residual MECL [105-114] (Figure 8).
Gene expression profiling of cells overlying focally disrupted MECLs. Formalin-fixed and paraffin-embedded human breast tissue sections are double immune-stained for SMA (red) and ER (brown). Arrows identify the residual MECL near focal disruptions. Circles identify ER-negative cells overlying focal MECL disruptions. Squares identify ER positive cells still enclosed by residual MECL. Please note that ER-negative cells overlying focally disrupted MECL have a different LOH and gene expression profiles compared to their ER-positive counterparts within the tumor core.
6. Focal MECL disruptions are associated with MMP- and ER-negative cell clusters
Our previous studies have extensively studied the impact of focal MECL disruptions on the expression of a wide variety of cancer invasion-related molecules in confirmed DCIS cases [105-114]. Figure 9 shows the expression status of 9-consecutive sections of a DCIS case double immune-stained for SMA, MMP-26, MMP-4, MMP-9, ER, and PR, respectively.
Focal MECL disruptions are associated with MMPs- and ER-negative cell clusters. A set of 9 consecutive sections of a DCIS were double immune-stained for SMA (brown or red) and MMP-26, MMP-4, MMP-9, ER, or PR. Circles identify focal MECL disruptions and overlying cells. Arrows identify residual MECLs. Red stars identify vascular-like structures. Please note that: (1) all cell clusters overlying focal MECL disruptions are MMPs- negative, but cells within the tumor core surrounded by the residual MECL are MMPs positive, and (2) all cell clusters overlying focally disrupted MECL are ER- and PR-negative, whereas their counterparts within the tumor core are ER- and PR-positive.
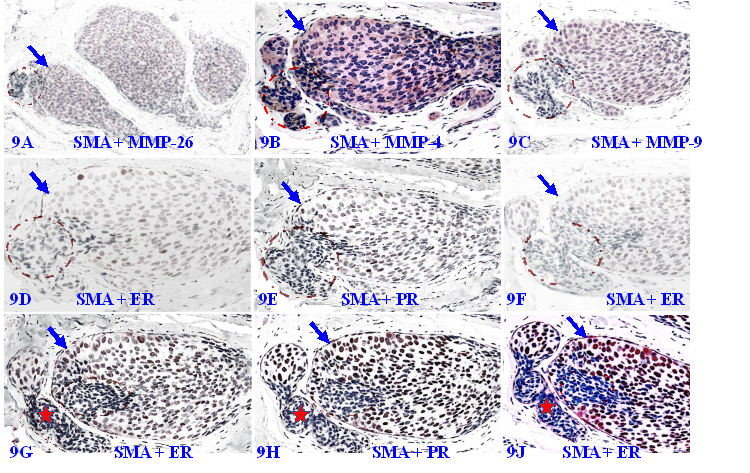
It can be seen that a small DCIS harbors a relatively large focal disruption on its MECL (circles), in which overlying cells and their adjacent counterparts still surrounded by the residual MECL exhibit different immunohistochemical features. In the first 3-sections, cells surrounded by the residual MECL are positive for MMPs, while cells overlying focal MECL disruptions are devoid of all MMPs. In the remaining 6-sections, cells surrounded by the residual MECL show distinct ER- and PR-positivity, while cells overlying disrupted MECL are ER- and PR-negative.
In Figure 9C to 9F, cells overlying focal MECL disruptions are arranged as tongue-like protrusions in which all MMP-9-, ER, and PR-negative cells are physically joined together into a single unit that is morphologically and immunohistochemically indistinguishable. Please note that, in Figure 9G to 9J, ER-negative cells within the stroma appear to be within vascular-like structures, suggesting that cells overlying focally disrupted MECL are able to join with similar cells and vascular structures from adjacent tissues to form metastatic lesions.
7. Focal MECL disruptions are associated with elevated immune-cell infiltration
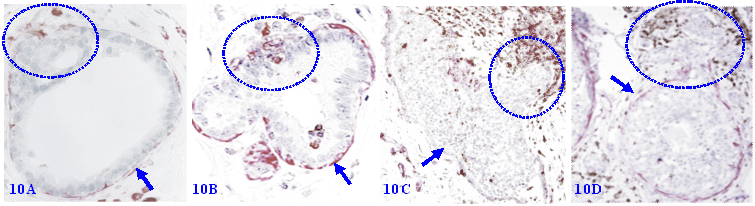
Focal MECL disruptions among normal, hyperplastic, and pre-invasive cancers are almost always associated with elevated infiltration of immune cells, which are consistently located at or near disruptions. Non-disrupted MECLs are barely with immune-cell infiltration (Figure 10).
Focal MECL disruptions are associated with elevated immune-cell infiltration. Formalin-fixed and paraffin-embedded human breast tissue sections from 4 different cases were double immune-stained for SMA (red) and leukocyte common antigen (LCA; brown). Arrows identify the residual MECL. Circles identify infiltrating immune cells overlying focal MECL disruptions. Please note that the non-disrupted MECL is largely devoid of infiltrated immune cells.
Collectively, these findings strongly suggest that the morphological integrity of the MECL has the crucial functions on the proliferation, growth, gene expression, migration of associated EP cells. These findings also strongly suggest that a focal MECL disruption has the potential to trigger a cascade of reactions of cancer invasion- and metastasis-related events.
B. The MECL also suffers from a wide variety of pathologic alterations
On the other hand, in addition to the focal disruptions seen above, the MECL also suffers from a wide variety of pathologic or immunohistochemical alterations:
1. The loss of phenotypic marker SMS in morphologically distinct MECLs
SMA is a normal and primary constitute of the MECL and BM, functioning as the core of the capsule of all EP-derived cancers, and double immunohistochemistry with SMA and cytokeratin has been routinely used to confirm early invasive breast and other cancers [115,116]. However, a subset of cases harbor MECLs that are morphologically distinct, while are devoid of the SMA expression. As shown in Figure 11A - 11B, all EP cells surrounded by the SMA negative MECL are devoid of ER-expression, whereas their adjacent counterparts are largely ER-positive.
Loss of phenotypic marker in morphologically distinct MECLs. Formalin-fixed and paraffin-embedded human breast tissue sections from a DCIS case are double immune-stained for SMA (red) and ER or Ki-67 (black). Thick arrows identify the residual MECL. Thin arrows identify morphologically distinct MECLs that are devoid of the SMA expression. Circles identify ER-negative cells or Ki-67-positive EP cells. Please note that all SMA negative ME cells completely lack the expression of the proliferating cell specific marker Ki-67.
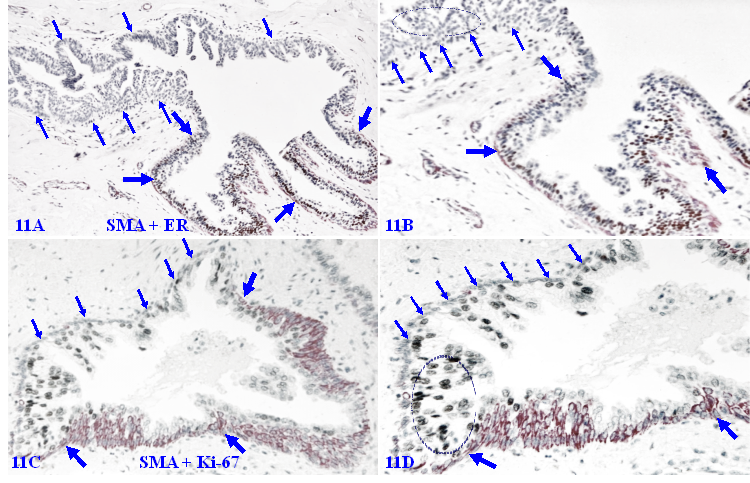
Figure 11C - 11D show an adjacent section from the same case double immune-stained for SMA and Ki-67, a cell proliferation specific marker. A vast majority of EP cells surrounded by SMA-negative MECL are strongly positive for Ki-67, while all SMA-negative ME cells are completely devoid of the Ki-67 expression, suggesting that these SMA-negative ME cells may have lost the power, or have had a unique system, of the self-cellular replenishment.
2. Elevated apoptosis in focally disrupted MECL
Among morphologically comparable EP structures, MECLs with focal disruptions also have a significantly higher rate of apoptosis. A vast majority of apoptotic ME cells are located at or near the focal MECL disruptions, and no distinct apoptotic EP cell is seen (Figure 12).
Elevated apoptosis in focally disrupted MECLs. Formalin-fixed and paraffin-embedded human breast tissue section was double immune-stained for SMA (red) and an apoptotic marker (brown). Thick arrows identify the residual MECL. Thin arrows identify apoptotic ME cells. Squares identify focal MECL disruptions. Please note that all apoptotic ME cells are located at or near focal MECL disruptions.

3. Elevated Tenascin expression in focally disrupted MECLs
Tenascin C is an extracellular matrix glycoprotein, which paves the paths and facilitates the migration and metastasis of breast cancer cells [117-119]. Our previous studies have shown that Tenascin C is also highly expressed at distinct degenerating prostate basal cells, and EP cells in the vicinity of areas with elevated Tenascin C often lose the cohesion [120-125] (Figure 13).
Elevated Tenascin expression at focally disrupted MECLs. Formalin-fixed and paraffin-embedded human breast tissue section was double immune-stained for SMA (red) and Tenascin (brown). Thick arrows identify non-disrupted or residual MECL. Thin arrows show the Tenascin positivity. Please note that a high level of Tenascin expression is seen at or near focally disrupted MECL, whereas the non-disrupted MECL is completely devoid of any distinct Tenascin expression.

4. Immunohistochemically altered MECLs in normal and hyperplastic tissues
In a vast majority of autopsy, biopsy, and surgically resected breast tissues, the MECL is physically continuous with a high level of tumor suppressor expression as those seen in figures 3 and 4 above. However, about 10-15% of cases harbor normal or hyperplastic tissue clusters that display several forms of morphological and immunohistochemical alterations in MECLs. These altered MECLs are generally distributed in the entire structures of a given small lobule.
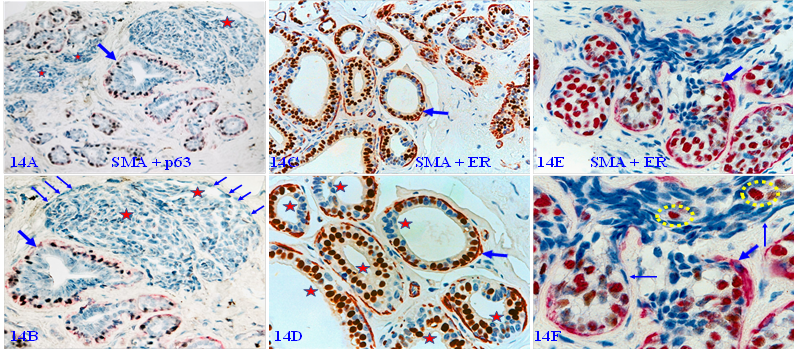
Figure 14A - 14B shows that all MECLs in the entire normal and hyperplastic structures are either focally disrupted or are devoid of the SMA expression in morphologically distinct ME cells of multiple solid tumor nests. Figure 14C - 14D shows that all MECLs in all normal and hyperplastic ducts within a lobule are focally disrupted with ER-negative cell clusters overlying focal MECL disruptions. Figure 14E - 14F shows that all MECLs in normal-appearing structures are focally disrupted with ER-negative and positive cells migrating into vascular-like structures.
Immunohistochemically altered MECLs in normal and hyperplastic tissues. Formalin-fixed and paraffin-embedded human breast tissue sections from 3-cases were double immune-stained for SMA (red) and p63 (black), or ER (brown). Thick arrows identify residual MECLs. Thin arrows identify immunohistochemically altered ME cells. Stars identify solid tumor nests or ducts with focally disrupted MECL and ER-negative cells clusters. Circles identify ER- positive cells within a vascular-like structure. The MECLs in all structures of all 3-cases are immunohistochemically altered.
5. Mixed normal appearing ME and EP cell clusters with cytoplasmic Her-2 expression
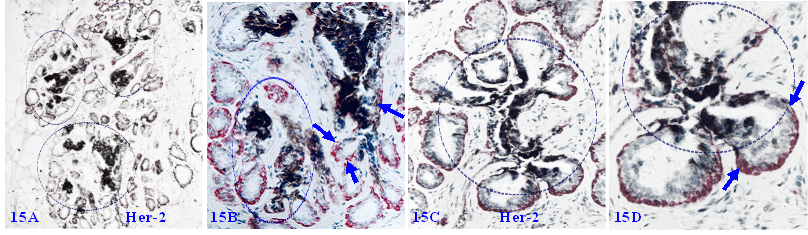
Some focal MECL disruptions are also associated with cytoplasmic Her-2 expression. As shown in Figure 15, these cytoplasmic Her-2 expressing cells are exclusively located at or near focal MECL disruptions, and often extending into tongue-like protrusions invading the stroma or vascular-like structures. These clusters and their derivatives are immunohistochemically and morphologically comparable to those seen in DCIS cases, suggesting that these cell clusters are very likely developed from the same progenitors and also have the same clinic significance.
Mixed normal appearing ME and EP cell clusters with cytoplasmic Her-2 expression. Formalin-fixed and paraffin-embedded human breast tissue sections from two cases were double immune-stained for SMA (red) and Her-2 (black). Arrows identify normal appearing MECL. Circles identify focal MECL disruptions and overlying Her-2 positive cell clusters. Please note that these Her-2-positive cells are arranged as tongue-like protrusions invading into the stroma or vascular-appearing structures.
Collectively, these findings suggest that the ME cell population belongs to a self-renewal population, which must constantly undergo cell proliferation and differentiation to replace aged or injured cells. Consequentially, any internal or external pathological insult on the ME cell population or its progenitors would undoubtedly have the potential to result in a wide variety of morphological and immunohistochemical alterations.
C. Our detailed hypothesis of MECL-mediated cancer invasion
Based on above findings, we have hypothesized that breast cancer progression and invasion are triggered by the aberrant morphological and immunohistochemical alterations in the MECL via the following specific mechanisms and steps:
1. The predisposition of genetic defects in ME cell replenishment-related genes results in elevated focal degeneration in some ME cells (Figure 11-13);
2. The degradation products of ME cells attract the infiltration of immunoreactive cells into the affected sites to clean-up the degenerated ME cells and the BM (Figure 10);
3. The destruction and cleaning-up of degenerated ME cells and the BM results in a focal disruption in the MECL (Figure 10);
4. A focal MECL disruption results in a focal reduction or loss of tumor suppressors, which favors the proliferation of stem cells or an more aggressive cell clone (Figures 5-8);
5. A focal MECL disruption also results in the increase of oxygen, nutrients, and growth factors that promote stem cell-mediated EP cell proliferation;
6. The apoptosis and degeneration of ME cells promote the expression of Tenascin and Her-2, which facilitate the migration and invasion of proliferating EP cells (Figures 12-13);
7. The proliferation of a give stem cells generates a cluster of unmatured cells that lack the expression of cell age-dependent proteins, including ER, PR, and MMPs (Figure 9);
8. A focal MECL disruption lifts the confinement of the capsule, which selectively favors proliferating stem cells to migrate into the stroma (Figure 5-9);
9. A focal MECL disruption favors multipotent stem cells to form a mixed cell population with the capability to make its own lymphatic or vascular structures facilitating adjacent tumor cells to metastasize (Figure 14).
Compared to previously proposed theories of breast cancer invasion [20-53], our hypothesis is distinct in two main respects: (1) our hypothesis is entirely based on morphological, molecular, and immunohistochemical data from studies on human breast tissues with detailed clinic records and follow-up data, and (2) our hypothesis can reasonably elucidate the cellular and molecular mechanisms for cancer cells to overcome potential physical and functional barriers for invasion.
In comparison to the proteolytic enzyme theory of cancer invasion [69-88], our hypothesis is also noteworthy in that (1) our studies have consistently revealed that EP cells overlying focally disrupted MECL and their stromal derivatives are devoid of MMPs, while cells within the tumor core surrounded by the residual MECL have a high level of MMPs (Figure 9), and (2) “invasive” growth patterns are also seen in normal or hyperplastic appearing structures (Figures 5 and 14).
Our hypothesis also differs from the "clonal evolution and cancer heterogeneity theory", which proposes that "most neoplasms arise from a single cell of origin, and tumor progression results from acquired genetic variability within the original clone allowing sequential selection of more aggressive sublines" [13]. Although the main concept of this theory has been universally accepted and supported by several lines of experimental and clinic data [126-135], it leaves four unanswered questions: (1) is carcinogenesis initiated by a normal or cancer stem cell? (2) is "sequential selection of more aggressive sublines" alone sufficient to accomplish carcinogenesis and progression? (3) can an aberrant microenvironment change the fate of "acquired genetic variability within the original clone"? (4) is the progression of different tumors triggered by the same or different factor(s)?
Our hypothesis is very likely to be applicable to the invasion cascade observed in all EP-derived malignancies. Our previous studies of human prostate, lung, gastric, colorectal, skin, salivary gland, and cervical tumor tissues have consistently seen that the capsules of these tissues (especially the prostate tumor) harbor morphologically and immunohistochemically similar focal disruptions, which are associated with EP cell alterations that are essentially identical as those seen in the breast [109, 110, 120-125].
A recent article compared our hypothesis with the enzyme theory, and has concluded that “…the FMCLD theory has some advantages over proteolytic theory because it focuses on the interaction of the different types of cells present in the tumor microenvironment [136]. The localized death of myoepithelial cells causes the release of its inner contents like the proteolytic enzymes and growth factors. The resulting immunoreactions that accompany an external environmental insult or internal genetic alterations are triggering factors for further disruptions of the myoepithelial cell layer, BM degradation, and subsequent tumor progression and invasion”. Many other articles have also reported the advantages of our hypothesis and results [137-142].
D. Specific applications of MECL in detection and interventions of cancer invasion
Based on above findings, it is apparent that the MECL not only decisively controls the proliferation rate and migration direction of adjacent EP cells, but also accurately reflects their functional status. More importantly, the MECL is intimately intermixed with the BM, forming the only physical barrier to inhibit the invasion of malignant EP cells into the stroma. As the morphologic, pathological, and immunohistochemical profiles of the ME cell population is far more easily recognizable and definable than its EP counterpart, the MECL appears to have the following specific clinic implications and applications:
1. To use maspin and ER expression as independent risk factors for identifying more aggressive lesions and corresponding therapeutics. Previous studies have shown that the expression of maspin is correlated with breast cancer invasion, brain metastases, and cancer recurrence [143-146]. Previous studies have revealed that the ER-expression level is a reliable indicator for cancer therapeutic responses [147-150]. Therefore, to assess the mRNA expression level of masoin and ER in the serum is likely to facilitate the detection of more aggressive lesions and the corresponding therapeutics.
2. To use p63 as risk factor for a population-based screening to detect predisposition of cancer susceptibility or tumor suppressing genes. Since p63 belongs to the p53 tumor suppressor family, and is normally expressed in the nucleus of the ME cells [151,152], an aberrant expression level or subcellular localization of p63 may signify the predisposition of cancer susceptibility or mutated suppressing genes. Previous studies have consistently revealed that the loss or cytoplasmic expression of p63 is associated with elevated stem cells, enhanced cell migration and metastasis, and increased mortality [153-155].
3. To use ME cell-derived tumor suppressor-related signatures for early non-invasive detection of breast cancer. Previous studies have consistently shown that several forms of tumor suppressor-related signatures are detectable in the blood sample and body fluid of patients with breast cancer [156-162]. Thus, a statistical comparison of the expression levels of p63, maspin, and other tumor suppressors in the blood samples and body fluid may be used as a non-invasive clinic test or a population-based screening method for the detection of the specific individuals with or at a higher risk for early breast cancer.
4. To search exfoliated ME cells or p63-related signatures in ductal lavage to identify women at increased genetic risk of breast cancer. Previous studies have consistently shown that a variable number of exfoliated epithelial cells can be retrieved from ductal lavage for different assays for the identification of BRCA1/BRCA2 mutations [163-166]. The search for exfoliated ME cells or p63-related signatures in ductal lavage may lead to the development of a new non-invasive cancer detection method.
5. To use the MECL physical integrity (disrupted vs non-disrupted) as a clinic marker for the differentiation diagnosis. As the disruption of the MECL is a prerequisite for breast cancer invasion and metastasis, the physical integrity of the MECL in patients with and without focal disruptions could effectively differentiate between non-invasive and invasive breast cancer.
6. To use Tenascin expression in focally disrupted MECL as a routine clinic test of breast biopsy. Previous studies have consistently demonstrated that aberrant Tenascin expression is exclusively seen at or near focally disrupted MECL and is also significantly correlated with breast cancer invasion and metastasis [117-119]. Thus, the assessment of MECL-associated Tenascin expression may lead to the identification of the specific cases at increased risk for breast cancer progression.
7. To use focal MECL disruptions as a localizer to identify cancer-stem cell clusters/ specific precursors of invasive cancer. Our previous studies of multiple cancers have consistently shown that a focal disruptions of tumor capsules selectively facilitate clonal proliferation of overlying cancer stem cells to form distinct cell clusters. These newly formed clusters have a significantly higher level of cancer stem cell markers and invasion and metastasis-related gene expression than their morphologically comparable counters still enclosed by the non-disrupted tumor capsules [105-114] (Figure 8). It is very likely that these cell clusters may represent the direct precursors of invasive lesions. However, it is very difficult, if not impossible, to detect small MECL disruptions and associated ER negative cell clusters (Figure 6A -6B). Therefore, double immunohistochemistry with ER and SMA may be used as a reliable localizer to identify these potential stem cell clusters.
8. To use MECL-associated immune cell infiltration to monitor the tumor progression and treatment responses. Our previous studies have consistently shown that immune-cell infiltration is significantly associated with breast tumor capsule disruptions, which lead to the subsequent invasion and metastasis (Figure 10) [107, 109, 110, 120, 123]. A number of recent studies have not only confirmed our previous reports and conclusions, but have also consistently shown that immune-cell infiltration is significantly correlated with the treatment responses in multiple cancer types [167-171]. Therefore, a double immunohistochemistry with SMA and LCA to assess the physical integrity and extent of associated infiltrating immune cells of the MECL in the breast and the tumor capsule in other cancer types in biopsy samples may provide a novel clinic method to differentiate aggressive from indolent cancers, and also to monitor treatment responses of immuno- therapies.
9. To use anti-inflammatory drug aspirin or statin to repair ME degeneration-related tumor capsule disruptions. Previous studies have consistently demonstrated that aspirin or statin could significantly alter the chronic inflammation milieu of a variety of human cancers and prevent cancer progression [172-176]. Thus, the administration of aspirin or statin to individuals with focally disrupted MECL associated with significant infiltration of immune cells (as shown in Figure 10) may potentially reduce the extent of infiltrating immune cells. It is likely that the reduction of the immune cell infiltration may facilitate the repairing of focally disrupted tumor capsules and consequently prevents the invasion of associated EP malignancies.
10. To administer stem cell specific molecules, inducers, or stimulators to burst normal replenishment of MECL. Previous studies have shown that a number of biomolecules, including CD24, CD44, CD133, Oct4, Gli1, ALDH1, Notch-1, Nectin-4, Neuregulin-1, Musashi-1, SSEA-3, DDX53, O-Acetyl-GD2, and DEAD-box helicase 27, are breast stem cell-related biomolecules, which are essential for the maintenance of the normal cellular replenishment or the regeneration processes after the internal or external insults [177-191]. Thus, the administration of these molecules, or stem cell specific inducers and stimulators to patients with a high frequency of degenerations and focal disruptions (Figure 11, 14) may facilitate the restoration of the normal replenishment and functions of the MECLs. On the other hand, the administration of specific antagonists to those biomolecules may inhibit the aberrant EP cell proliferation and cancer stem cells-mediated invasion or metastasis.
11. To use MECL lacking phenotypic markers or physically conjoined with vascular structures to identify novel cell proliferation pathways or cell cycle regulators. As a subset of morphologically distinct and non-disrupted MECL is completely devoid of the expression tumor suppressors, phenotypic and proliferation specific markers (Figure 11) or is physically conjoined with vascular structures (Figure 14), it is likely that these ME cells are derived from or regulated by a previously unidentified mechanism or pathway [192-199]. Thus, microdissection of these MECL for the gene expression profiling may lead to the identification of a novel cell proliferation pathway and cell cycle regulators.
12. To use profiles of cells overlying focal MECL to explore therapeutic applications of "clonal evolution and cancer heterogeneity theory"[13]. Although the main concept of the clonal evolution theory has been universally accepted and supported, it has failed to address four essential issues: (1) a normal or malignant nature of "single cell of origin" for carcinogenesis; (2) a partial or complete role of the EP in cancer development and progression; (3) a partial or complete role of microenvironment in cancer development and progression; (4) an all-shared or independent triggering factor for the invasion of all EP-derived cancers. The lack of definite answers for these fundamental issues makes it very difficult, if not impossible, to develop effective therapeutic strategies and agents for the early detection and intervention of the cancer invasion. Therefore, a systematic gene expression profiling and comparison of the profiles of the cells overlying focal MECL disruptions may lead novel findings that have the potential to address the unanswered issues and to develop more effective therapeutic strategies and agents.
In summary, the above findings strongly suggest that a focal disruption in the MECL of the breast or equivalent cell type and capsule in other EP-derived tissues is the most likely triggering factor for the cancer invasion. However, the impact of these EP-surrounding structures has been largely ignored since the previous efforts of the basic scientific researches on cancer invasion are primarily focused on cell lines or animal models. It is apparent that human tissue-derived basic research data may provide a more realistic and feasible roadmap to permit the observations of the direct interactions among different cell types, and thereby avoid potentially misleading pitfalls or shortcomings of in vitro and animal studies.
Acknowledgements
All the studies were financially supported in part by grants from the Armed Forces Institute of Pathology and American Registry of Pathology (AFIP & ARP 05AA), The US Military Cancer Institute and Henry M. Jackson Foundation (USMCI2008-02), The US Congressionally Directed Medical Research Programs (DAMD17-01-1-0129, DAMD17-01-1-0130, and PC0513080), The Susan G. Komen Breast Cancer Foundation (BCTR0706983), The Ministry of Chinese Science and Technology Department (2006CB910505), and The Natural Science Foundation of China (NSFC30801176) under the category of “Cancer Early Detection” to Dr. Yan-gao Man.
All pictures presented in the current commentary were taken from the above studies on human breast tissues with detailed clinic parameters and following-up from the AFIP & ARP, which were the world's largest and most comprehensive second-opinion consultation center with the world's largest human tissue repository. As the basic medical scientific studies are traditionally conducted on cancer cell lines or animal models, which are impossible to completely mirror or mimic the human in vivo status, we hope that these pictures would more realistically elucidate the interactions among different tissue components and serve as standards for judging the value of the experimental findings from cell lines or cell line-derived animal models.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Idikio HA. Human cancer classification: a systems biology- based model integrating morphology, cancer stem cells, proteomics, and genomics. J Cancer. 2011;2:107-15
2. Almagro J, Messal HA, Elosegui-Artola A, van Rheenen J, Behrens A. Tissue architecture in tumor initiation and progression. Trends Cancer. 2022;8(6):494-505
3. Martínez-Climent JA, Andreu EJ, Prosper F. Somatic stem cells and the origin of cancer. Clin Transl Oncol. 2006;8(9):647-63
4. Méndez-López LF. Revisiting Epithelial Carcinogenesis. Int J Mol Sci. 2022Jul4;23(13):7437
5. Nakamura R, Oyama T, Inokuchi M, Ishikawa S, Hirata M, Kawashima H, Ikeda H, Dobashi Y, Ooi A. Neural EGFL like 2 expressed in myoepithelial cells and suppressed breast cancer cell migration. Pathol Int. 2021;71(5):326-336
6. Ghannam SF, Rutland CS, Allegrucci C, Mongan NP, Rakha E. Defining invasion in breast cancer: the role of basement membrane. J Clin Pathol. 2022:jcp-2022-208584.
7. Liu A, Wei L, Gardner WA, Deng CX, Man YG. Correlated alterations in prostate basal cell layer and basement membrane. Int J Biol Sci. 2009;5(3):276-85
8. Ye P, Gao Y, Wei T, Yu GY, Peng X. Absence of myoepithelial cells correlates with invasion and metastasis of Carcinoma ex pleomorphic adenoma. Int J Oral Maxillofac Surg. 2017;46(8):958-964
9. Shekhar MP, Werdell J, Santner SJ, Pauley RJ, Tait L. Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: implications for tumor development and progression. Cancer Res. 2001;61(4):1320-6
10. Mierke CT. The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep Prog Phys. 2019;82(6):064602
11. Perryman SV, Sylvester KG. Repair and regeneration: opportunities for carcinogenesis from tissue stem cells. J Cell Mol Med. 2006;10(2):292-308
12. Farnie G, Clarke RB. Mammary stem cells and breast cancer-role of Notch signalling. Stem Cell Rev. 2007;3(2):169-75
13. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23-8
14. Krøigård AB, Larsen MJ, Lænkholm AV, Knoop AS, Jensen JD, Bak M. et al. Identification of metastasis driver genes by massive parallel sequencing of successive steps of breast cancer progression. PLoS One. 2018;13(1):e0189887
15. Cocker R, Oktay MH, Sunkara JL, Koss LG. Mechanisms of progression of ductal carcinoma in situ of the breast to invasive cancer. A hypothesis. Med Hypotheses. 2007;69(1):57-63
16. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7-33
17. Li N, Deng Y, Zhou L, Tian T, Yang S, Wu Y. et al. Global burden of breast cancer and attributable risk factors in 195 countries and territories, from 1990 to 2017: results from the Global Burden of Disease Study 2017. J Hematol Oncol. 2019;12(1):140
18. Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac J Cancer Prev. 2016;17(S3):43-6
19. Ghoncheh M, Momenimovahed Z, Salehiniya H. Epidemiology, Incidence and Mortality of Breast Cancer in Asia. Asian Pac J Cancer Prev. 2016;17(S3):47-52
20. Platet N, Cathiard AM, Gleizes M, Garcia M. Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol. 2004;51(1):55-67
21. Allison KH, Hammond MEH, Dowsett M, McKernin SE, Carey LA, Fitzgibbons PL. et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Guideline Update. Arch Pathol Lab Med. 2020;144(5):545-563
22. Mouttet D, Laé M, Caly M, Gentien D, Carpentier S, Peyro-Saint-Paul H. et al. Estrogen-Receptor, Progesterone-Receptor and HER2 Status Determination in Invasive Breast Cancer. Concordance between Immuno-Histochemistry and MapQuant™ Microarray Based Assay. PLoS One. 2016;11(2):e0146474
23. Karnik P, Paris M, Williams BR, Casey G, Crowe J, Chen P. Two distinct tumor suppressor loci within chromosome 11p15 implicated in breast cancer progression and metastasis. Hum Mol Genet. 1998;7(5):895-903
24. Lee S, Stewart S, Nagtegaal I, Luo J, Wu Y, Colditz G. et al. Differentially expressed genes regulating the progression of ductal carcinoma in situ to invasive breast cancer. Cancer Res. 2012;72(17):4574-86
25. Zou Z, Gao C, Nagaich AK, Connell T, Saito S, Moul JW, Seth P, Appella E, Srivastava S. p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem. 2000;275(9):6051-4
26. Maass N, Hojo T, Zhang M, Sager R, Jonat W, Nagasaki K. Maspin-a novel protease inhibitor with tumor-suppressing activity in breast cancer. Acta Oncol. 2000;39(8):931-4
27. Moelans CB, de Weger RA, Monsuur HN, Vijzelaar R, van Diest PJ. Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol. 2010;23(7):1029-39
28. Révillion F, Bonneterre J, Peyrat JP. ERBB2 oncogene in human breast cancer and its clinical significance. Eur J Cancer. 1998;34(6):791-808
29. Pakneshan P, Szyf M, Rabbani SA. Methylation and inhibition of expression of uPA by the RAS oncogene: divergence of growth control and invasion in breast cancer cells. Carcinogenesis. 2005;26(3):557-64
30. Gabbert H, Wagner R, Moll R, Gerharz CD. Tumor dedifferentiation: an important step in tumor invasion. Clin Exp Metastasis. 1985;3(4):257-79
31. May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA. Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res. 2011;13(1):202
32. Wang H, Unternaehrer JJ. Epithelial-mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev Dyn. 2019;248(1):10-20
33. Zhou C, He X, Tong C, Li H, Xie C, Wu Y. et al. Cancer-associated adipocytes promote the invasion and metastasis in breast cancer through LIF/CXCLs positive feedback loop. Int J Biol Sci. 2022;18(4):1363-1380
34. Tewari D, Bawari S, Sharma S, DeLiberto LK, Bishayee A. Targeting the crosstalk between canonical Wnt/β-catenin and inflammatory signaling cascades: A novel strategy for cancer prevention and therapy. Pharmacol Ther. 2021;227:107876
35. Naseer F, Ahmed M, Majid A, Kamal W, Phull AR. Green nanoparticles as multifunctional nanomedicines: Insights into anti-inflammatory effects, growth signaling and apoptosis mechanism in cancer. Semin Cancer Biol. 2022;86(Pt 2):310-324
36. Cocker R, Oktay MH, Sunkara JL, Koss LG. Mechanisms of progression of ductal carcinoma in situ of the breast to invasive cancer. A hypothesis. Med Hypotheses. 2007;69(1):57-63
37. Dey M, Ayan B, Yurieva M, Unutmaz D, Ozbolat IT. Studying Tumor Angiogenesis and Cancer Invasion in a Three-Dimensional Vascularized Breast Cancer Micro-Environment. Adv Biol (Weinh). 2021;5(7):e2100090
38. Yaman S, Gumuskaya B, Ozkan C, Aksoy S, Guler G, Altundag K. Lymphatic and capillary invasion patterns in triple negative breast cancer. Am Surg. 2012;78(11):1238-42
39. Paredes J, Milanezi F, Reis-Filho JS, Leitão D, Athanazio D, Schmitt F. Aberrant P-cadherin expression: is it associated with estrogen-independent growth in breast cancer? Pathol Res Pract. 2002;198(12):795-801
40. Turashvili G, McKinney SE, Goktepe O, Leung SC, Huntsman DG, Gelmon KA. et al. P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod Pathol. 2011;24(1):64-81
41. Paredes J, Albergaria A, Oliveira JT, Jerónimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res. 2005;11(16):5869-77
42. Yang SS, Ma S, Dou H, Liu F, Zhang SY, Jiang C, Xiao M, Huang YX. Breast cancer-derived exosomes regulate cell invasion and metastasis in breast cancer via miR-146a to activate cancer associated fibroblasts in tumor microenvironment. Exp Cell Res. 2020;391(2):111983
43. Yan Z, Sheng Z, Zheng Y, Feng R, Xiao Q, Shi L. et al. Cancer-associated fibroblast-derived exosomal miR-18b promotes breast cancer invasion and metastasis by regulating TCEAL7. Cell Death Dis. 2021;12(12):1120
44. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR. et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol Cancer. 2019;18(1):91
45. Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018 Sep;18(9):533-548. doi: 10.1038/s41568-018-0038-z. Erratum in: Nat Rev Cancer. 2019;19(3):179
46. Annis MG, Ouellet V, Rennhack JP, L'Esperance S, Rancourt C, Mes-Masson AM. et al. Integrin-uPAR signaling leads to FRA-1 phosphorylation and enhanced breast cancer invasion. Breast Cancer Res. 2018;20(1):9
47. Chung J, Yoon SO, Lipscomb EA, Mercurio AM. The Met receptor and alpha 6 beta 4 integrin can function independently to promote carcinoma invasion. J Biol Chem. 2004;279(31):32287-93
48. Novikov NM, Zolotaryova SY, Gautreau AM, Denisov EV. Mutational drivers of cancer cell migration and invasion. Br J Cancer. 2021;124(1):102-114
49. Krishnamurti U, Silverman JF. HER2 in breast cancer: a review and update. Adv Anat Pathol. 2014;21(2):100-7
50. Bergholtz H, Kumar S, Wärnberg F, Lüders T, Kristensen V, Sørlie T. Comparable cancer-relevant mutation profiles in synchronous ductal carcinoma in situ and invasive breast cancer. Cancer Rep (Hoboken). 2020;3(3):e1248
51. Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A. et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell. 2022;185(2):299-310.e18
52. Khalaf K, Hana D, Chou JT, Singh C, Mackiewicz A, Kaczmarek M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front Immunol. 2021;12:656364
53. Burks H, Pashos N, Martin E, Mclachlan J, Bunnell B, Burow M. Endocrine disruptors and the tumor microenvironment: A new paradigm in breast cancer biology. Mol Cell Endocrinol. 2017;457:13-19
54. Ingthorsson S, Hilmarsdottir B, Kricker J, Magnusson MK, Gudjonsson T. Context-Dependent Function of Myoepithelial Cells in Breast Morphogenesis and Neoplasia. Curr Mol Biol Rep. 2015;1(4):168-174
55. Pandey PR, Saidou J, Watabe K. Role of myoepithelial cells in breast tumor progression. Front Biosci (Landmark Ed). 2010;15(1):226-36
56. Nakamura R, Oyama T, Inokuchi M, Ishikawa S, Hirata M, Kawashima H, Ikeda H, Dobashi Y, Ooi A. Neural EGFL like 2 expressed in myoepithelial cells and suppressed breast cancer cell migration. Pathol Int. 2021;71(5):326-336
57. Gaiko-Shcherbak A, Fabris G, Dreissen G, Merkel R, Hoffmann B, Noetzel E. The Acinar Cage: Basement Membranes Determine Molecule Exchange and Mechanical Stability of Human Breast Cell Acini. PLoS One. 2015;10(12):e0145174
58. Wisdom KM, Indana D, Chou PE, Desai R, Kim T, Chaudhuri O. Covalent cross-linking of basement membrane-like matrices physically restricts invasive protrusions in breast cancer cells. Matrix Biol. 2020;85-86:94-111
59. Nazari SS, Doyle AD, Yamada KM. Mechanisms of Basement Membrane Micro-Perforation during Cancer Cell Invasion into a 3D Collagen Gel. Gels. 2022;8(9):567
60. Brennan K, Offiah G, McSherry EA, Hopkins AM. Tight junctions: a barrier to the initiation and progression of breast cancer? J Biomed Biotechnol. 2010;2010:460607
61. Lauko A, Mu Z, Gutmann DH, Naik UP, Lathia JD. Junctional Adhesion Molecules in Cancer: A Paradigm for the Diverse Functions of Cell-Cell Interactions in Tumor Progression. Cancer Res. 2020;80(22):4878-4885
62. Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008;68(7):2194-203
63. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, Zhang B, Meng Q, Yu X, Shi S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131
64. Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, Yang SR, Kurian A, Van Valen D, West R, Bendall SC, Angelo M. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell. 2018;174(6):1373-1387.e19
65. Shen L, Zhou Y, He H, Chen W, Lenahan C, Li X, Deng Y, Shao A, Huang J. Crosstalk between Macrophages, T Cells, and Iron Metabolism in Tumor Microenvironment. Oxid Med Cell Longev. 2021;2021:8865791
66. Economopoulou P, Kaklamani VG, Siziopikou K. The role of cancer stem cells in breast cancer initiation and progression: potential cancer stem cell-directed therapies. Oncologist. 2012;17(11):1394-401
67. Zheng Y, Karnoub AE. Endocrine regulation of cancer stem cell compartments in breast tumors. Mol Cell Endocrinol. 2021;535:111374
68. Raimondi M, Marcassa E, Cataldo F, Arnandis T, Mendoza-Maldonado R, Bestagno M, Schneider C, Demarchi F. Calpain restrains the stem cells compartment in breast cancer. Cell Cycle. 2016;15(1):106-16
69. Liotta LA, Thorgeirsson UP, Garbisa S. Role of collagenases in tumor cell invasion. Cancer Metastasis Rev. 1982;1(4):277-88
70. Liotta LA, Rao CN, Barsky SH. Tumor invasion and the extracellular matrix. Lab Invest. 1983;49(6):636-49
71. Liotta LA. Tumor invasion and metastases: role of the basement membrane. Warner-Lambert Parke-Davis Award lecture. Am J Pathol. 1984;117(3):339-48
72. Goldfarb RH, Liotta LA. Proteolytic enzymes in cancer invasion and metastasis. Semin Thromb Hemost. 1986;12(4):294-307
73. Khokha R, Denhardt DT. Matrix metalloproteinases and tissue inhibitor of metalloproteinases: a review of their role in tumorigenesis and tissue invasion. Invasion Metastasis. 1989;9(6):391-405
74. Cottam D, Rees R. Regulation of matrix metalloproteinases - their role in tumor invasion and metastasis. Int J Oncol. 1993Jun;2(6):861-72
75. DeClerck YA, Yean TD, Chan D, Shimada H, Langley KE. Inhibition of tumor invasion of smooth muscle cell layers by recombinant human metalloproteinase inhibitor. Cancer Res. 1991;51(8):2151-7
76. McDonnell S, Fingleton B. Role of matrix metalloproteinases in invasion and metastasis: biology, diagnosis and inhibitors. Cytotechnology. 1993;12(1-3):367-84
77. Brown PD. Matrix metalloproteinase inhibitors: a novel class of anticancer agents. Adv Enzyme Regul. 1995;35:293-301
78. Duffy MJ, McCarthy K. Matrix metalloproteinases in cancer: prognostic markers and targets for therapy (review). Int J Oncol. 1998;12(6):1343-8
79. Otani Y, Fujii M, Kubota T, Kitajima M, Okada Y. [Recent progress of matrix metalloproteinase (MMP) research and its clinical application for cancer therapy]. Gan To Kagaku Ryoho. 1998;25(7):957-64
80. Maquoi E, Noël A, Frankenne F, Angliker H, Murphy G, Foidart JM. Inhibition of matrix metalloproteinase 2 maturation and HT1080 invasiveness by a synthetic furin inhibitor. FEBS Lett. 1998;424(3):262-6
81. Yu AE, Hewitt RE, Connor EW, Stetler-Stevenson WG. Matrix metalloproteinases. Novel targets for directed cancer therapy. Drugs Aging. 1997;11(3):229-44
82. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387-92
83. Matrisian LM, Sledge GW Jr, Mohla S. Extracellular proteolysis and cancer: meeting summary and future directions. Cancer Res. 2003;63(19):6105-9
84. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161-74
85. Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2(9):657-72
86. Bonomi P. Matrix metalloproteinases and matrix metalloproteinase inhibitors in lung cancer. Semin Oncol. 2002;29(1 Suppl 4):78-86
87. Leighl NB, Paz-Ares L, Douillard JY, Peschel C, Arnold A, Depierre A. et al. Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J Clin Oncol. 2005;23(12):2831-9
88. DeClerck YA, Mercurio AM, Stack MS, Chapman HA, Zutter MM, Muschel RJ, Raz A, Matrisian LM, Sloane BF, Noel A, Hendrix MJ, Coussens L, Padarathsingh M. Proteases, extracellular matrix, and cancer: a workshop of the path B study section. Am J Pathol. 2004;164(4):1131-9
89. Winer A, Adams S, Mignatti P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol Cancer Ther. 2018;17(6):1147-1155
90. Bonomi P. Matrix metalloproteinases and matrix metalloproteinase inhibitors in lung cancer. Semin Oncol. 2002;29(1 Suppl 4):78-86
91. Gabbert H. Mechanisms of tumor invasion: evidence from in vivo observations. Cancer Metastasis Rev. 1985;4(4):293-309
92. Zhu J, Li K, Dong X, Zhou P, Li P, Bi J. Metaplastic breast carcinoma composed of epithelial-myoepithelial carcinoma and squamous cell carcinoma: A case report. Medicine (Baltimore). 2018;97(15):e0364
93. Buza N, Zekry N, Charpin C, Tavassoli FA. Myoepithelial carcinoma of the breast: a clinicopathological and immunohistochemical study of 15 diagnostically challenging cases. Virchows Arch. 2010;457(3):337-45
94. Kwon SY, Bae YK, Cho J, Kang SH. Myoepithelial carcinoma with contralateral invasive micropapillary carcinoma of the breast. J Korean Surg Soc. 2011;81(3):211-5
95. Yang Y, Wang Y, He J, Pan G, Tuo X, Jiang A, Bian L. Malignant adenomyoepithelioma combined with adenoid cystic carcinoma of the breast: a case report and literature review. Diagn Pathol. 2014;9:148
96. Ohtake H, Iwaba A, Kato T, Ohe R, Maeda K, Matsuda M, Izuru K, Morimoto K, Katagiri S, Yamakawa M. Myoepithelial carcinoma of the breast with focal rhabdoid features. Breast J. 2013;19(1):100-3
97. Ye RP, Liao YH, Xia T, Kuang R, Long HA, Xiao XL. Breast mucoepidermoid carcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2020;13(12):3192-3199
98. Reddy KB, McGowen R, Schuger L, Visscher D, Sheng S. Maspin expression inversely correlates with breast tumor progression in MMTV/TGF-alpha transgenic mouse model. Oncogene. 2001;20(45):6538-43
99. Duivenvoorden HM, Rautela J, Edgington-Mitchell LE, Spurling A, Greening DW, Nowell CJ. et al. Myoepithelial cell-specific expression of stefin A as a suppressor of early breast cancer invasion. J Pathol. 2017;243(4):496-509
100. Xu Z, Wang W, Deng CX, Man YG. Aberrant p63 and WT-1 expression in myoepithelial cells of pregnancy-associated breast cancer: implications for tumor aggressiveness and invasiveness. Int J Biol Sci. 2009;5(1):82-96
101. Yamamoto T, Oda K, Miyazaki K, Ichigotani Y, Takenouchi Y, Kamei T, Shirafuji N, Nimura Y, Hamaguchi M, Matsuda S. p73 is highly expressed in myoepithelial cells and in carcinomas with metaplasia. Int J Oncol. 2001;19(2):271-6
102. Moreira JM, Ohlsson G, Rank FE, Celis JE. Down-regulation of the tumor suppressor protein 14-3-3sigma is a sporadic event in cancer of the breast. Mol Cell Proteomics. 2005;4(4):555-69
103. Khazai L, Agosto-Arroyo E, Rosa M. P40 Immunostain Does Not Outperform p63 as a Myoepithelial Cell Marker in the Daily Practice of Breast Pathology. Appl Immunohistochem Mol Morphol. 2018;26(8):599-604
104. Nakamura R, Oyama T, Inokuchi M, Ishikawa S, Hirata M, Kawashima H, Ikeda H, Dobashi Y, Ooi A. Neural EGFL like 2 expressed in myoepithelial cells and suppressed breast cancer cell migration. Pathol Int. 2021;71(5):326-336
105. Man YG, Tai L, Barner R, Vang R, Saenger JS, Shekitka KM, Bratthauer GL, Wheeler DT, Liang CY, Vinh TN, Strauss BL. Cell clusters overlying focally disrupted mammary myoepithelial cell layers and adjacent cells within the same duct display different immunohistochemical and genetic features: implications for tumor progression and invasion. Breast Cancer Res. 2003;5(6):R231-41
106. Man YG, Zhang Y, Shen T, Zeng X, Tauler J, Mulshine JL, Strauss BL. cDNA expression profiling reveals elevated gene expression in cell clusters overlying focally disrupted myoepithelial cell layers: implications for breast tumor invasion. Breast Cancer Res Treat. 2005;89(2):199-208
107. Man YG. Focal degeneration of aged or injured myoepithelial cells and the resultant auto-immunoreactions are trigger factors for breast tumor invasion. Med Hypotheses. 2007;69(6):1340-57
108. Man YG. Tumor cell budding from focally disrupted tumor capsules: a common pathway for all breast cancer subtype derived invasion? J Cancer. 2010;1:32-7
109. Man YG, Mason J, Harley R, Kim YH, Zhu K, Gardner WA. Leukocyte-mediated cell dissemination and metastasis: findings from multiple types of human tumors. J Cell Biochem. 2011;112(4):1154-67
110. Jiang B, Mason J, Jewett A, Liu ML, Chen W, Qian J, Ding Y, Ding S, Ni M, Zhang X, Man YG. Tumor-infiltrating immune cells: triggers for tumor capsule disruption and tumor progression? Int J Med Sci. 2013;10(5):475-97
111. Hsiao YH, Chou MC, Fowler C, Mason JT, Man YG. Breast cancer heterogeneity: mechanisms, proofs, and implications. J Cancer. 2010;1:6-13
112. Man YG, Sang QX. The significance of focal myoepithelial cell layer disruptions in human breast tumor invasion: a paradigm shift from the “protease-centered” hypothesis. Exp Cell Res. 2004;301(2):103-18
113. Zhang X, Hashemi SS, Yousefi M, Ni J, Wang Q, Gao L, Gong P, Gao C, Sheng J, Mason J, Man YG. Aberrant c-erbB2 expression in cell clusters overlying focally disrupted breast myoepithelial cell layers: a trigger or sign for emergence of more aggressive cell clones? Int J Biol Sci. 2008;4(5):259-69
114. Man YG, Shen T, Weisz J, Berg PE, Schwartz AM, Mulshine JL, Sang QX, Nieburgs HE. A subset of in situ breast tumor cell clusters lacks expression of proliferation and progression related markers but shows signs of stromal and vascular invasion. Cancer Detect Prev. 2005;29(4):323-31
115. Prasad ML, Hyjek E, Giri DD, Ying L, O'Leary JJ, Hoda SA. Double immunolabeling with cytokeratin and smooth-muscle actin in confirming early invasive carcinoma of breast. Am J Surg Pathol. 1999;23(2):176-81
116. Ishizaki M, Ashida K, Higashi T, Nakatsukasa H, Kaneyoshi T, Fujiwara K, Nouso K, Kobayashi Y, Uemura M, Nakamura S, Tsuji T. The formation of capsule and septum in human hepatocellular carcinoma. Virchows Arch. 2001;438(6):574-80
117. Jahkola T, Toivonen T, Virtanen I, von Smitten K, Nordling S, von Boguslawski K, Haglund C, Nevanlinna H, Blomqvist C. Tenascin-C expression in invasion border of early breast cancer: a predictor of local and distant recurrence. Br J Cancer. 1998;78(11):1507-13
118. Tastekin D, Tas F, Karabulut S, Duranyildiz D, Serilmez M, Guveli M, Vatansever S. Clinical significance of serum tenascin-C levels in breast cancer. Tumour Biol. 2014;35(7):6619-25
119. Wawrzyniak D, Grabowska M, Głodowicz P, Kuczyński K, Kuczyńska B, Fedoruk-Wyszomirska A, Rolle K. Down-regulation of tenascin-C inhibits breast cancer cells development by cell growth, migration, and adhesion impairment. PLoS One. 2020;15(8):e0237889
120. Man YG, Gardner WA. Focal degeneration of basal cells and the resultant auto-immunoreactions: a novel mechanism for prostate tumor progression and invasion. Med Hypotheses. 2008;70(2):387-408
121. Liu A, Wei L, Gardner WA, Deng CX, Man YG. Correlated alterations in prostate basal cell layer and basement membrane. Int J Biol Sci. 2009;5(3):276-85
122. Man YG, Fu SW, Liu AJ, Stojadinovic A, Izadjoo MJ, Chen L, Gardner WA. Aberrant expression of chromogranin A, miR-146a, and miR-146b-5p in prostate structures with focally disrupted basal cell layers: an early sign of invasion and hormone-refractory cancer? Cancer Genomics Proteomics. 2011;8(5):235-44
123. Man YG, Shen T, Zhao Y, Amy Sang QX. Focal prostate basal cell layer disruptions and leukocyte infiltration are correlated events: A potential mechanism for basal cell layer disruptions and tumor invasion. Cancer Detect Prev. 2005;29(2):161-9
124. Man YG, Gardner WA. Bad seeds produce bad crops: a single stage-process of prostate tumor invasion. Int J Biol Sci. 2008;4(4):246-58
125. Man YG, Mannion C, Jewett A, Hsiao YH, Liu A, Semczuk A. et al. The most effective but largely ignored target for prostate cancer early detection and intervention. J Cancer. 2022;13(13):3463-3475
126. Kuukasjärvi T, Karhu R, Tanner M, Kähkönen M, Schäffer A, Nupponen N. et al. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57(8):1597-604
127. Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle. 2007;6(19):2332-8
128. Hernandez L, Wilkerson PM, Lambros MB, Campion-Flora A, Rodrigues DN, Gauthier A. et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227(1):42-52
129. Fountzilas E, Kotoula V, Zagouri F, Giannoulatou E, Kouvatseas G, Pentheroudakis G. et al. Disease evolution and heterogeneity in bilateral breast cancer. Am J Cancer Res. 2016;6(11):2611-2630
130. Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA. et al. Lobular Carcinomas In situ Display Intralesion Genetic Heterogeneity and Clonal Evolution in the Progression to Invasive Lobular Carcinoma. Clin Cancer Res. 2019;25(2):674-686
131. Kalinowski L, Saunus JM, McCart Reed AE, Lakhani SR. Breast Cancer Heterogeneity in Primary and Metastatic Disease. Adv Exp Med Biol. 2019;1152:75-104
132. Taurin S, Alkhalifa H. Breast cancers, mammary stem cells, and cancer stem cells, characteristics, and hypotheses. Neoplasia. 2020;22(12):663-678
133. Black JRM, McGranahan N. Genetic and non-genetic clonal diversity in cancer evolution. Nat Rev Cancer. 2021;21(6):379-392
134. Kavan S, Kruse TA, Vogsen M, Hildebrandt MG, Thomassen M. Heterogeneity and tumor evolution reflected in liquid biopsy in metastatic breast cancer patients: a review. Cancer Metastasis Rev. 2022;41(2):433-446
135. Kujala J, Hartikainen JM, Tengström M, Sironen R, Auvinen P, Kosma VM, Mannermaa A. Circulating Cell-Free DNA Reflects the Clonal Evolution of Breast Cancer Tumors. Cancers (Basel). 2022;14(5):1332
136. Pandey PR, Saidou J, Watabe K. Role of myoepithelial cells in breast tumor progression. Front Biosci (Landmark Ed). 2010;15(1):226-36
137. Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6(4):281-91
138. Russell TD, Jindal S, Agunbiade S, Gao D, Troxell M, Borges VF, Schedin P. Myoepithelial cell differentiation markers in ductal carcinoma in situ progression. Am J Pathol. 2015;185(11):3076-89
139. Polyak K, Hu M. Do myoepithelial cells hold the key for breast tumor progression? J Mammary Gland Biol Neoplasia. 2005;10(3):231-47
140. Polyak K. Molecular markers for the diagnosis and management of ductal carcinoma in situ. J Natl Cancer Inst Monogr. 2010;2010(41):210-3
141. Rakha EA, Miligy IM, Gorringe KL, Toss MS, Green AR, Fox SB. et al. Invasion in breast lesions: the role of the epithelial-stroma barrier. Histopathology. 2018;72(7):1075-1083
142. Hayward MK, Allen MD, Gomm JJ, Goulding I, Thompson CL, Knight MM, Marshall JF, Jones JL. Mechanostimulation of breast myoepithelial cells induces functional changes associated with DCIS progression to invasion. NPJ Breast Cancer. 2022;8(1):109
143. Umekita Y, Ohi Y, Iwaya O, Souda M, Sagara Y, Tamada S, Yotsumoto D, Tanimoto A. Maspin mRNA expression in sentinel lymph nodes predicts non-SLN metastasis in breast cancer patients with SLN metastasis. Histopathology. 2018;73(6):916-922
144. Stark AM, Schem C, Maass N, Hugo HH, Jonat W, Mehdorn HM, Held-Feindt J. Expression of metastasis suppressor gene maspin is reduced in breast cancer brain metastases and correlates with the estrogen receptor status. Neurol Res. 2010;32(3):303-8
145. Helal DS, El-Guindy DM. Maspin expression and subcellular localization in invasive ductal carcinoma of the breast: Prognostic significance and relation to microvessel density. J Egypt Natl Canc Inst. 2017;29(4):177-183
146. Sakabe T, Wakahara M, Shiota G, Umekita Y. Role of cytoplasmic localization of maspin in promoting cell invasion in breast cancer with aggressive phenotype. Sci Rep. 2021;11(1):11321
147. Juliansyah A, Rahman S, Indra I, Nelwan B, Prihantono P. Association of ERα-36 expression with the de novo resistance of tamoxifen in ER-positive breast cancer. Breast Dis. 2021;40(S1):S123-S127
148. Reinert T, Cascelli F, de Resende CAA, Gonçalves AC, Godo VSP, Barrios CH. Clinical implication of low estrogen receptor (ER-low) expression in breast cancer. Front Endocrinol (Lausanne). 2022;13:1015388
149. Ma S, Tang T, Probst G, Konradi A, Jin C, Li F, Gutkind JS, Fu XD, Guan KL. Transcriptional repression of estrogen receptor alpha by YAP reveals the Hippo pathway as therapeutic target for ER+ breast cancer. Nat Commun. 2022;13(1):1061
150. Wei G, Teng M, Rosa M, Wang X. Unique ER PR expression pattern in breast cancers with CHEK2 mutation: a hormone receptor and HER2 analysis based on germline cancer predisposition genes. Breast Cancer Res. 2022;24(1):11
151. Barbareschi M, Pecciarini L, Cangi MG, Macrì E, Rizzo A, Viale G, Doglioni C. p63, a p53 homologue, is a selective nuclear marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25(8):1054-60
152. Galoczová M, Nenutil R, Coates P, Vojtěšek B. Possible Usage of p63 in Bioptic Diagnostics. Klin Onkol. 2018 31(Suppl 2):27-31
153. Xu Z, Wang W, Deng CX, Man YG. Aberrant p63 and WT-1 expression in myoepithelial cells of pregnancy-associated breast cancer: implications for tumor aggressiveness and invasiveness. Int J Biol Sci. 2009;5(1):82-96
154. Hsiao YH, Su YA, Tsai HD, Mason JT, Chou MC, Man YG. Increased invasiveness and aggressiveness in breast epithelia with cytoplasmic p63 expression. Int J Biol Sci. 2010;6(5):428-42
155. Choi J, Kim D, Koo JS. Secretory carcinoma of breast demonstrates nuclear or cytoplasmic expression in p63 immunohistochemistry. Int J Surg Pathol. 2012;20(4):367-72
156. Bernardo MM, Dzinic SH, Matta MJ, Dean I, Saker L, Sheng S. The Opportunity of Precision Medicine for Breast Cancer With Context-Sensitive Tumor Suppressor Maspin. J Cell Biochem. 2017;118(7):1639-1647
157. Ashekyan O, Abdallah S, Shoukari AA, Chamandi G, Choubassy H, Itani ARS, Alwan N, Nasr R. Spotlight on Exosomal Non-Coding RNAs in Breast Cancer: An In Silico Analysis to Identify Potential lncRNA/circRNA-miRNA-Target Axis. Int J Mol Sci. 2022;23(15):8351
158. Dean I, Dzinic SH, Bernardo MM, Zou Y, Kimler V, Li X, Kaplun A, Granneman J, Mao G, Sheng S. The secretion and biological function of tumor suppressor maspin as an exosome cargo protein. Oncotarget. 2017;8(5):8043-8056
159. Apostolaki S, Vlachonikolis I, Lianidou E, Georgoulias V. Molecular detection of cancer cells in the peripheral blood of patients with breast cancer: comparison of CK-19, CEA and maspin as detection markers. Anticancer Res. 2003;23(2C):1883-90
160. Herman-Saffar O, Boger Z, Libson S, Lieberman D, Gonen R, Zeiri Y. Early non-invasive detection of breast cancer using exhaled breath and urine analysis. Comput Biol Med. 2018;96:227-232
161. Loke SY, Lee ASG. The future of blood-based biomarkers for the early detection of breast cancer. Eur J Cancer. 2018;92:54-68
162. Bitisik O, Saip P, Saglam S, Derin D, Dalay N. Mammaglobin and maspin transcripts in blood may reflect disease progression and the effect of therapy in breast cancer. Genet Mol Res. 2010;9(1):97-106
163. Loud JT, Thiébaut AC, Abati AD, Filie AC, Nichols K, Danforth D, Giusti R, Prindiville SA, Greene MH. Ductal lavage in women from BRCA1/2 families: is there a future for ductal lavage in women at increased genetic risk of breast cancer? Cancer Epidemiol Biomarkers Prev. 2009;18(4):1243-51
164. Mitchell G, Antill YC, Murray W, Kirk J, Salisbury E, Lindeman GJ, Di Iulio J, Milner AD, Devereaux L, Phillips KA. Nipple aspiration and ductal lavage in women with a germline BRCA1 or BRCA2 mutation. Breast Cancer Res. 2005;7(6):R1122-31
165. Kurian AW, Mills MA, Jaffee M, Sigal BM, Chun NM, Kingham KE, Collins LC, Nowels KW, Plevritis SK, Garber JE, Ford JM, Hartman AR. Ductal lavage of fluid-yielding and non-fluid-yielding ducts in BRCA1 and BRCA2 mutation carriers and other women at high inherited breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14(5):1082-9
166. Loud JT, Beckjord EB, Nichols K, Peters J, Giusti R, Greene MH. Tolerability of breast ductal lavage in women from families at high genetic risk of breast cancer. BMC Womens Health. 2009;9:20
167. Goff SL, Danforth DN. The Role of Immune Cells in Breast Tissue and Immunotherapy for the Treatment of Breast Cancer. Clin Breast Cancer. 2021;21(1):e63-e73
168. Perrone M, Talarico G, Chiodoni C, Sangaletti S. Impact of Immune Cell Heterogeneity on HER2+ Breast Cancer Prognosis and Response to Therapy. Cancers (Basel). 2021;13(24):6352
169. Gatti G, Betts C, Rocha D, Nicola M, Grupe V, Ditada C, Nuñez NG, Roselli E, Araya P, Dutto J, Boffelli L, Fernández E, Coussens LM, Maccioni M. High IRF8 expression correlates with CD8 T cell infiltration and is a predictive biomarker of therapy response in ER-negative breast cancer. Breast Cancer Res. 2021;23(1):40
170. Song S, Zhang M, Xie P, Wang S, Wang Y. Comprehensive analysis of cuproptosis-related genes and tumor microenvironment infiltration characterization in breast cancer. Front Immunol. 2022;13:978909
171. Huang G, Zhou J, Chen J, Liu G. Identification of pyroptosis related subtypes and tumor microenvironment infiltration characteristics in breast cancer. Sci Rep. 2022;12(1):10640
172. Liberale L, Carbone F, Montecucco F, Sahebkar A. Statins reduce vascular inflammation in atherogenesis: A review of underlying molecular mechanisms. Int J Biochem Cell Biol. 2020;122:105735
173. Hurwitz LM, Kulac I, Gumuskaya B, Valle JABD, Benedetti I, Pan F. et al. Use of Aspirin and Statins in Relation to Inflammation in Benign Prostate Tissue in the Placebo Arm of the Prostate Cancer Prevention Trial. Cancer Prev Res (Phila). 2020;13(10):853-862
174. Beckwitt CH, Brufsky A, Oltvai ZN, Wells A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018;20(1):144
175. Sankaranarayanan R, Kumar DR, Altinoz MA, Bhat GJ. Mechanisms of Colorectal Cancer Prevention by Aspirin-A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways. Int J Mol Sci. 2020;21(23):9018
176. Elwood P, Protty M, Morgan G, Pickering J, Delon C, Watkins J. Aspirin and cancer: biological mechanisms and clinical outcomes. Open Biol. 2022;12(9):220124
177. Li S, Ma J, Zheng A, Song X, Chen S, Jin F. DEAD-box helicase 27 enhances stem cell-like properties with poor prognosis in breast cancer. J Transl Med. 2021;19(1):334
178. Louhichi T, Ziadi S, Saad H, Dhiab MB, Mestiri S, Trimeche M. Clinicopathological significance of cancer stem cell markers CD44 and ALDH1 expression in breast cancer. Breast Cancer. 2018;25(6):698-705
179. Panigoro SS, Kurnia D, Kurnia A, Haryono SJ, Albar ZA. ALDH1 Cancer Stem Cell Marker as a Prognostic Factor in Triple-Negative Breast Cancer. Int J Surg Oncol. 2020;2020:7863243
180. Siddharth S, Goutam K, Das S, Nayak A, Nayak D, Sethy C, Wyatt MD, Kundu CN. Nectin-4 is a breast cancer stem cell marker that induces WNT/β-catenin signaling via Pi3k/Akt axis. Int J Biochem Cell Biol. 2017;89:85-94
181. Jeong H, Kim J, Lee Y, Seo JH, Hong SR, Kim A. Neuregulin-1 induces cancer stem cell characteristics in breast cancer cell lines. Oncol Rep. 2014;32(3):1218-24
182. Troschel FM, Palenta H, Borrmann K, Heshe K, Hua SH, Yip GW, Kiesel L, Eich HT, Götte M, Greve B. Knockdown of the prognostic cancer stem cell marker Musashi-1 decreases radio-resistance while enhancing apoptosis in hormone receptor-positive breast cancer cells via p21WAF1/CIP1. J Cancer Res Clin Oncol. 2021;147(11):3299-3312
183. Ni W, Yang Z, Qi W, Cui C, Cui Y, Xuan Y. Gli1 is a potential cancer stem cell marker and predicts poor prognosis in ductal breast carcinoma. Hum Pathol. 2017;69:38-45
184. Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. 2017;8(22):36305-36318
185. Joseph C, Arshad M, Kurozomi S, Althobiti M, Miligy IM, Al-Izzi S, Toss MS, Goh FQ, Johnston SJ, Martin SG, Ellis IO, Mongan NP, Green AR, Rakha EA. Overexpression of the cancer stem cell marker CD133 confers a poor prognosis in invasive breast cancer. Breast Cancer Res Treat. 2019;174(2):387-399
186. Kim H, Kim Y, Jeoung D. DDX53 Promotes Cancer Stem Cell-Like Properties and Autophagy. Mol Cells. 2017;40(1):54-65
187. Cheung SK, Chuang PK, Huang HW, Hwang-Verslues WW, Cho CH, Yang WB. et al. Stage-specific embryonic antigen-3 (SSEA-3) and β3GalT5 are cancer specific and significant markers for breast cancer stem cells. Proc Natl Acad Sci U S A. 2016;113(4):960-5
188. Pal D, Kolluru V, Chandrasekaran B, Baby BV, Aman M, Suman S, Sirimulla S, Sanders MA, Alatassi H, Ankem MK, Damodaran C. Targeting aberrant expression of Notch-1 in ALDH+ cancer stem cells in breast cancer. Mol Carcinog. 2017;56(3):1127-1136
189. Chekhun SV, Zadvorny TV, Tymovska YO, Anikusko MF, Novak OE, Polishchuk LZ. СD44+/CD24- markers of cancer stem cells in patients with breast cancer of different molecular subtypes. Exp Oncol. 2015;37(1):58-63
190. Cheng JY, Hung JT, Lin J, Lo FY, Huang JR, Chiou SP, Wang YH, Lin RJ, Wu JC, Yu J, Yu AL. O-Acetyl-GD2 as a Therapeutic Target for Breast Cancer Stem Cells. Front Immunol. 2022;12:791551
191. Chekhun SV, Zadvorny TV, Tymovska YO, Anikusko MF, Novak OE, Polishchuk LZ. СD44+/CD24- markers of cancer stem cells in patients with breast cancer of different molecular subtypes. Exp Oncol. 2015;37(1):58-63
192. Xia P, Liu Y, Chen J, Cheng Z. Cell Cycle Proteins as Key Regulators of Postmitotic Cell Death. Yale J Biol Med. 2019;92(4):641-650
193. Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36(3):131-49
194. Khan MGM, Wang Y. Cell Cycle-Related Clinical Applications. Methods Mol Biol. 2022;2579:35-46
195. Caglar HO, Biray Avci C. Alterations of cell cycle genes in cancer: unmasking the role of cancer stem cells. Mol Biol Rep. 2020;47(4):3065-3076
196. Chen Z, Guo Q, Song G, Hou Y. Molecular regulation of hematopoietic stem cell quiescence. Cell Mol Life Sci. 2022;79(4):218
197. Spurlock B, Tullet J, Hartman JL 4th, Mitra K. Interplay of mitochondrial fission-fusion with cell cycle regulation: Possible impacts on stem cell and organismal aging. Exp Gerontol. 2020;135:110919
198. Gupta N, Huang TT, Horibata S, Lee JM. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor-resistant cancer. Pharmacol Res. 2022;178:106162
199. Lin M, Xie SS, Chan KY. An updated view on the centrosome as a cell cycle regulator. Cell Div. 2022;17(1):1
Author contact
![]() Corresponding authors: Yan-gao Man., MD., PhD. E-mail: ymanorg or Yanmanncom. Ciaran Mannion., MD. E-mail: cjmannion50com. Department of Pathology, Hackensack Meridian Health-Hackensack University Medical Center, Hackensack, New Jersey, NJ, USA
Corresponding authors: Yan-gao Man., MD., PhD. E-mail: ymanorg or Yanmanncom. Ciaran Mannion., MD. E-mail: cjmannion50com. Department of Pathology, Hackensack Meridian Health-Hackensack University Medical Center, Hackensack, New Jersey, NJ, USA