Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2023; 14(8):1336-1349. doi:10.7150/jca.83556 This issue Cite
Research Paper
PERK/ATF3-Reduced ER Stress on high potassium environment in the suppression of tumor ferroptosis
Xufeng Pu1*, Li Li3*, Zhenhui Chen1*, Aihua Gong3, Jiao Lei4, Lirong Zhang1 ![]() , Hsiang-I Tsai1,2
, Hsiang-I Tsai1,2 ![]()
1. Institute of Medical Imaging and Artificial Intelligence, 212001, Zhenjiang, Jiang Su Province, China.
2. Department of Medical Imaging, The Affiliated Hospital of Jiangsu University, 212001, Zhenjiang, China.
3. Department of Cell Biology, School of Medicine, Jiangsu University, 212001, Zhenjiang, Jiang Su Province, China.
4. Nanjing Lishui People's Hospital, Zhongda Hospital Lishui Branch, Southeast University, Nanjing, Jiang Su Province, China.
*Equal contributors
Received 2023-2-15; Accepted 2023-5-8; Published 2023-5-15
Abstract

Potassium (K+) is a vital intracellular cation. In the human body, it regulates membrane potential, electrical excitation, protein synthesis, and cell death. Recent studies revealed that dying cancer cells release potassium into the tumor microenvironment (TME), thereby influencing cell survival-related events. Several investigations reported that potassium channels and high potassium levels influence apoptosis. Increasing extracellular potassium and inhibiting K+ efflux channels significantly block the apoptotic machinery. However, it is unknown whether a high-potassium environment also affects other types of cell death such as ferroptosis. In the present study, cell counting kit (CCK-8), colony formation ability, and 5-ethynyl-2′-deoxyuridine (EdU) assays demonstrated that a high-potassium environment reverses erastin-induced ferroptosis. RNA sequencing (RNA-Seq) and Kyoto Encyclopedia of Genes and Genomes (KEGG) and gene ontology (GO) analyses indicated that high potassium levels attenuated the unfolded protein response that is characteristic of endoplasmic reticulum (ER) stress. The ER transmembrane proteins PRKR-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) are recognized as ER stress sensors. Here, the PERK blocker GSK2606414 significantly rescued ferroptosis. The present work also disclosed that the ER-related gene activating transcription factor 3 (ATF3) played a vital role in regulating ferroptosis in a high-potassium environment. The foregoing results revealed the roles of potassium and the TME in cancer cell ferroptosis and provided a potential clinical therapeutic strategy for cancer.
Introduction
Homeostasis of metal ions such as copper, iron, zinc, and calcium are crucial for the cell survival, function and normal physiological activity of the brain. However, tumorigenesis upsets metal cation balance [1]. Previous research demonstrated that the concentrations of Fe, K, Mg, Na, Rb, Se, and Zn were significantly higher in neoplastic than in normal tissues [2, 3]. Recent studies revealed that the potassium ion content increases mainly in the extracellular milieu in response to cell death. High extracellular potassium levels favor cell survival by suppressing T-cell function [4]. Nevertheless, the mechanism by which a high-potassium environment influences cancer cells is unknown. A few studies indicated that high extracellular potassium levels inhibit apoptosis. Nevertheless, the effects of elevated extracellular K+ on other types of cell death have not yet been determined [5].
Unlike other forms of apoptosis, ferroptosis is driven by iron-dependent phospholipid peroxidation and oxidative stress [6] and is regulated by mitochondrial activity, redox homeostasis, and amino acid metabolism [7]. Ferroptosis is associated with high oxidative stress and is, therefore, a novel therapeutic target in refractory tumors. For example, as ferroptosis induction targets GPX4[8], it might be a therapeutic strategy for the luminal androgen receptor subtype of triple-negative breast cancers. Several authors reported that the TME influences ferroptosis-inducing pathways such as hypoxia, lactate, and low glucose in cancer cells [9-11]. Nevertheless, it remains to be determined how a high-potassium environment affects ferroptosis.
In the present study, we confirmed that a high-potassium environment protects cells against ferroptosis. RNA sequencing disclosed that high potassium levels alleviated ER stress associated with ferroptosis induction. We also discovered that inhibiting the ER-related gene ATF3 played a vital role in a ferroptosis-inducted high-potassium environment.
Methods
Reagents
(1S,3R)-RSL3 (RSL3, #HY-100218A), erastin (#HY-15763), ferrostatin-1 (#HY-100579), Z-VAD-FMK (#HY-16658B), necrostastin-1 (#HY-15760), necrostatin-2 (#HY-14622), GSK2606414 (#HY-18072), and piperazine erastin (#HY-100887) were purchased from MedChemExpress (MCE; Monmouth Junction, NJ, USA). Ceapin-A7 (T9110) and 4μ8C (T6363) were purchased from MedChemExpress.
Cell culture and stable cell line establishment
HT1080, HepG2, and HEK293T cells were purchased from the Cell Bank of the China Academy of Sciences (Shanghai, China). Four target sequences (Table S1) were inserted into lenticrispr v2 and transfected HEK293T cells to produce a lentivirus for the generation of ATF3 knockout cells. The HT1080 cells were infected with the lentivirus for 48 h and cultured for 48 h in DMEM containing 2 µg/mL puromycin. ATF3-overexpressing cells were established according to the preceding protocol. ATF3-overexpressing plasmids were purchased from Guang Zhou JiDan, Guangdong. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM); Nanjing KeyGen Biotech, Nanjing, China) supplemented with 10% (v/v) fetal bovine serum (FBS; ExCell Biotech Co. Ltd., Taicang, China) in an incubator at 37 °C and under a 5% CO2 atmosphere.
Cell viability and colony formation assays
Cell viability was measured with a CCK-8 (Dojindo Laboratories, Kumamoto, Japan). HT1080 and Hepg2 cells were cultured overnight in a 96-well plate (Yongjin, Guangzhou, China) at densities of 2 × 103/well and 4 × 103/well. The cells were subjected to the preceding agents for 48 h and then 90 µL fresh medium and 10 µL CCK-8 solution (EdU; 10 µM) were added to them. Optical densities (OD) were measured in a microplate reader (BioTek Instruments, Winooski, VT, USA) at 450 nm. The EdU images were obtained by fluorescence microscopy, magnification 10x (Leica, Germany). Colony formation assays were used to evaluate cell viability. About 1,000 cells were placed in six-well plates and cultured in complete DMEM. The latter was changed every 2 d. After 1 wk, the colonies were washed twice with phosphate-buffered saline (PBS), incubated with 4% (v/v) paraformaldehyde (PFA; Beyotime Biotechnology, Shanghai, China) for 30 min, and stained with crystal violet solution (Beyotime Biotechnology).
RNA sequencing analysis
HT1080 cells were seeded into a six-well plate at a density of 4 × 104/well. The following day, the cells were subjected to KCl, erastin, GSK'414, KCl + erastin, or GSK'414 + erastin for 48 h and analyzed by Su Zhou Jin Weizhi.
Western blotting (WB)
Briefly, the cells were lysed in radioimmunoprecipitation (RIPA) buffer (150 mM NaCl, 1.0% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 0.1% (v/v) sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH 8.0), and Roche Complete Mini EDTA-free) for 30 min. They were then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for WB. The following primary antibodies were used: anti-ATF3 (1:1,000; #18665; Cell Signaling Technology (CST), Danvers, MA, USA), anti-PERK (1:1,000; #5683; CST), anti-ATF6 (1:1,000; #65880; CST), and anti-IRE1α (1:1,000; #3294; CST).
Immunohistochemistry (IHC)
Tissue microarrays were obtained from the Affiliated Hospital of Jiangsu University (Zhengjiang, China), subjected twice to xylene for 10 min each time, and hydrated with 100% (v/v), 95% (v/v), and 70% (v/v) ethanol for 5 min per step. Sodium citrate solution (pH 6.0) was used for antigen retrieval and the tissue microarrays were blocked in 5% (v/v) bovine serum albumin (BSA) for 1 h. The slides were incubated with anti-ATF3 (1:1,00; Abmart, Shanghai, China) and anti-COX2 (1:100; Abcam, Cambridge, UK) antibodies at 4°C overnight. They were then stained with secondary antibody (Aobose, Shandong, China) for 1 h, incubated with horseradish peroxidase (HRP)-conjugated Streptomyces ovalbumin (Aobose, Shandong, China) for 30min, stained with 3,3'-diaminobenzidine (DAB, Servicebio, Wuhan, China) for 5 min to monitor primary antibody levels, and finally stained with hematoxylin to disclose the nuclei.
Xenograft tumor models
Male BALB/c-nu mice age 4-6 weeks were purchased from the Animal Center of Jiangsu University (Zhenjiang, China). All animal experiments were performed under standard conditions. HT1080 cells were cultured with or without KCl for 1 week and injected subcutaneously into the mice. When the tumor volume reached 50 mm3, (piperazine erastin (PE) 20 mg/kg) or dimethyl sulfoxide (DMSO) was injected every 2 day for 2 weeks and the tumors were analyzed 2 day after the final injection. Tumor volumes were calculated as shown in Eq. 1 below:
V = L × W2 × ½ (1)
where V is tumor volume, L is tumor length, and W is tumor width.
Lipid peroxidation detection
Malondialdehyde (MDA) and glutathione (GSH) assay kits were purchased from Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China. A reactive oxygen species (ROS) detection kit was purchased from Dalian Meilunbio, Dalian, China. Cells were cultured in 100-mm plates (Yongjin, Guangzhou, China) and subjected to 35 mM K+, 3µM GSK'414, and 2.5 µM erastin for 48 h and subjected to MDA, GSH, and ROS detection.
Transmission electron microscopy analysis
HT1080 cells were seeded into a six-well plate at a density of 4 × 104/well. The following day, the cells were subjected to KCl, erastin, GSK'414, KCl + erastin, or GSK'414 + erastin for 48 h. Then, fixed with glutaraldehyde, and analyzed by Nangjing Shiyanjia Lab.
Statistical analysis
All data were normalized as means ± standard error of the mean (SEM). Significant differences between groups were identified by one-way analysis of variance (ANOVA) and Student's t-test. A value of p < 0.05 reflected a statistically significant difference.
Results
A high-potassium environment confers ferroptosis resistance in cancer cells
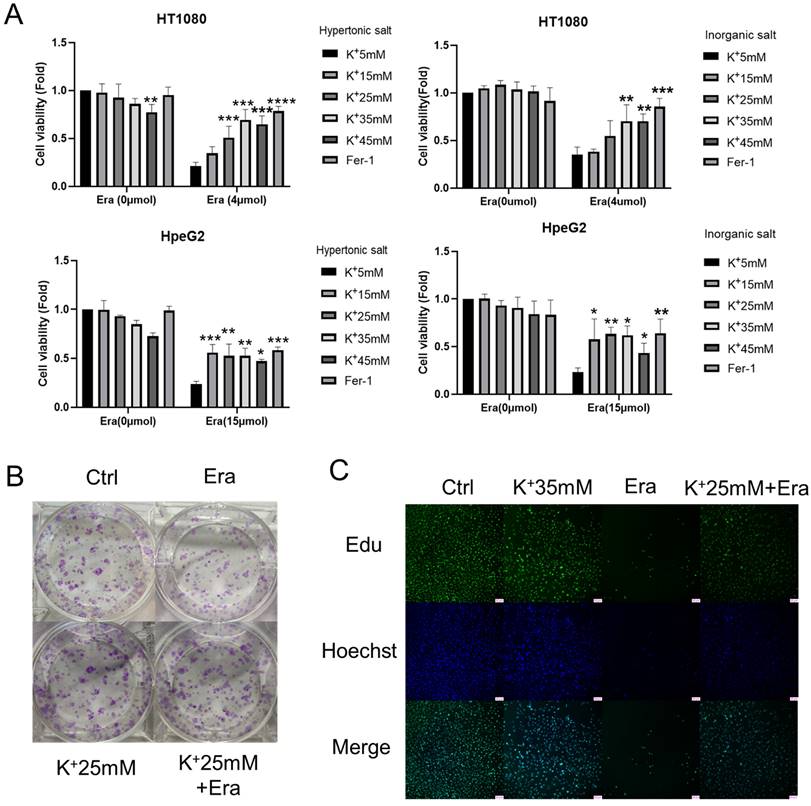
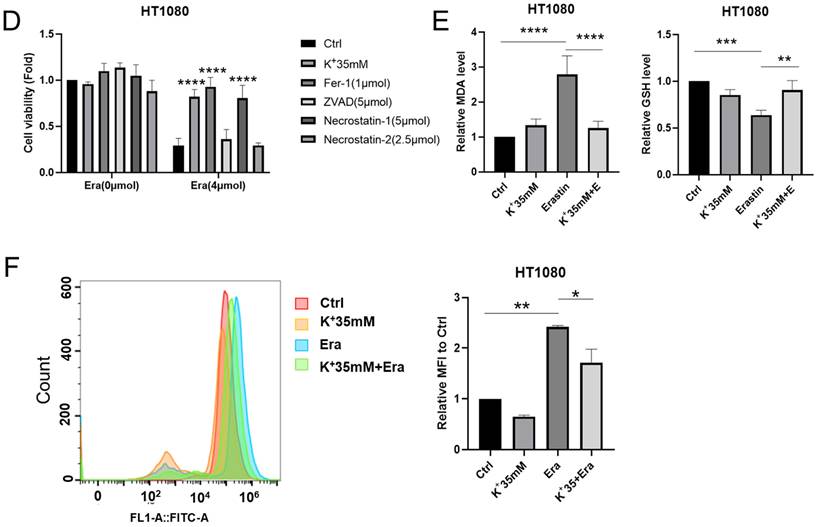
We set up hyperosmotic and isotonic potassium concentrations to assess the relationships between high-potassium conditions and ferroptosis. The CCK-8 assay revealed that 35 mM potassium cation significantly reversed the ferroptosis induced by erastin but not that which was induced by RSL3 in HT1080 and HepG2 cells (Figures 1A and S1A). In contrast, sodium supplementation failed to rescue ferroptosis (Figure S1B). The colony formation assay and EdU incorporation revealed that potassium-treated cells proliferated in the presence of erastin compared to the erastin control (Figures 1B, 1C, and S1C). Necrostastin-1 suppressed necroptosis by inhibiting receptor-interacting serine/threonine-protein kinase 1 (RIPK1). However, Hanna Yuk showed that necrostastin-1 also reversed erastin-induced ferroptosis. In the present work, we used necrostastin-2 for the express purpose of inhibiting necroptosis [12]. The CCK-8 assay confirmed that whereas neither carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK) nor necrostastin-2 regulated ferroptosis, extra K+, necrostatin-1, and Fer-1 significantly prevented ferroptosis after erastin treatment (Figures 1D and S1D). MDA, GSH, and ROS flow cytometry were also used to reveal ferroptosis. In response to erastin treatment, the potassium-treated cells had lower MDA and ROS and higher GHS levels than the normal cells (Figures 1E, 1F, and S1E). The preceding results suggest that a high-potassium environment plays a critical role in ferroptosis induction.
High potassium environment confers cancer cells resistance to ferroptosis. (A) CCK-8 assay for cell viability of HT1080 and HepG2 cells treated with Erastin (4 µmol, 15 µmol) in a high potassium environment for 48 h, respectively. The relative viability was normalized to the K+ 5mM group. (B) Colony-forming ability of HT1080 cells cultured with K+ 25mM medium and Erastin (1 µmol) for one week. (C) HT1080 cells were cultured with K+ 35mM medium and Erastin (4 µmol) in 96 wells for 48 h, then incubated with 10 µmol Edu. Images were obtained through a fluorescence microscope. (D) The cell viability of HT1080 with the treatments DMSO, Erastin (4 µmol) for 48 h combined with Ferrostain-1 (1 µmol), Z-VAD-FMK (5 µmol), Necrostatin-1(5 µmol) and Necrostastin-2 (2.5 µmol) were monitored using a CCK-8 assay. The relative viability was normalized to K+ 5 mM group. (E-F) MDA, GSH, and ROS levels in HT1080 cell treated with Erastin (2.5 µmol) in a high potassium environment for 48 h. ∗P< 0.05. ∗∗P< 0.01. ∗∗∗P< 0.001. ∗∗∗∗P< 0.0001.
A high-potassium environment is associated with ER stress
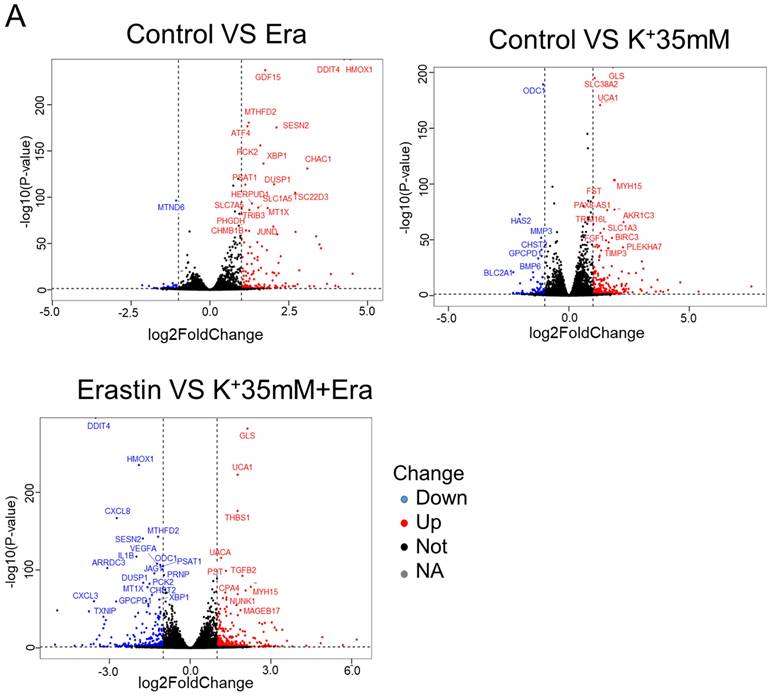
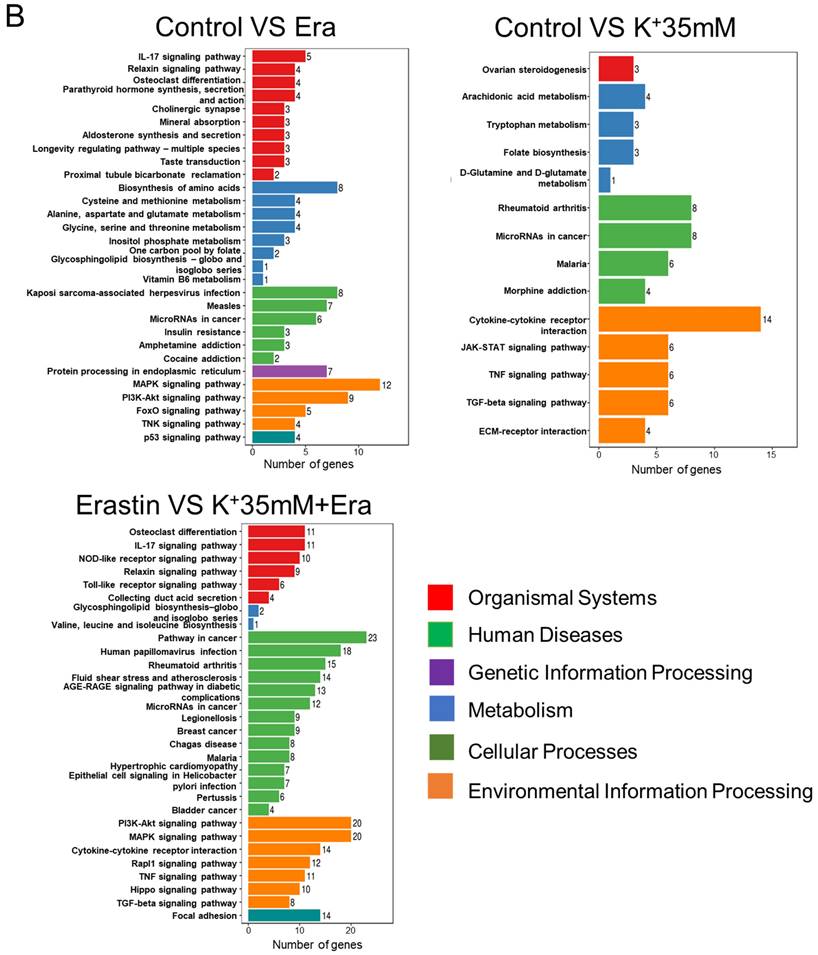
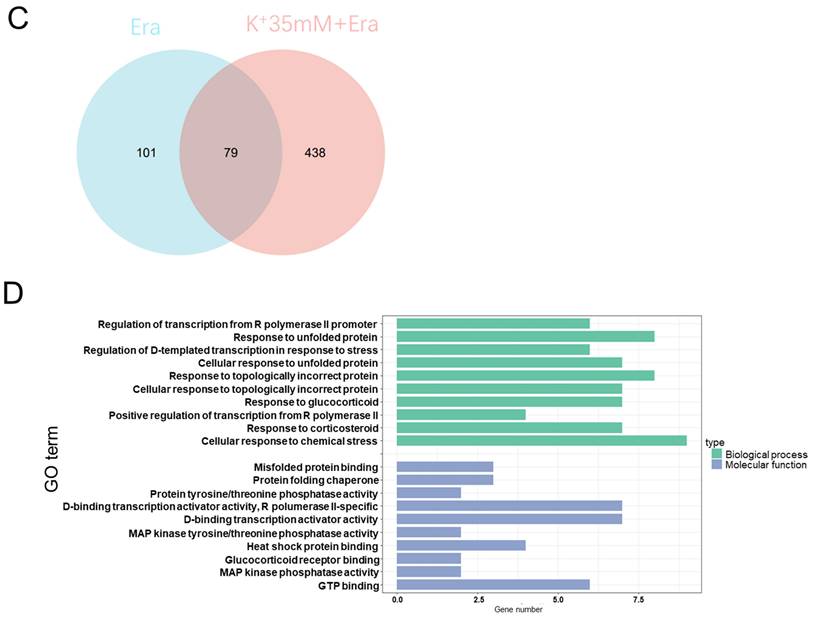
RNA sequencing detected changes in cellular RNA level following treatment with 5 mM K+, erastin, 35 mM K+, or 35 mM K+ + erastin to identify correlations between a high-potassium environment and ferroptosis. The results revealed that high extracellular potassium significantly modulated amino acid metabolism. The RNA levels of glutaminase (GLS) and the amino acid transporters SLC38A2, SLC2A12, and SLC1A3 were significantly increased. The KEGG and GO analyses disclosed that the high-potassium treatment significantly altered the cytokine-cytokine receptor interaction pathway (Figures 2A, 2B, and S2B). The results of both the present study and previous investigations showed that the erastin treatment upregulated ER-related genes [13]. The expression levels of DDIT4, HMOX1, ATF4, XBP1, HERPUD1, and ATF3 were substantially higher for the erastin treatment than for the control. The KEGG and GO analyses confirmed significant alteration of the ER pathway. Nevertheless, additional extracellular potassium significantly decreased the RNA levels of HMOX1, DDIT4, TXNIP, and XBP1 (Figures 2A, 2B, S2A, and S2C). To elucidate the mechanisms underlying these responses, we analyzed the foregoing genes and found that the expression levels of 79 of them had changed in the elastin and combination groups relative to the control (Figure 2C). The GO analysis associated these genes with the terms “molecular function”, “cellular component”, and “biological process”. Analyses of the terms “biological process” and “molecular function” revealed that the genes were involved mainly in misfolded protein binding associated with ER stress (Figure 2D).
RNA sequencing indicated that a high potassium environment is associated with ER stress. (A-B) Volcano plot and KEGG analysis of RNA sequencing data from HT1080 cells treated with K+ 35mM, Erastin, and a combination of K+ 35mM and Erastin. (C-D) Venn diagram and GO analysis of Erastin group and K+ 35mM+Erastin group.
A high-potassium environment alleviates erastin-induced ER stress
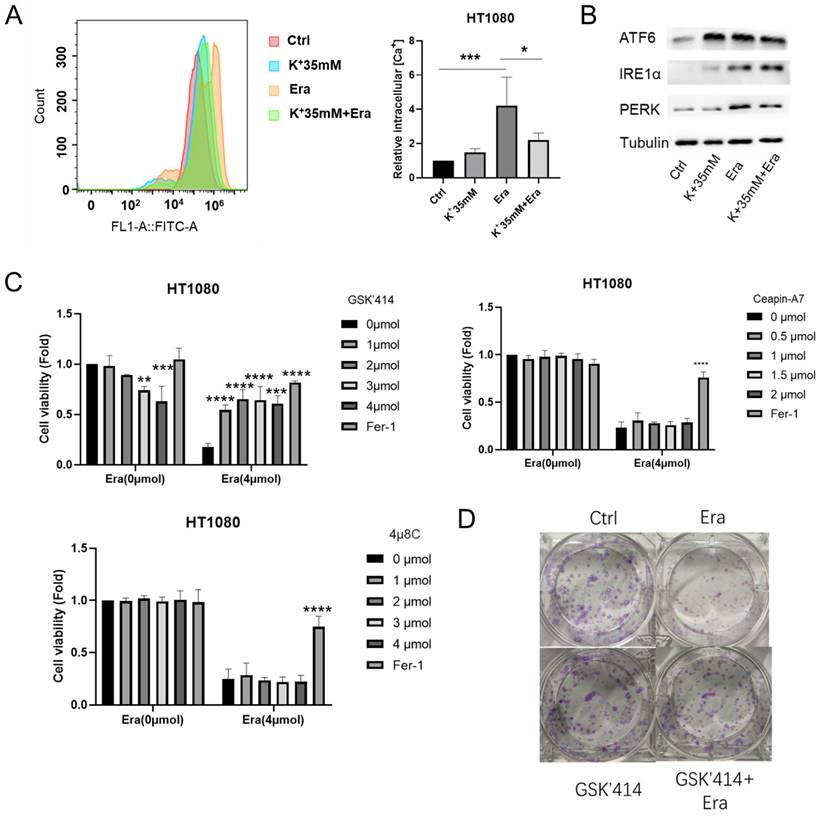
DaeYong Lee confirmed that perturbation of intracellular potassium homeostasis induced severe ER stress and apoptosis [14]. However, the optimal balance between extracellular potassium and ER stress has not yet been identified. Hence, we applied Fura-3, AM and measured the intracellular Ca2+ level as an ER stress marker. A high-potassium environment drastically reduced intracellular Ca2+ accumulation following erastin treatment (Figure 3A). In humans, three ER transmembrane proteins (PERK, IRE1α, and ATF6) operate as ER stress sensors [15]. Here, a high-potassium environment and erastin treatment upregulated IRE1α and ATF6. However, the expression levels of PERK and ATF6 were not higher in the combination treatment than in the erastin treatment (Figure 3B).
High potassium environment relieved ER stress induced by Erastin. (A) Intracellular Ca2+ level of HT1080 treated with Erastin (2.5 µmol) in a high potassium environment for 48 h. (B) The expression level of ER-associated genes ATF6, IRE1α, and PERK. (C) CCK-8 assay assessed the cell viability of HT1080 treated with Erastin (4 µmol) combination of GSK'414, Ceapin-A7, and 4u8C for 48 h, respectively. The relative viability was normalized to control. (D) Colony forming ability of HT1080 cells cultured with GSK'414 (2 µmol) and Erastin (1 µmol) for one week. (E) HT1080 cells were cultured with GSK'414 (2 µmol) and Erastin (4 µmol) for 48 h, then incubated with 10 µmol Edu. (F-G) MDA, GSH, and ROS levels in HT1080 cells treated with Erastin (2.5 µmol) and GSK'414 (2 µmol) for 48 h. (H) TEM analysis of mitochondrion and endoplasmic reticulum of HT1080 cells treated with K+ 35 mM, GSK'414, and Erastin.
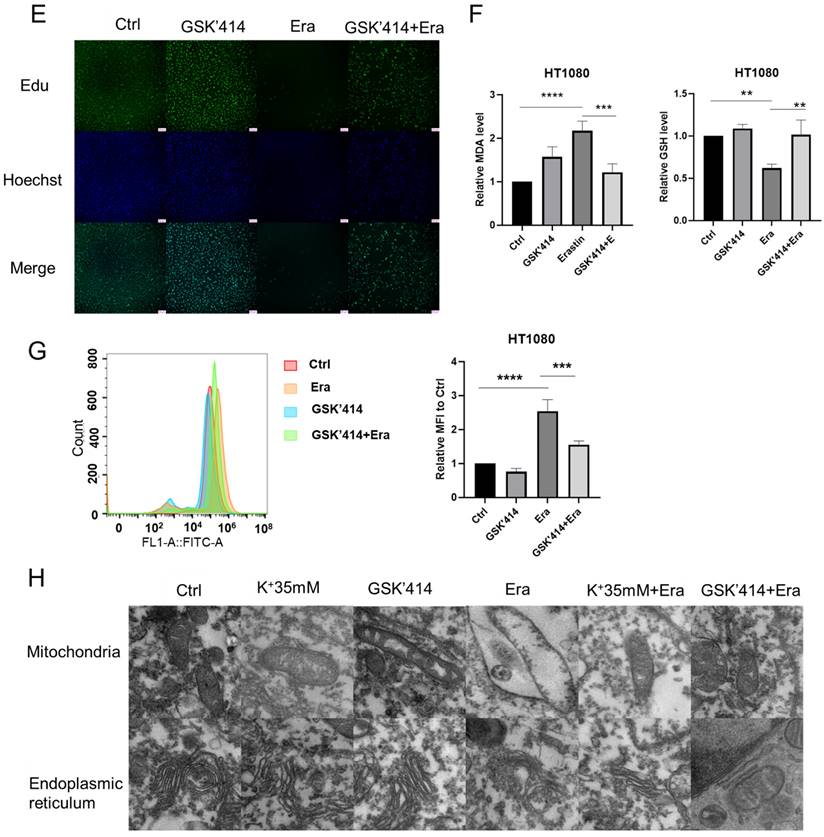
We then inhibited PERK, ATF6, and IRE1α with GSK2606414 (GSK'414), ceapin-A7, and 4u8C, respectively [16-18]. The CCK-8 and colony formation assays and EdU incorporation revealed that only the PERK inhibitor GSK2606414 significantly reversed erastin-induced ferroptosis (Figures 3C-3E). The MDA, GSH, and ROS levels confirmed this reversal. The GSK'414 treatment decreased the MDA and ROS levels and increased the GSH level following the erastin treatment (Figures 3F and 3G). Transmission electron microscopy disclosed that a high-potassium environment and GSK'414 reversed erastin-induced mitochondrial and ER damage (Figure 3H). The preceding results imply that a high-potassium environment reverses ferroptosis through the PERK pathway.
High potassium inhibited ferroptosis rescue by regulating ATF3 expression
We then performed RNA sequencing on the GSK'414-treated cells to identify the factors explaining the rescue of ferroptosis mediated by a high-potassium environment. The ER-related proteins DDIT4, ATF3, XBP1, and HSPA1B were upregulated in response to GSK'414 but downregulated following the erastin combination treatment (Figure 4A). The KEGG analysis showed that the GSK'414 treatment and the erastin combination affected mainly the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) and mitogen-activated protein kinase (MAPK) signaling pathways, respectively (Figures S3A and S3B). The GO analysis demonstrated that the GSK'414 treatment influenced the PERK-mediated unfolded protein response while the erastin combination was related mainly to the inflammatory response (Figures S3C and S3D).
ATF3 plays a significant role in ferroptosis in a high potassium environment. (A) Volcano plot analysis of RNA sequencing results of HT1080 treated with GSK'414 and a combination of GSK'414 and Erastin. (B) Hub genes analysis of Erastin, K+ 35mM+Erastin, GSK'414+Erastin. (C) Venn diagram analysis indicated that ATF3 played a vital role in ferroptosis. (D) The expression level of ATF3 when treated with K+35mM, GSK'414 (3µmol), and Erastin (2.5 µmol) for 48 h. (E-G) Knockdown ATF3 attenuated sensitivity to ferroptosis and MDA, confirmed by the GSH level. (H-I) Overexpression ATF3 enhanced sensitivity to ferroptosis.
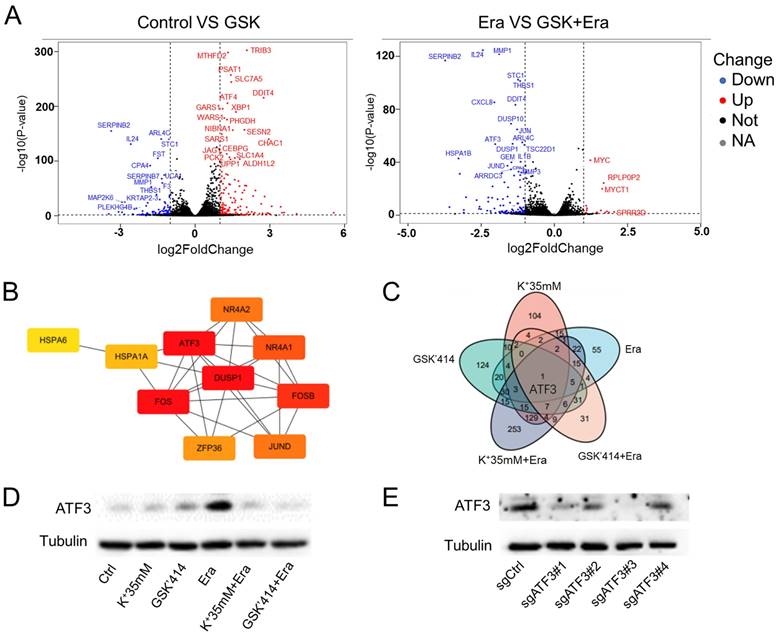
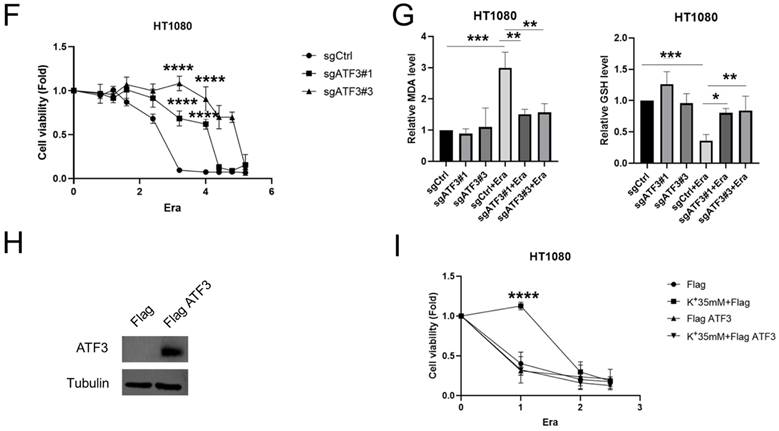
We analyzed the foregoing genes to identify the factors playing crucial roles in reversing cell ferroptosis. The relative expression levels of these genes changed in the erastin, 35 mM K+ + erastin, and GSK'414 + erastin groups. Twenty-four genes were altered in all three groups and ATF3, DUSP1, FOS, and FOSB might be the hub genes regulating ferroptosis (Figures 4B, S3E, and S3F). Nevertheless, RNA sequencing of the 35 mM K+- and GSK'414-treated cells indicated that ATF3 might be the pivotal gene (Figure 4C).
HT1080 cells were treated with high potassium concentrations, GSK'414, and erastin for 2 days in an attempt to clarify the relationship between ATF3 in ferroptosis and a high-potassium environment. WB demonstrated that erastin significantly upregulated ATF3 whereas the combination of 35 mM K+ or GSK'414 plus erastin downregulated ATF3 to the expression level of the control (Figure 4D). We used the CRISPR/Cas9 tool to generate ATF3 knockout HT1080 cells and corroborate the preceding results. We constructed four sgRNA sequences and observed that both sgATF3#1 and sgATF3#3 successfully silenced ATF3 (Figures 4E and S3G). The CCK-8 assay revealed that ATF3 knockout prevented ferroptosis after the 4 µM erastin treatment (Figure 4F). The MDA and GSH measurements confirmed that ATF3 knockout reversed ferroptosis-induced damage. ATF3 knockout substantially decreased the MDA and increased the GSH levels relative to those of the erastin-treated control (Figure 4G). We also overexpressed ATF3 to verify that a high-potassium environment reverses ferroptosis by inhibiting ATF3 (Figure 4H). High potassium-mediated ferroptosis repression under the condition of ATF3 overexpression indicates that other heretofore unidentified factors are also involved in ferroptosis. However, the CCK-8 assay showed that a high-potassium environment could not reverse ferroptosis in response to ATF3 overexpression in the presence of erastin (Figure 4I). These results confirmed that a high-potassium environment inhibits ferroptosis by repressing ATF3.
Cells subjected to high potassium prevented ferroptosis in vivo
We subcutaneously injected HT1080 cells into BALB/c-nu mice to validate the observation that a high-potassium environment prevents ferroptosis in vitro. The mice were peritumorally injected with KCl and PE when the tumor volume reached 50 mm3. However, the mice died following the KCl injection. Eil reported abundant cancer cells in a high-potassium environment (Figure 5A) [4]. Here, we cultured HT1080 cells with or without KCl for 1 week and then subcutaneously injected them into BALB/c-nu mice. We injected PE every 2 d for 2 weeks and measured the tumors 2 days after the final injection.
High potassium-treated cells prevented ferroptosis in vivo. (A) Schematic illustration of the therapeutic scheme in vivo. HT1080 cells were pretreated with or without KCL (35 mM) for one week and then injected subcutaneously into BALB/c-nu mice. The mice were treated with Piperazine Erastin (10 mg/kg/s.c.) by peritumoral injection every other day for two weeks. (B-C) The tumor image of each group taken at the end of the study and tumor volumes in mice indicated treatment. (D) MDA level in the indicated tumor tissues. (E-F) Western blotting and IHC images of COX2 and ATF3 in indicated treatment mice tumors.
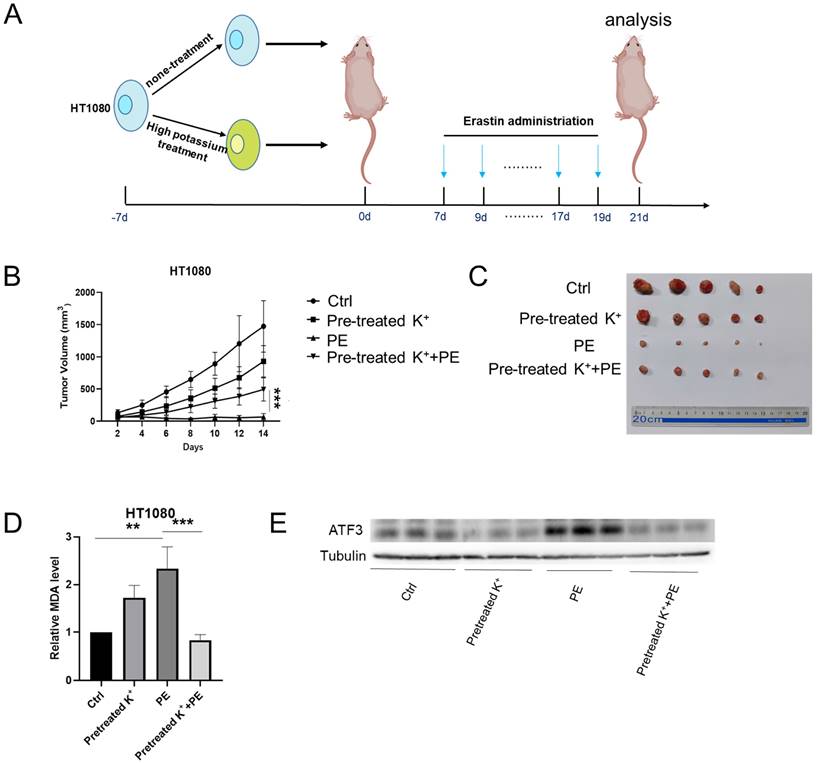
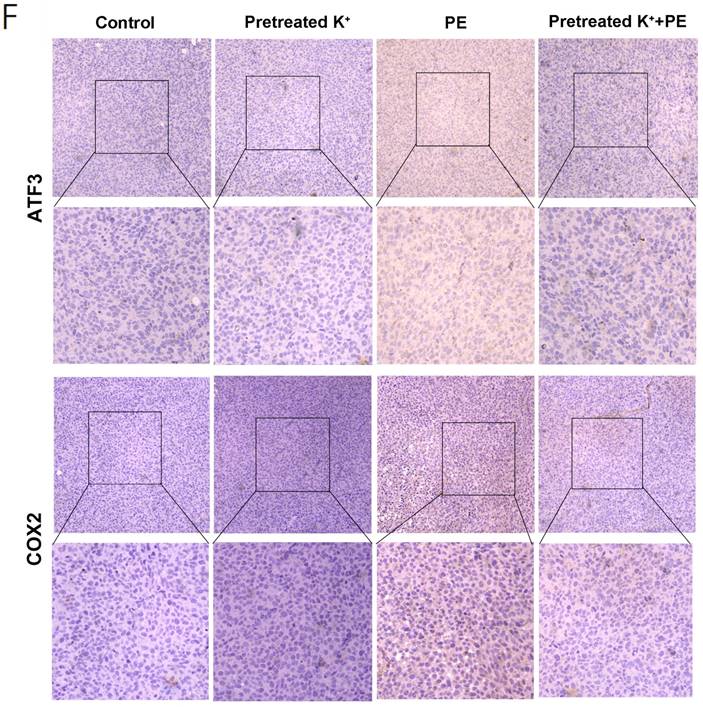
Compared with the control, the KCl pretreatment and PE treatment mildly and strongly inhibited tumor growth, respectively. Nevertheless, PE treatment following KCl pretreatment protected the cells against ferroptosis (Figures 5B and 5C). The MDA measurements confirmed that KCl pretreatment alleviated PE-induced ferroptosis (Figure 5D). WB disclosed that KCl pretreatment followed by PE treatment downregulated ATF3 to a greater extent than the PE treatment alone (Figure 5E). The IHC showed that the levels of ATF3 and the ferroptosis indicator COX2 were significantly lower in the combination group than in the PE group (Figure 5F). Taken together, the preceding results indicated that a high-potassium environment protects cells against ferroptosis.
Discussion
It was previously shown that ionic homeostasis in general, and potassium homeostasis in particular, regulate apoptosis. Increasing the extracellular K+ concentration or inhibiting K+ efflux via channel blockers suppresses cellular apoptosis [5, 19, 20]. However, it is unknown whether K+ also influences other types of cell death such as ferroptosis. The latter is iron-dependent regulated cell death and it morphologically and biochemically differs from apoptosis [21, 22]. Several studies have implied that various ions including Fe2+, Ca2+, and Zn2+ are implicated in ferroptosis [21, 23-25]. The present study demonstrated that a high-potassium environment seems to prevent ferroptosis in cancer cells.
Mitochondria, ER, lysosomes, lipid droplets, peroxisomes, nuclei, the Golgi apparatus, and other subcellular organelles are involved in ferroptosis. They generate signals driving lipid biosynthesis, iron accumulation, and lipid peroxidation [26]. The present work revealed that a high-potassium environment alleviates ferroptosis through ER stress. Several investigations have shown that the TME is significantly involved in ER stress, hypoxia, low pH/acidosis, and nutrient deprivation [27-29]. Adaptive ER stress favors cellular survival and reprogramming. Persistent, robust ER stress may induce cell death. As ER stress is higher in cancer cells than normal ones, it is promising as a target for anticancer agents. However, the present study revealed that a high-potassium environment might alleviate induced ER stress. Hence, future studies should endeavor to explore whether high extracellular K+ affects the therapeutic efficacy of drugs targeting the ER. Previous research also disclosed that high extracellular K+ levels may influence mitochondrial metabolism [30]. Nevertheless, our RNA sequencing results failed to corroborate this finding. Therefore, it remains to be determined whether high K+ concentrations affect other subcellular organelles.
Ferroptosis also regulates neurodegenerative conditions such as Alzheimer's disease (AD) and Parkinson's disease (PD) [31, 32]. Though these disorders have imposed substantial socioeconomic and healthcare burdens, there are few therapeutic strategies for them. Several studies have reported that elevated oxidative and ER stress comprise the main molecular mechanisms of neurodegenerative diseases. Prior research reported that secondary increases in calcium ion levels also lead to ferroptosis [24, 33, 34]. Other earlier work demonstrated that PERK inhibitors prevented neurodegeneration in an animal PD model. However, these drugs could also cause secondary pancreatic toxicity [35]. The present study confirmed that the PERK inhibitor GSK'414 protected cells against ferroptosis and demonstrated that high K+ concentrations attenuate ER stress and inhibit increases in Ca2+. Future investigations should aim to determine whether elevated cerebral K+ concentrations critically regulate neurodegenerative diseases.
Abbreviations
KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; ER: Endoplasmic Reticulum; GSK'414: GSK'2606414; PERK: PRKR-like ER kinase; IRE1α: inositol-requiring enzyme 1α; ATF6: activating transcription factor 6; ATF3: activating transcription factor 3.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (number: 82071984). Social Development Foundation of Jiangsu Province (number: BK20191223), Young Medical Talents of Jiangsu (number: QNRC2016833). Nanjing Health Science and Technology Development Special Fund project (YKK20176).
Ethics Statement
Animal Studies: All procedures in the manuscript were reviewed in advance by UJS IACUC and also met the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The number is UJS-IACUC-2023010801.
Author Contributions
Lirong Zhang andcHsiang-I Tsai are corresponding authors and organized this study. Xufeng Pu, Zhenhui Chen and Li Li performed experiments. Aihua Gong and Jiao Lei improved the images. All authors read and approved the final manuscript. Xufeng Pu, and Li Li contributed equally to this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang L, Yin YL, Liu XZ, Shen P, Zheng YG, Lan XR. et al. Current understanding of metal ions in the pathogenesis of Alzheimer's disease. Transl Neurodegener. 2020;9:10
2. Ranade SS, Panday VK. Major metals in human cancer: calcium, magnesium, sodium and potassium. Sci Total Environ. 1985;41:79-89
3. Wu CW, Wei YY, Chi CW, Lui WY, P'Eng FK, Chung C. Tissue potassium, selenium, and iron levels associated with gastric cancer progression. Dig Dis Sci. 1996;41:119-25
4. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH. et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539-43
5. Karki P, Seong C, Kim JE, Hur K, Shin SY, Lee JS. et al. Intracellular K(+) inhibits apoptosis by suppressing the Apaf-1 apoptosome formation and subsequent downstream pathways but not cytochrome c release. Cell Death Differ. 2007;14:2068-75
6. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266-82
7. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107-25
8. Jiang L, Gao XM, Cao J. The Achilles heel of TNBCs: Ferroptosis heterogeneity. Cell Metab. 2023;35:1-2
9. Fuhrmann DC, Mondorf A, Beifuß J, Jung M, Brüne B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020;36:101670
10. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z. et al. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020;33:108487
11. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225-34
12. Yuk H, Abdullah M, Kim DH, Lee H, Lee SJ. Necrostatin-1 Prevents Ferroptosis in a RIPK1- and IDO-Independent Manner in Hepatocellular Carcinoma. Antioxidants (Basel). 2021 10
13. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523
14. Lee D, Lee SH, Noh I, Oh E, Ryu H, Ha J. et al. A Helical Polypeptide-Based Potassium Ionophore Induces Endoplasmic Reticulum Stress-Mediated Apoptosis by Perturbing Ion Homeostasis. Adv Sci (Weinh). 2019;6:1801995
15. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421-38
16. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW. et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012;55:7193-207
17. Gallagher CM, Garri C, Cain EL, Ang KK, Wilson CG, Chen S. et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife. 2016 5
18. Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM. et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869-78
19. Dallaporta B, Hirsch T, Susin SA, Zamzami N, Larochette N, Brenner C. et al. Potassium leakage during the apoptotic degradation phase. J Immunol. 1998;160:5605-15
20. Nietsch HH, Roe MW, Fiekers JF, Moore AL, Lidofsky SD. Activation of potassium and chloride channels by tumor necrosis factor alpha. Role in liver cell death. J Biol Chem. 2000;275:20556-61
21. Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1292-r7
22. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486-541
23. Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J. et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021;28:1644-57
24. Angelova PR, Choi ML, Berezhnov AV, Horrocks MH, Hughes CD, De S. et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020;27:2781-96
25. Chen PH, Wu J, Xu Y, Ding CC, Mestre AA, Lin CC. et al. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis. 2021;12:198
26. Chen X, Kang R, Kroemer G, Tang D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021;28:2843-56
27. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer. 2021;21:71-88
28. Vandewynckel YP, Laukens D, Geerts A, Bogaerts E, Paridaens A, Verhelst X. et al. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013;33:4683-94
29. Walczak A, Gradzik K, Kabzinski J, Przybylowska-Sygut K, Majsterek I. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxid Med Cell Longev. 2019;2019:5729710
30. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH. et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019 363
31. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49
32. Heemels MT. Neurodegenerative diseases. Nature. 2016;539:179
33. Esmaeili Y, Yarjanli Z, Pakniya F, Bidram E, Łos MJ, Eshraghi M. et al. Targeting autophagy, oxidative stress, and ER stress for neurodegenerative disease treatment. J Control Release. 2022;345:147-75
34. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13:477-91
35. Mercado G, Castillo V, Soto P, López N, Axten JM, Sardi SP. et al. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol Dis. 2018;112:136-48
Author contact
![]() Corresponding authors: Lirong Zhang, the Affiliated Hospital of Jiangsu University 212001, Zhenjiang, Jiang Su Province, China. Email: tianchen861edu.cn (L.Z.); Tel.: +86-13376149021. Hsiang-I Tsai, the Affiliated Hospital of Jiangsu University, 212001, Zhenjiang, Jiang Su Province, China; Email: Tsaihsiangi88com; Tel: +86-13247606832.
Corresponding authors: Lirong Zhang, the Affiliated Hospital of Jiangsu University 212001, Zhenjiang, Jiang Su Province, China. Email: tianchen861edu.cn (L.Z.); Tel.: +86-13376149021. Hsiang-I Tsai, the Affiliated Hospital of Jiangsu University, 212001, Zhenjiang, Jiang Su Province, China; Email: Tsaihsiangi88com; Tel: +86-13247606832.