Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2023; 14(10):1751-1762. doi:10.7150/jca.83954 This issue Cite
Research Paper
Super Enhancer Driven Hyaluronan Synthase 3 Promotes Malignant Progression of Nasopharyngeal Carcinoma
Quanzhu Chen1,2,3,4, Qian Peng1,2,3,4, Jing Cai5, Ying Liu1,2,3,4, Xingxing Lu1,2,3,4, Wei Xiong1,2,3,4, Zhaoyang Zeng1,2,3,4, Guiyuan Li1,2,3,4, Xiaoling Li1,2,3,4, Xiayu Li4, Bo Xiang1,2,3,4, Mei Yi6 ![]() , Pan Chen1
, Pan Chen1 ![]()
1. Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha 410013 Hunan, China.
2. The Key Laboratory of Carcinogenesis of the Chinese Ministry of Health, Cancer Research Institute and School of Basic Medical Sciences, Central South University, Changsha 410008 Hunan, China.
3. The Key Laboratory of Carcinogenesis and Cancer Invasion of the Chinese Ministry of Education, Cancer Research Institute and School of Basic Medical Sciences, Central South University, Changsha 410078 Hunan, China.
4. Hunan Key Laboratory of Nonresolving Inflammation and Cancer, The Third Xiangya Hospital, Central South University, Changsha 410013 Hunan, China.
5. Department of Pathology, the Second Xiangya Hospital, Central South University, Changsha 410000 Hunan, China.
6. Department of Dermatology; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008 Hunan, China.
Received 2023-3-1; Accepted 2023-5-23; Published 2023-6-12
Abstract

Nasopharyngeal carcinoma (NPC) is a malignant tumor of the head and neck with high metastatic and invasive nature. Super enhancers (SEs) control the expression of cell identity genes and oncogenes during tumorigenesis. As a glycosaminoglycan in the tumor microenvironment, hyaluronan (HA) is associated with cancer development. High expression of hyaluronan synthase 3 (HAS3) resulted in HA deposition, which promoted the growth of cancer cell. However, its role in NPC development remains elusive. We demonstrated that the levels of HAS3 mRNA or protein were increased in NPC cell lines. Transcription of HAS3 is associated with SE. Disruption of SE by bromodomain containing 4 (BRD4) inhibitor JQ1 resulted in downregulation of HAS3 and inhibition of cell proliferation and invasiveness in NPC cells. Inhibition of HA synthesis by HAS inhibitor 4-MU suppressed cell growth and invasion of NPC cells, whereas HA treatment exerted opposite effects. Genetically silencing HAS3 in HK1 and FaDu NPC cells attenuated cell proliferation and mobility, while re-expression of HAS3 enhanced malignant potential of CNE1 and CNE2 NPC cells. Furthermore, loss of HAS3 impaired metastatic potential of HK1 cells in nude mice. Mechanistically, inhibition of HA synthesis by chemical inhibitor or silencing HAS3 led to reduction of the levels of phosphorylation of EGFR, AKT, and ERK proteins. In contrast, exogenous HA treatment or forced expression of HAS3 activated EGFR/AKT/ERK signaling cascade. This study suggested that HAS3 is driven by SE and overexpressed in NPC. High expression of HAS3 promotes the malignant features of NPC via activation of EGFR/AKT/ERK signaling pathway.
Keywords: nasopharyngeal carcinoma, super-enhancer, hyaluronan, hyaluronan synthase 3, EGFR/AKT/ERK signaling pathway
Introduction
Nasopharyngeal carcinoma (NPC) is a malignant tumor of head and neck cancer occurring in the top and lateral walls of the nasopharyngeal cavity. Owing to this unique anatomical structure of nasopharyngeal cavity, NPC can be difficult to make an early diagnosis [1]. In addition, NPC is characterized by high invasion and early metastasis, 70-80% of NPC patients have developed cervical lymph node metastases at the first diagnosis [2-5]. However, the underlying mechanism of NPC metastasis is still not clear.
NPC is characterized as having comparatively low mutation rate, widespread hypermethylation, high copy number alterations and chromosome abnormalities by Genome-wide studies [6-14]. Compared with inactivation of tumor suppressor genes, tumorigenic mutation is a very rare cause for inherited mutations of NPC, such as the mutation of K-RAS gene or epidermal growth factor receptor (EGFR) gene [8, 15, 16]. Furthermore, it has been demonstrated that chromatin remodeling factors, such as ARID1A, MLL2, and MLL3 have shown high mutation frequency in NPC [8, 16]. Thus, the epigenetic alterations may play a crucial role in NPC initiation and development. It will be highly desirable if we study the activation of oncogenes in the processes evolve on NPC from the perspective of epigenetics.
Super enhancers (SEs) are related to epigenetic regulatory mechanisms, which involve in the regulation of gene expression [17]. As the large clusters of cis-elements, SEs are characterized by intensive H3K27ac modification, and significantly enriched with the master transcription factors (TFs) and mediator. Compared with the typical enhancers (TEs), SEs possess stronger capability of driving genes expression, and are highly sensitive to JQ1 (a SE inhibitor) [18]. In many types of cancer, SEs act as key oncogenic drivers during tumorigenesis [19-21]. Therefore, in NPC development, understanding the role of the SEs landscape will provide a novel approach for identifying oncogenic factors. Through SE profile and RNA-Seq, we found hyaluronan synthase 3 (HAS3) was driven by SE.
As a core mucopolysaccharide component of extracellular matrix (ECM), hyaluronan (HA) involves in many biological processes, such as malignant cell migration [22-24], and also promotes the proliferation and tumorigenicity of CNE2 cell lines [25]. HA is synthesized by three hyaluronan synthase (HAS) enzymes, HAS1, HAS2, and HAS3 [26]. High expression of HAS3 results in HA deposition, which induces extracellular matrix remodeling and promotes the growth of colon, prostate and pancreatic cancer cell [27-29]. However, HAS3 knockdown or inhibition of HA suppresses cell proliferation and survival, decreases esophageal xenograft tumorigenesis, and suppresses epidermal growth factor receptor (EGFR)/AKT/ERK signaling pathway in esophageal squamous cell carcinoma (ESCC) and lung cancer [30, 31].
Previous research has confirmed that the overexpression of EGFR is common in NPC and the EGFR signaling pathway plays the significant role in the pathogenesis of NPC [32, 33]. EGFR and downstream signaling proteins affect many cellular functions, such as proliferation, differentiation, migration, and apoptosis [34, 35]. It was found that through activating EGFR-SRC signaling, HA and HAS3 could induce oncogenic actions [36-38]. However, the precise mechanisms that mediate HAS3-dependent malignant cell migration remain unclear. In this study, we demonstrated that the levels of HAS3 mRNA or protein were increased in NPC cell lines. Inhibition of HA synthesis suppressed cell growth and invasion of NPC cells, whereas HA treatment exerted opposite effects. Genetically silencing HAS3 in NPC cells attenuated cell proliferation, mobility, and invasiveness in vitro, and metastasis in vivo, while re-expression of HAS3 enhanced malignant potential of NPC cells. Mechanistically, inhibition of HA synthesis or silencing HAS3 led to reduction of the levels of phosphorylation of EGFR, AKT and ERK proteins. In contrast, exogenous HA treatment or forced expression of HAS3 activated EGFR/AKT/ERK signaling cascade. This study suggested that HAS3 is driven by SE and overexpressed in NPC. High expression of HAS3 promotes the malignant features of NPC via activation of EGFR/AKT/ERK signaling pathway.
Material and Methods
Cell lines and cell culture
NP69, HK1, C666-1, CNE1, CNE2, and FaDu cell lines were used in this study. NP69 is an immortalized nasopharyngeal epithelial cell line, which maintained in our lab [39]. The human NPC cell lines HK1, C666-1, CNE1 and CNE2 are maintained in our lab [40-43]. FaDu is a hypopharyngeal carcinoma cell line purchased from the National Infrastructure of Cell Line Resource (Shanghai, China) [44, 45]. All these cells were cultured in 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), RPMI 1640 medium (Gibco, Grand Island, NY, USA) at 37℃, 5% CO2 [46].
RNA isolation and real-time reverse-transcription PCR (qPCR)
Total cellular RNAs were extracted using Trizol reagent (Life Technologies, Grand Island, NY, USA). cDNAs were synthesized from the RNAs using RNase-free DNase I (Takara, Beijing, China) and RevertAid First Strand cDNA Synthesis Kits (Thermo Fisher Scientific, Beijing, China). Quantitative PCR mixtures were prepared according to the manufacturer's manual of SYBR Green (Bimake, Shanghai, China) and reactions were run in a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Richmond, CA, USA) [47]. 2-ΔΔCT methods were adopted for genes relative expression levels. Primers sequences are as follows: HAS3 forward primer ACTACATCCAGGTGTGCGAC; HAS3 reverse primer CAGCCAAAGTAGGACTGGCA. GAPDH forward primer AACGGATTTGGTCGTATTGG; GAPDH reverse primer TTGATTTTGGAGGGATCTCG.
Western blotting
Western blotting was performed to measure protein expression levels. NPC cells were lysed with RIPA buffer (Beyotime, Jiangsu, China). Equal cellular protein sample amounts from different extracts were separated by 10% SDS-PAGE gels and transferred onto PVDF membranes (Millipore, Billerica, MA, USA) [48]. After transfer, membranes were blocked with 5% non-fat milk in Tris buffer saline containing 0.1% Tween-20 for 1 hour at room temperature and then incubated with primary antibodies at 4 °C overnight. The following antibodies against HAS3, p-EGFR (Y992), p-AKT (S473), p-ERK1/2 (ABclonal Technology, Woburn, MA, USA), EGFR (Proteintech, Chicago, IL, USA), AKT, ERK1/2 (Cell Signaling Technology, Beverly, MA, USA) and GAPDH (ABclonal Technology) were used.
Cell viability assay
Cell Counting Kit-8 (CCK-8, Beyotime) was performed as previously described [49, 50]. Briefly, NPC cells (1 × 103 cells/mL) were plated into a 96-well plate in each well in quintuplicate and treated with different dose of JQ1, 4-Methylumbelliferone (4-MU, Selleck Chemicals, Houston, TX, USA), or HA (8000 Da, Sigma-Aldrich, St Louis, MO, USA) for 24 h. Cells were incubated with 10 μl of CCK8 for 2h at 37℃. The absorbance of sample was measured at 450 nm using a microplate reader. Data were presented as means ± SD from five wells per experiment.
Cell migration and invasion assays
Tumor cell migration and invasion were determined by Transwell (Corning‐Costar, Cambridge, MA, USA) migration and invasion assays as previously described [42]. In serum-free medium, cell suspensions were seeded onto 8-μm-pore Transwell upper inserts precoated without (migration assay) or with (invasion assay) 15 μL Matrigel (BD Biosciences, Bedford, MA, USA) at a density of 105 cells/well, and then the inserts were held in a lower chamber with 700 μl of RPMI 1640 medium containing 15% FBS. After culturing for 10-48 h at 37℃, cells that migrated to the lower surface of the membrane were fixed and stained with 0.1% crystal violet as previously described [45, 51]. The number of tumor cells were counted in five random fields to calculate the average number of cells.
Stable knockdown and overexpression experiments
For knockdown experiments, tumor cells were infected with shRNA (GenePharma Co., Ltd., Shanghai, China) expressing lentivirus and selected by puromycin. For stable overexpression, tumor cells were infected with HAS3 expressing lentivirus and selected by puromycin. Sequences of shRNAs are as follows: shHAS3#1 GCTCTACAACTCTCTGTGGTTC; shHAS3#2 CCATTGCTACCATCAACAAAT.
In vivo metastasis study
The animal protocols used were reviewed and approved by the ethical review committee of The Central South University of China. The animal experiment followed ARRIVE guidelines. The suspended cells (1 × 106/0.2 mL) were injected into the lateral tail vein of 5-week-old male BALB/c nude mice (Hunan Slack King Laboratory Animal Co. Ltd., Changsha, China), and they were sacrificed 6 weeks after tumor cell inoculation [52]. Lung tissues were fixed in 4% saline-buffered formalin, embedded in paraffin, sectioned at 5 μm, and then stained with hematoxylin & eosin (H&E) as previously described [53]. A minimum of 15 sections was examined per mouse under a light microscope.
Statistical analysis
SPSS v17.0 software (SPSS, Chicago, IL, USA) was employed for statistical analysis, and Prism 5.0 (Graphpad Software, CA, USA) was used for Statistical histograms. Student's t-test, Pearson's χ2 test, two-way ANOVA were used. Experiments were repeated in triplicate. Significance was defined as P < 0.05 (*), P < 0.01 (**), or P < 0.001 (***).
Results
HAS3 is highly expressed in HNSC and correlates with poor survival
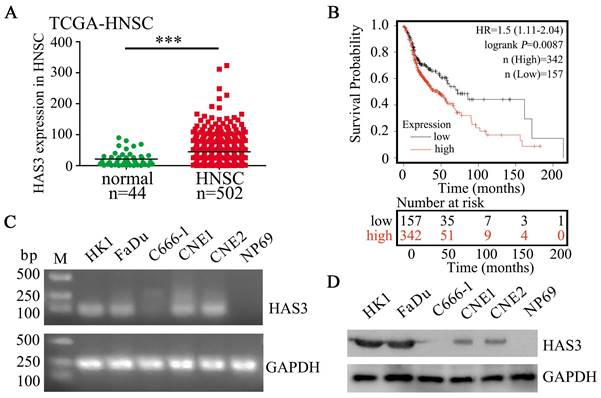
Based on analyzing The Cancer Genome Atlas (TCGA) cohort, HAS3 showed up-regulated in head and neck squamous cell carcinomas (HNSC) samples when compared with normal control (P < 0.001, Figure 1A), suggesting it might serve an oncogenic role in HNSC development and progression. In addition, Kaplan-Meier plotter analysis showed that the overall survival of HNSC patients with high HAS3 expression was correlated with poor survival (P = 0.0087, Figure 1B). Verification experiment using RT-PCR and western blotting indicated that HAS3 was highly expressed in HK1 and FaDu cells, but not expressed in C666-1 cell and normal nasopharyngeal epithelial cell line NP69 (Figure 1C and D).
HAS3 was highly expressed and predicted poor survival in HNSC. (A) Relative expression of HAS3 in HNSC tissues in comparison with adjacent non‐tumour tissues. (B) Kaplan-Meier overall survival curves of HNSC patients based on HAS3 expression (low vs. high, log‐rank test). Kaplan-Meier plots were generated using Kaplan-Meier Plotter (http://kmplot.com/). (C) HAS3 mRNA level in various cell lines was determined by RT-PCR. (D) HAS3 protein level in various cell lines was determined by western blotting.
JQ1 disrupted HAS3-SE-associated transcription and inhibited NPC cells proliferation and mobility
In previous work (Jing Cai et al. 2019), the SE landscapes and gene transcriptomic between HK1 and C666-1 cells were established by H3K27ac and H3K4me1 ChIP-Seq and RNA-Seq. We defined HAS3 as a SE-driven gene by ROSE algorithm. And we found the region of HAS3 upstream bears high-level H3K4me1 and H3K27ac modifications in HK1 cell lines, but C666-1 cell lines do not present the same modifications (Figure 2A).
HAS3 was defined as a SE (HAS3-SE), JQ1 disrupted HAS3-SE-associated transcription and inhibited NPC cells proliferation and invasiveness. (A) ChIP-seq profiles of H3K4me1, H3K27ac modifications at HAS3-SE in C666-1 and HK1 NPC cells. (B) HAS3 mRNA level in HK1 and FaDu cells was inhibited by JQ1 in a dose-dependent manner. Mean ± SD, n = 3. (C) CCK8 assay showed that JQ1 suppressed the growth of HK1 and FaDu cells. Mean ± SD, n = 3. (D) The migration and invasion of HK1 and FaDu cells treated by JQ1 at various concentrations (× 20). Mean ± SD, n = 3. The control group treated with DMSO. *P < 0.05, **P < 0.01, or ***P < 0.001. ns, no significance.
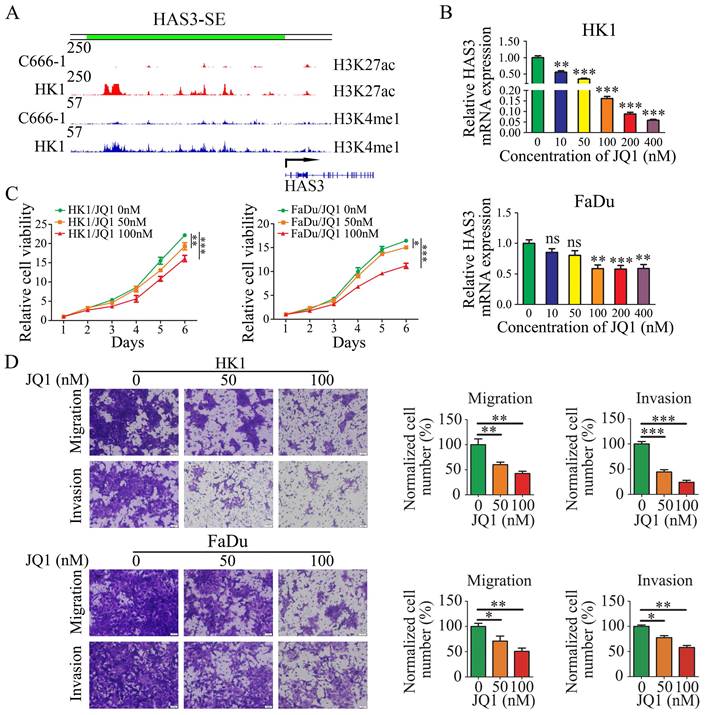
As a wide spectrum inhibitor of SE, JQ1 binds to and inhibits the bromodomain containing 4 (BRD4) and preferentially affects coding genes with SEs [54]. The BRD4 inhibitor JQ1 has been shown to suppress the tumor cell proliferation [55, 56]. Therefore, we examined the effect of JQ1 on the HAS3-SE-associated transcription and NPC cells proliferation. At first, we detected HAS3 mRNA level in HK1 and FaDu cells after treated with JQ1. Quantitative PCR analysis indicated that JQ1 treatment decreased the levels of SE-associated transcript HAS3, in HK1 and FaDu cells in a dose-dependent manner (Figure 2B). Secondly, we compared the proliferation of JQ1 treated cells and control cells. CCK8 assays revealed that JQ1 inhibited the growth of the HK1 and FaDu cell lines in a dose- and time-dependent manner (Figure 2C). Moreover, migration and invasion assays showed that with increasing of dose and time, JQ1 decreased the amount of HK1 and FaDu cells migrated and invaded from the Transwell upper inserts to the lower surface of the membrane significantly (Figure 2D). These results point to the importance of SE-associated transcription for NPC cell proliferation and invasiveness.
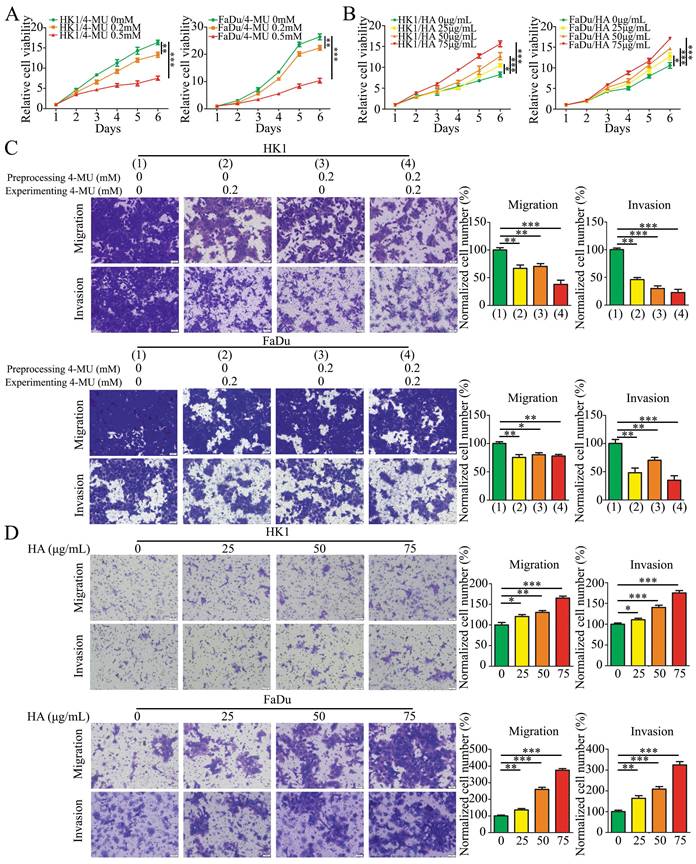
HA promoted NPC cells proliferation, migration, and invasion
To examine whether the HA promotes the malignant biological behaviors of NPC cells, we tested the effects of HA and 4-MU by CCK-8 and Transwell migration and invasion assays. 4-MU is an HA synthesis inhibitor that acts by depleting a common substrate for HASs, the cellular UDP-glucuronic acid (UDP-GlcUA). The effects of 4-MU on cell proliferation were shown that 4-MU inhibited cell proliferation in a concentration-dependent manner in HK1 and FaDu cells (Figure 3A). In addition, HK1 and FaDu cells were treated with 4-MU at 0.2 mM concentration before or/and during the migration and invasion assays experiment. The results showed that 4-MU suppressed the amount of HK1 and FaDu cells migration and invasion (Figure 3C). On the contrary, the results for HA were completely different. The growth of HK1 and FaDu cells was significantly promoted by HA in a concentration-dependent manner (Figure 3B). We also demonstrated that HA in media promoted additional migration and invasion in a concentration-dependent manner of HK1 and FaDu cells (Figure 3D).
HA promoted NPC cells proliferation, migration, and invasion. (A and B) CCK8 assay showed that 4-MU (A) inhibited cell growth, while HA (B) promoted cell growth. Mean ± SD, n = 3. (C) The migration and invasion of HK1 and FaDu cells treated by 4-MU pre-cell seeding or/and post-cell seeding (× 20). Mean ± SD, n = 3. (D) The migration and invasion of HK1 and FaDu cells treated by HA with different concentrations (× 20). Mean ± SD, n = 3. *P < 0.05, **P < 0.01, or ***P < 0.001.
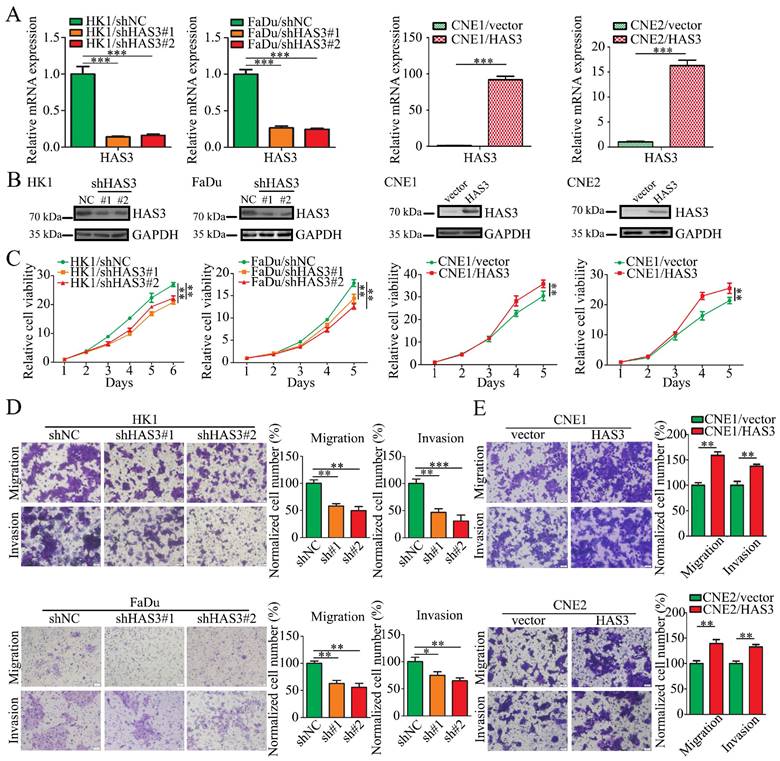
HAS3 promoted malignant potential of NPC cell in vitro
To determine the role of HAS3 protein in NPC cells, endogenous HAS3 expression was depleted in HK1 or FaDu cells by shRNAs expressing lentivirus. Alternatively, a HAS3 cDNA-expressing lentivirus was used to establish gain-of-function models in CNE1 and CNE2 cells. The mRNA expression levels of HAS3 in the gain- or loss-of-function models were measured by qPCR (Figure 4A). And protein levels in lentivirus-infected cells were determined using western blotting (Figure 4B). The effect of HAS3 expression on NPC cells growth, motility and invasiveness was measured. As evidenced by the CCK-8 assay, silencing HAS3 in HK1 and FaDu cells slowed cell growth in vitro, but re-expression of HAS3 in CNE1 and CNE2 NPC cell lines promoted cell proliferation (Figure 4C). We also performed Boyden Chamber migration and invasion assays and demonstrated that genetically silencing HAS3 in HK1 and FaDu cells inhibited cell metastatic and invasiveness (Figure 4D), while re-expression of HAS3 enhanced malignant potential of CNE1 and CNE2 NPC cells in vitro (Figure 4E).
Loss or gain of function of HAS3 in NPC cell. (A) The mRNA levels of HAS3 in loss-of-function or gain-of-function NPC cell models were measured using real-time qPCR. Mean ± SD, n = 3. (B) The protein levels of HAS3 in loss-of-function or gain-of-function NPC cell models were measured using western blotting. (C) Cell growth and proliferation measured using CCK8 assay. Mean ± SD, n = 3. (D and E) Cell migration and invasion measured using Transwell migration and invasion assay (× 20). Mean ± SD, n = 3. *P < 0.05, **P < 0.01, or ***P < 0.001.
Loss of HAS3 decreased tumor metastasis in vivo
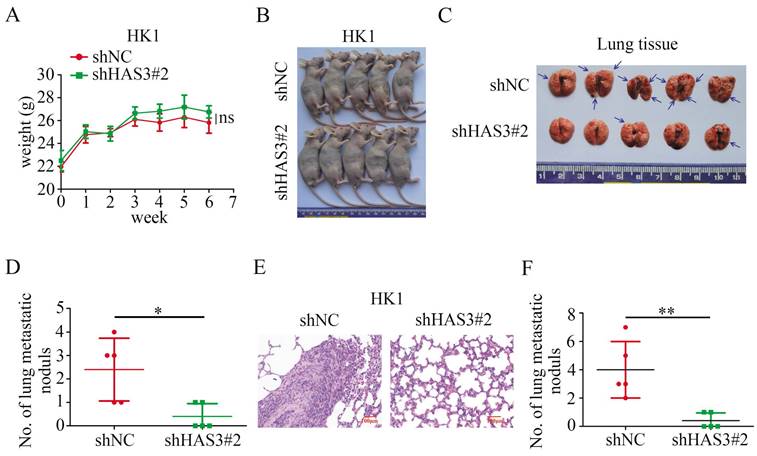
We then asked whether HAS3 promotes NPC cells metastatic potential in vivo. 1 × 106 HK1 cells stably transfected with shHAS3 or empty vector were injected into nude mice through tail vein to evaluate the lung metastatic potential for 6 weeks (Figure 5A and B). Compared with the control group, less and smaller tumor nodules in the lung of shHAS3 groups, indicating that loss of HAS3 suppressed the metastatic ability of HK1 cells (Figure 5C and D). Moreover, tumor formation in lung tissue was examined by H&E staining, which showed that loss of HAS3 inhibited HK1 NPC cell differentiation with a decreased nucleus-to-cytoplasm ratio, but control cells result in severe tumor formation in the lung (Figure 5E and F). These data indicate that HAS3 plays a role as a bona fide tumor promotor in NPC.
Loss of HAS3 suppressed NPC cell metastasis in vivo. (A) Body weight change curve in nude mice after tail vein injection. (B) The macroscopic view of nude mice at the endpoint of experiment. (C) Nude mice metastasis assay showed the macroscopic view of mice lung at 6 weeks after tail vein injection of shNC and shHAS3 NPC HK1 cells. Arrow: metastatic lung nodules. (D) The number of lung metastatic tumor nodules in mice were counted on the surface (n = 5, respectively). (E) Representative H&E staining of mice lung metastatic tumors are shown. (F) H&E staining of metastatic tumors in lung tissues and node counts. In the graph, the scatter diagram shows the number of metastatic nodules in 5 tissue sections from each group. ns, no significance, *P < 0.05, **P < 0.01.
JQ1 and 4-MU inhibited the expression of EGFR and downstream effectors on HK1 cell line
In HK1 cells, inhibition of phosphorylated (p)-EGFR was obtained exposure to JQ1, at a concentration of 50 nM. Concomitant with a downstream effect yields a decrease in p-AKT and p-ERK1/2 protein expression, but JQ1 treatment did not affect the total protein levels of EGFR, AKT and ERK1/2 (Figure 6A).
JQ1, 4-MU and HAS3 have influence on the level of EGFR and downstream effectors expression on NPC cell lines. (A) p-EGFR, EGFR, p-AKT, AKT, p-ERK1/2, and ERK1/2 expression in HK1 cellular extracts were treated with JQ1 at 50 nM and 100 nM concentration. (B) Western blotting was applied to detect the protein level of p-EGFR, EGFR, p-AKT, AKT, p-ERK1/2, and ERK1/2 after 4-MU treatment at 0.2 mM and 0.5 mM concentration in HK1 cells. (C) Protein expression of p-EGFR, EGFR, p-AKT, AKT, p-ERK1/2, and ERK1/2 were detected by western blotting in HK1 cells treated with or without 4-MU and HA. (D and E) HAS3, p-EGFR, EGFR, p-AKT, AKT, p-ERK1/2, and ERK1/2 protein levels in HAS3-silenced HK1 and FaDu NPC cells (D) and in HAS3 overexpression CNE1 and CNE2 NPC cells (E) were measured by western blotting.
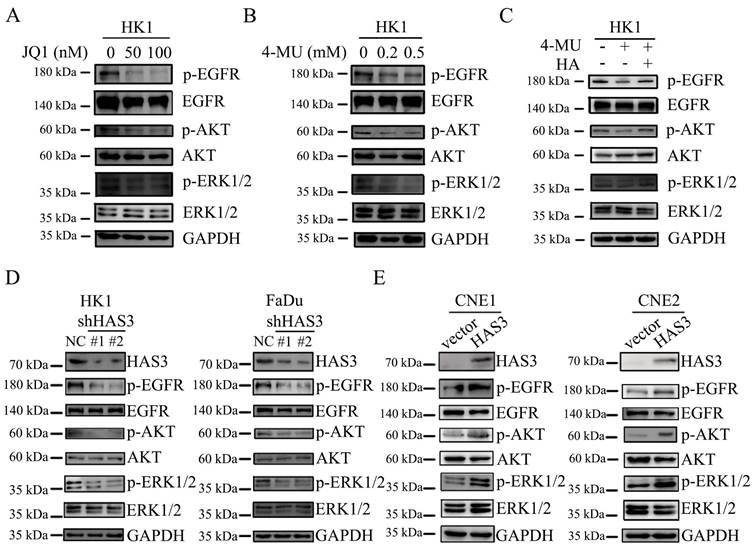
It has been established that HA promotes EGFR-mediated signaling and EGFR phosphorylation in HNSC [38]. Therefore, we utilized HA and HA synthesis inhibitor 4-MU to conduct subsequent experiments. Exposure of the NPC cell line HK1 to 4-MU, at a concentration of 0.2 mM and 0.5 mM resulted in a decrease in p-EGFR expression level. Concomitant with a downstream effect yields a decrease in p-AKT and p-ERK1/2 protein expression (Figure 6B). However, the addition of HA on 4-MU treated HK1 cells had effect on p-EGFR, p-AKT and p-ERK1/2, and restored protein expression (Figure 6C).
HAS3 promoted EGFR and downstream effectors expression on NPC cell lines
We next examined whether HAS3 expression affected the downstream signaling of EGFR. The p-EGFR, EGFR, p-AKT, AKT, p-ERK1/2 and ERK1/2 protein levels in loss or gain of function of HAS3 NPC cells were measured by western blotting assay. As shown in Figure 6, shRNA-dependent loss of function of HAS3 in HK1 and FaDu cells led to a reduction in the levels of p-EGFR and downstream p-AKT and p-ERK1/2 (Figure 6D). By contrast, HAS3 expression in CNE1 and CNE2 cells resulted in a significant elevation of p-EGFR, p-AKT, and p-ERK1/2 (Figure 6E). Neither loss nor gain of function of HAS3 affected the total protein levels of EGFR, AKT and ERK1/2 (Figure 6D and E).
Discussion
Our study showed that HAS3 is overexpressed in NPC and driven by SE. High expression of HAS3 promoted malignant progression of NPC in vitro and in vivo. Exogenous HA treatment or HAS3 also activated EGFR/AKT/ERK signaling pathway.
NPC is characterized by high invasion and early metastasis, 70-80% of NPC patients have developed cervical lymph node metastases at the first diagnosis [2-5]. Cancer research has been elucidating mechanisms of metastasis for decades [57, 58]. However, the underlying mechanism of NPC metastasis is still not clear. Representative characteristics of NPC include comparatively low mutation rate, widespread hypermethylation, high copy number alterations and chromosome abnormalities [6-14]. Thus, the exploration of epigenetic regulation is a promising direction in the field of the processes evolve on NPC.
Since in tumor development and progression, SEs often drive oncogene overexpression and may provide an opportunity to identify oncogenes behaviors [19-21]. Based on this strategy, in previous work (Jing Cai et al. 2019) [46], we built the SE landscapes in HK1 and C666-1 NPC cells, and identified HAS3 as a SE-driven oncogene from the analyzed data by stitching H3K27ac peaks using the ROSE algorithm. To prove the transcription of HAS3 is associated with SE, JQ1, the wide spectrum inhibitor of SE was used. We found that JQ1 treatment resulted in downregulation of HAS3 in HK1 and FaDu NPC cells in a dose-dependent manner and inhibition of cell proliferation and invasiveness in NPC cells. In addition, results showed that JQ1 treatment inhibited the activation of the EGFR/AKT/ERK pathway in HK1 NPC cells.
In several cancer types, HA is important for tumorigenesis and tumor progression [59, 60]. HA synthesis is one of the important mechanisms for HA accumulation, which is associated with HAS family members in tumor [61, 62]. It has previously been reported that the expression levels of HAS3 were higher than that of HAS1 and HAS2 in most oral cancers, and HAS3 was more catalytically active in HA synthesis than HAS1 and HAS2 [36]. In this study, we found that NPC cells expressed high levels of HAS3 mRNA and protein, which is in contrast to the normal nasopharynx epithelium (NPE) cell lines. Inhibition of HA synthesis by HAS inhibitor 4-MU suppressed cells growth and invasion of HK1 and FaDu NPC cells in vitro, whereas HA treatment exerted opposite effects. To some extent, these results are consistent with previous studies on that HA promotes tumor cells proliferation and invasiveness. Furthermore, HAS3 expression correlate to malignant transformation [63] and the overexpression of HAS3 in several cancer types, such as prostate cancer, osteosarcoma and colon carcinoma is known to be associated with higher malignancy or metastasis [64-66]. According to our observation, genetically silencing HAS3 in HK1 and FaDu NPC cells attenuated cell proliferation and mobility, while re-expression of HAS3 enhanced malignant potential of CNE1 and CNE2 NPC cells. And the effect of HAS3 overexpression was comparable to that of the enough additional HA supplementation. In addition, loss of HAS3 impaired metastatic potential of HK1 cells in nude mice. Thus, we demonstrated for the first time that HAS3 promotes malignant progression of NPC.
Consistent with the HA-mediated activation of EGFR signaling [38], previous research has demonstrated that HAS3 exerts its biological functions in tumor via activation of EGFR related pathways [36]. The mutation of EGFR gene tumorigenic mutation is a cause of NPC [8]. And the activation of EGFR signaling pathways promote carcinogenesis, by increasing tumor cell proliferation, migration, angiogenesis and apoptosis inhibition [67]. Therefore, in this study, we explored EGFR and its downstream signaling pathways in NPC. Western blotting analysis showed that 4-MU treatment did not affect the total protein levels of EGFR, AKT and ERK, but reduced the phosphorylated protein level of EGFR, AKT and ERK in a dose-dependent manner, respectively. In contrast, exogenous HA treatment activated EGFR/AKT/ERK signaling cascade. Furthermore, the effect of HAS3 on EGFR/AKT/ERK pathway was further investigated in this study through directly knockdown or overexpression of HAS3 expression. And the results showed that loss of HAS3 protein inhibited the expression of the phosphorylation of EGFR, AKT and ERK proteins in HK1 and FaDu cells, but HAS3 overexpression promoted EGFR/AKT/ERK signaling pathway in CNE1 and CNE2 cell lines. In summary, all these results demonstrated that HAS3 expression induces the activation of the EGFR/AKT/ERK pathway in NPC.
In conclusion, we depicted for the first time that HAS3 is driven by SE and overexpressed in NPC. High expression of HAS3 promotes the malignant features of NPC in vitro and in vivo. In addition, this study mechanistically explaining the role of HAS3 in cancer development. Exogenous HA or overexpression of HAS3 promotes activation of the EGFR/AKT/ERK pathway, but HA inhibition or HAS3 silencing attenuates activation of the EGFR/AKT/ERK pathway in NPC. In summary, this study highlights the role of SE-driven oncogene HAS3 which promotes malignant progression of NPC and complements the mechanism and provide a novel approach for identifying oncogenic factors in NPC development.
Abbreviations
NPC: nasopharyngeal carcinoma; EGFR: epidermal growth factor receptor; SEs: super enhancers; TFs: transcription factors; TEs: typical enhancers; HAS3: hyaluronan synthase 3; ECM: extracellular matrix; HA: hyaluronan; HAS: hyaluronan synthase; EGFR: epidermal growth factor receptor; ESCC: esophageal squamous cell carcinoma; EGF: epidermal growth factor; ROSE: ranking of super-enhancer; qPCR: real-time Reverse-transcription PCR; CCK-8: Cell Counting Kit-8; TCGA: The Cancer Genome Atlas; 4-MU: 4-Methylumbelliferone; NPE: normal nasopharynx epithelium.
Acknowledgements
This study was supported in part by grants from The National Natural Science Foundation of China (82173201, 82272631, 82072596, 81872278, 82173339, 82172766), the National “111” Project (Project #111-2-12), the Hunan Provincial Key Research and Development Program (2022SK2026), the Natural Science Foundation of Hunan Province, China (2020JJ4920, 2020JJ4838, 2020JJ4766, 2020JJ3055, 2022JJ10096), the Scientific Research Project of Hunan Provincial Health Commission (20201067, 20201040, 20201047), the Beijing Xisike Clinical Oncology Research Foundation (Y-HR2020ZD-0052), the Open Funds of State Key Laboratory of Oncology in south China (HN2021-07), and the Open Funds of Xuzhou Medical University (XYKF202118).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chua MLK, Wee JTS, Hui EP, Chan ATC. Nasopharyngeal carcinoma. Lancet. 2016;387:1012-1024
2. Sham JS, Choy D, Wei WI. Nasopharyngeal carcinoma: orderly neck node spread. Int J Radiat Oncol Biol Phys. 1990;19:929-933
3. Ho FC, Tham IW, Earnest A, Lee KM, Lu JJ. Patterns of regional lymph node metastasis of nasopharyngeal carcinoma: a meta-analysis of clinical evidence. BMC Cancer. 2012;12:98
4. Zhao Y, Liao X, Wang Y, Lan W, Ren J, Yang N. et al. Level Ib CTV delineation in nasopharyngeal carcinoma based on lymph node distribution and topographic anatomy. Radiother Oncol. 2022;172:10-17
5. Li DK, Chen XR, Wang LN, Wang JH, Wen YT, Zhou ZY. et al. Epstein-Barr Virus Induces Lymphangiogenesis and Lympth Node Metastasis via Upregulation of VEGF-C in Nasopharyngeal Carcinoma. Mol Cancer Res. 2022;20:161-175
6. Lo KW, Mok CH, Huang DP, Liu YX, Choi PH, Lee JC. et al. p53 mutation in human nasopharyngeal carcinomas. Anticancer Res. 1992;12:1957-1963
7. Spruck CH 3rd, Tsai YC, Huang DP, Yang AS, Rideout WM 3rd, Gonzalez-Zulueta M. et al. Absence of p53 gene mutations in primary nasopharyngeal carcinomas. Cancer Res. 1992;52:4787-4790
8. Lin DC, Meng X, Hazawa M, Nagata Y, Varela AM, Xu L. et al. The genomic landscape of nasopharyngeal carcinoma. Nat Genet. 2014;46:866-871
9. Dai W, Cheung AK, Ko JM, Cheng Y, Zheng H, Ngan RK. et al. Comparative methylome analysis in solid tumors reveals aberrant methylation at chromosome 6p in nasopharyngeal carcinoma. Cancer Med. 2015;4:1079-1090
10. Lo KW, Chung GT, To KF. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin Cancer Biol. 2012;22:79-86
11. Chen J, Fu L, Zhang LY, Kwong DL, Yan L, Guan XY. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin J Cancer. 2012;31:215-222
12. Chen YJ, Ko JY, Chen PJ, Shu CH, Hsu MT, Tsai SF. et al. Chromosomal aberrations in nasopharyngeal carcinoma analyzed by comparative genomic hybridization. Genes Chromosomes Cancer. 1999;25:169-175
13. Chien G, Yuen PW, Kwong D, Kwong YL. Comparative genomic hybridization analysis of nasopharygeal carcinoma: consistent patterns of genetic aberrations and clinicopathological correlations. Cancer Genet Cytogenet. 2001;126:63-67
14. Hui AB, Or YY, Takano H, Tsang RK, To KF, Guan XY. et al. Array-based comparative genomic hybridization analysis identified cyclin D1 as a target oncogene at 11q13.3 in nasopharyngeal carcinoma. Cancer Res. 2005;65:8125-8133
15. Li YY, Chung GT, Lui VW, To KF, Ma BB, Chow C. et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat Commun. 2017;8:14121
16. Zheng H, Dai W, Cheung AK, Ko JM, Kan R, Wong BW. et al. Whole-exome sequencing identifies multiple loss-of-function mutations of NF-kappaB pathway regulators in nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 2016;113:11283-11288
17. Yi M, Tan Y, Wang L, Cai J, Li X, Zeng Z. et al. TP63 links chromatin remodeling and enhancer reprogramming to epidermal differentiation and squamous cell carcinoma development. Cell Mol Life Sci. 2020;77:4325-4346
18. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307-319
19. Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW. et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature. 2018;553:515-520
20. Betancur PA, Abraham BJ, Yiu YY, Willingham SB, Khameneh F, Zarnegar M. et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat Commun. 2017;8:14802
21. Sanchez GJ, Richmond PA, Bunker EN, Karman SS, Azofeifa J, Garnett AT. et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018;46:1756-1776
22. Masellis-Smith A, Belch AR, Mant MJ, Turley EA, Pilarski LM. Hyaluronan-dependent motility of B cells and leukemic plasma cells in blood, but not of bone marrow plasma cells, in multiple myeloma: alternate use of receptor for hyaluronan-mediated motility (RHAMM) and CD44. Blood. 1996;87:1891-1899
23. Till KJ, Zuzel M, Cawley JC. The role of hyaluronan and interleukin 8 in the migration of chronic lymphocytic leukemia cells within lymphoreticular tissues. Cancer Res. 1999;59:4419-4426
24. Wu M, Cao M, He Y, Liu Y, Yang C, Du Y. et al. A novel role of low molecular weight hyaluronan in breast cancer metastasis. FASEB J. 2015;29:1290-1298
25. Zhang Qinming LX, Tang Weiping. Effect of hyaluronic acid on proliferation of CNE-2Z cells and the growth of implanted tumor in nude mice. Journal of Practical Oncology. 1999;14:14-17
26. Jacobson A, Brinck J, Briskin MJ, Spicer AP, Heldin P. Expression of human hyaluronan synthases in response to external stimuli. Biochem J. 2000 348 Pt 1: 29-35
27. Kultti A, Zhao C, Singha NC, Zimmerman S, Osgood RJ, Symons R. et al. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. Biomed Res Int. 2014;2014:817613
28. Liu N, Gao F, Han Z, Xu X, Underhill CB, Zhang L. Hyaluronan synthase 3 overexpression promotes the growth of TSU prostate cancer cells. Cancer Res. 2001;61:5207-5214
29. Bullard KM, Kim HR, Wheeler MA, Wilson CM, Neudauer CL, Simpson MA. et al. Hyaluronan synthase-3 is upregulated in metastatic colon carcinoma cells and manipulation of expression alters matrix retention and cellular growth. Int J Cancer. 2003;107:739-746
30. Twarock S, Freudenberger T, Poscher E, Dai G, Jannasch K, Dullin C. et al. Inhibition of oesophageal squamous cell carcinoma progression by in vivo targeting of hyaluronan synthesis. Mol Cancer. 2011;10:30
31. Song JM, Im J, Nho RS, Han YH, Upadhyaya P, Kassie F. Hyaluronan-CD44/RHAMM interaction-dependent cell proliferation and survival in lung cancer cells. Mol Carcinog. 2019;58:321-333
32. Chua DT, Nicholls JM, Sham JS, Au GK. Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:11-20
33. Leong JL, Loh KS, Putti TC, Goh BC, Tan LK. Epidermal growth factor receptor in undifferentiated carcinoma of the nasopharynx. Laryngoscope. 2004;114:153-157
34. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159-3167
35. Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637-643
36. Kuo YZ, Fang WY, Huang CC, Tsai ST, Wang YC, Yang CL. et al. Hyaluronan synthase 3 mediated oncogenic action through forming inter-regulation loop with tumor necrosis factor alpha in oral cancer. Oncotarget. 2017;8:15563-15583
37. Bourguignon LYW, Zhu HB, Shao LJ, Chen YW. CD44 interaction with c-Src kinase promotes cortactin-mediated cytoskeleton function and hyaluronic acid-dependent ovarian tumor cell migration. J Biol Chem. 2001;276:7327-7336
38. Wang SJ, Bourguignon LYW. Hyaluronan and the interaction between CD44 and epidermal growth factor receptor in oncogenic signaling and chemotherapy resistance in head and neck cancer. Arch Otolaryngol. 2006;132:771-778
39. Tsao SW, Wang X, Liu Y, Cheung YC, Feng H, Zheng Z. et al. Establishment of two immortalized nasopharyngeal epithelial cell lines using SV40 large T and HPV16E6/E7 viral oncogenes. Biochim Biophys Acta. 2002;1590:150-158
40. Chan SY, Choy KW, Tsao SW, Tao Q, Tang T, Chung GT. et al. Authentication of nasopharyngeal carcinoma tumor lines. Int J Cancer. 2008;122:2169-2171
41. Sun Y, Chen X, Zhou Y, Qiu S, Wu Y, Xie M. et al. Metformin reverses the drug resistance of cisplatin in irradiated CNE-1 human nasopharyngeal carcinoma cells through PECAM-1 mediated MRPs down-regulation. Int J Med Sci. 2020;17:2416-2426
42. Peng Q, Zhang L, Li J, Wang W, Cai J, Ban Y. et al. FOXA1 Suppresses the Growth, Migration, and Invasion of Nasopharyngeal Carcinoma Cells through Repressing miR-100-5p and miR-125b-5p. J Cancer. 2020;11:2485-2495
43. Cheung ST, Huang DP, Hui AB, Lo KW, Ko CW, Tsang YS. et al. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int J Cancer. 1999;83:121-126
44. Cai J, Yi M, Tan Y, Li X, Li G, Zeng Z. et al. Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-IotaIota. J Exp Clin Cancer Res. 2021;40:190
45. Chen S, Youhong T, Tan Y, He Y, Ban Y, Cai J. et al. EGFR-PKM2 signaling promotes the metastatic potential of nasopharyngeal carcinoma through induction of FOSL1 and ANTXR2. Carcinogenesis. 2020;41:723-733
46. Cai J, Chen S, Yi M, Tan Y, Peng Q, Ban Y. et al. DeltaNp63alpha is a super enhancer-enriched master factor controlling the basal-to-luminal differentiation transcriptional program and gene regulatory networks in nasopharyngeal carcinoma. Carcinogenesis. 2020;41:1282-1293
47. Li J, Zhang Y, Wang L, Li M, Yang J, Chen P. et al. FOXA1 prevents nutrients deprivation induced autophagic cell death through inducing loss of imprinting of IGF2 in lung adenocarcinoma. Cell Death Dis. 2022;13:711
48. Cai J, Liu C, Yi M, Tan Y, Chen S, Ren N. et al. The tumor suppressor NOR1 suppresses cell growth, invasiveness, and tumorigenicity in glioma. Neoplasma. 2020;67:851-860
49. Ban Y, Tan P, Cai J, Li J, Hu M, Zhou Y. et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol Oncol. 2020;14:1282-1296
50. Li J, Wang W, Chen S, Cai J, Ban Y, Peng Q. et al. FOXA1 reprograms the TGF-beta-stimulated transcriptional program from a metastasis promoter to a tumor suppressor in nasopharyngeal carcinoma. Cancer Lett. 2019;442:1-14
51. Ban Y, Tan Y, Li X, Li X, Zeng Z, Xiong W. et al. RNA-binding protein YBX1 promotes cell proliferation and invasiveness of nasopharyngeal carcinoma cells via binding to AURKA mRNA. J Cancer. 2021;12:3315-3324
52. Wu P, Hou X, Peng M, Deng X, Yan Q, Fan C. et al. Circular RNA circRILPL1 promotes nasopharyngeal carcinoma malignant progression by activating the Hippo-YAP signaling pathway. Cell Death Differ. 2023
53. Mo Y, Wang Y, Wang Y, Deng X, Yan Q, Fan C. et al. Circular RNA circPVT1 promotes nasopharyngeal carcinoma metastasis via the beta-TrCP/c-Myc/SRSF1 positive feedback loop. Mol Cancer. 2022;21:192
54. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320-334
55. Dong X, Hu X, Chen J, Hu D, Chen LF. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 2018;9:203
56. Wang JY, Liu ZT, Wang ZQ, Wang SB, Chen ZH, Li ZW. et al. Targeting c-Myc: JQ1 as a promising option for c-Myc-amplified esophageal squamous cell carcinoma. Cancer Lett. 2018;419:64-74
57. Liu P, Wang Z, Ou X, Wu P, Zhang Y, Wu S. et al. The FUS/circEZH2/KLF5/ feedback loop contributes to CXCR4-induced liver metastasis of breast cancer by enhancing epithelial-mesenchymal transition. Mol Cancer. 2022;21:198
58. Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H. et al. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350:985-990
59. Sironen RK, Tammi M, Tammi R, Auvinen PK, Anttila M, Kosma VM. Hyaluronan in human malignancies. Exp Cell Res. 2011;317:383-391
60. Knudson W, Biswas C, Li XQ, Nemec RE, Toole BP. The role and regulation of tumour-associated hyaluronan. Ciba Found Symp. 1989;143:150-159 discussion 159-169, 281-155
61. Triggs-Raine B, Natowicz MR. Biology of hyaluronan: Insights from genetic disorders of hyaluronan metabolism. World J Biol Chem. 2015;6:110-120
62. Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin Cancer Biol. 2008;18:288-295
63. Skandalis SS, Karalis T, Heldin P. Intracellular hyaluronan: Importance for cellular functions. Semin Cancer Biol. 2020;62:20-30
64. Bharadwaj AG, Rector K, Simpson MA. Inducible hyaluronan production reveals differential effects on prostate tumor cell growth and tumor angiogenesis. J Biol Chem. 2007;282:20561-20572
65. Tofuku K, Yokouchi M, Murayama T, Minami S, Komiya S. HAS3-related hyaluronan enhances biological activities necessary for metastasis of osteosarcoma cells. Int J Oncol. 2006;29:175-183
66. Kim HR, Wheeler MA, Wilson CM, Iida J, Eng D, Simpson MA. et al. Hyaluronan facilitates invasion of colon carcinoma cells in vitro via interaction with CD44. Cancer Res. 2004;64:4569-4576
67. Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25-79
Author contact
![]() Corresponding authors: Mei Yi, Department of Dermatology; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008 Hunan, China. E-mail: yi_meiedu.cn. Pan Chen, Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha 410013 Hunan, China. E-mail: chenpanorg.cn.
Corresponding authors: Mei Yi, Department of Dermatology; National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008 Hunan, China. E-mail: yi_meiedu.cn. Pan Chen, Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha 410013 Hunan, China. E-mail: chenpanorg.cn.