Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2024; 15(17):5643-5654. doi:10.7150/jca.99990 This issue Cite
Research Paper
Causal association of plasma lipidome with lung carcinoma and mediating role of inflammatory proteins: evidence from Mendelian randomization analysis
Haihao Yan1, Jiao Feng1, Xiao Jin1, Yuanyuan Zhang1, Cui Bao1, Chenghua Zhu2, ![]() , Ganzhu Feng1,
, Ganzhu Feng1, ![]()
1. Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Nanjing Medical University, Nanjing 210011, China.
2. Department of Respiratory Medicine, Nanjing Pukou Hospital of TCM, Pukou Hospital of Chinese Medicine affiliated to China Pharmaceutical University, Nanjing 210000, China.
Received 2024-6-23; Accepted 2024-8-22; Published 2024-9-3
Abstract

The evidence from clinical studies suggests that lung carcinoma (LC) patients exhibit dysregulation in lipid metabolism. However, the causal relationship between plasma lipidome and LC, and whether inflammatory proteins mediate, remains to be determined. Genetic data for 179 plasma lipids and 91 inflammatory proteins were obtained from the latest published genome-wide association studies. Genetic data on LC and subtypes were from the largest available meta-analysis. The causal relationship between plasma lipidome and LC was determined by the two-sample Mendelian randomization (MR) method. Mediation MR analysis was employed to ascertain whether inflammatory proteins mediate the impact of plasma lipidome on LC. We identified 39 causal relationships between genetically predicted plasma lipidome and LC and subtypes. These relationships involve the influence of phosphatidylcholines, phosphatidylethanolamines, diacylglycerols, triacylglycerols, sphingomyelins, and Sterol esters. Additionally, the mediating role of 5 inflammatory proteins in the causal relationship between plasma lipidome and LC and subtypes was determined. Our results highlight the complex network of plasma lipidome and inflammatory proteins regulating LC. Integrating plasma lipidome and inflammatory proteins into clinical practice may open new avenues for the prevention and treatment of LC.
Keywords: plasma lipidome, inflammatory proteins, lung carcinoma, Mendelian randomization
Introduction
Lung carcinoma (LC) is a highly lethal systemic invasive disease often associated with systemic metabolic dysregulation. Currently, LC remains a leading cause of cancer-related deaths globally, ranking second in incidence among all cancers [1].
Lipids are a highly diverse class of molecules, including phosphatidylcholines (PC), phosphatidylethanolamines (PE), diacylglycerols (DAG), triacylglycerols (TAG), sphingomyelins (SM), phosphatidylinositols (PI), cholesterol esters (CE), and sterol esters (SE). Lipids regulate and perform various biological functions, such as energy storage, maintenance of normal cell structure, involvement in cell signaling pathways, and regulation of inflammatory responses [2]. Many studies have confirmed the presence of lipid metabolism dysregulation in LC patients. Circulating levels of PC in early-stage non-small cell lung carcinoma (NSCLC) patients show significant differences compared to healthy control groups [3]. Heterogeneity in circulating lipidomic profiles has also been observed among patients with different subtypes of LC, such as lung adenocarcinoma (LADC), squamous cell carcinoma (SqCLC), or small cell lung carcinoma (SCLC) [4]. Moreover, a significant correlation exists between the lipidomic profiles and the genome of lipid-associated proteins [4].
One of the key roles of lipids in maintaining metabolic homeostasis is regulating inflammatory responses. Different lipids may either promote or attenuate the inflammatory process (such as prostaglandins, leukotrienes, and endocannabinoid signaling). Observational evidence suggests that chronic inflammation is associated with the development of LC [5, 6]. Prospective cohort studies indicate that specific circulating inflammatory proteins, such as Interleukin (IL)-6, IL-8, and C-reactive protein (CRP), are associated with an increased LC risk [5, 7, 8]. Elevated serum concentrations of monokine induced by gamma interferon (MIG) and serum amyloid A (SAA) prior to diagnosis are also positively correlated with the risk of LC [9].
The evidence of the above observational studies suggests that lipids and inflammatory proteins may be associated with the LC risk. Inflammatory protein may be the mediating factor of lipids affecting LC. However, traditional observational designs are susceptible to residual confounding and reverse causality. Even with appropriate statistical methods, the impact of these biases remains unavoidable. Therefore, translating the results of observational studies into practical cancer control strategies still presents significant challenges.
Mendelian Randomization (MR) utilizes germline genetic variants as genetic instruments to proxy lifelong exposure to risk factors (such as lipids and inflammatory proteins) [10, 11]. One of the advantages of MR is its ability to overcome the limitations of traditional epidemiology, thereby strengthening the evidence for potential causal effects of risk factors on cancer risk. Another advantage of MR is its consideration of the long-term effects of risk factors on cancer risk, as there may be a considerable exposure period from exposure to specific risk factors to the occurrence of cancer.
This study used an MR design to explore the causal relationship between plasma lipidome and LC and subtypes, including LADC, SqCLC, and SCLC. Mediated MR was used to evaluate whether inflammatory proteins serve as mediators of lipids affecting LC. Our study will expand insights into the impact of lipids and inflammatory proteins on the etiology of LC and support the development of LC prevention strategies.
Materials and methods
Study design
Our study meets the STROBE-MR criteria [12] (Supplementary Table S1) and consists of three main parts. Firstly, we determined whether 179 plasma lipids are associated with the risk of LC and subtypes. Secondly, we analyzed whether LC has a reverse causal effect on plasma lipidome. Finally, mediation MR analysis was conducted to ascertain whether inflammatory proteins mediate the causal relationship between plasma lipidome and LC. The overall design of this study is illustrated in Figure 1.
Mendelian randomization analysis flow chart. β, the total effect of lipids on LC, obtained using the IVW method, is denoted as β; β1, the effect of lipids on inflammatory proteins is denoted as β1; β2, the effect of inflammatory proteins on LC is denoted as β2. Abbreviations: SNPs, single nucleotide polymorphisms; GWAS, genome-wide association studies; MR, mendelian randomization; MR-PRESSP, the MR Pleiotropy RESidual Sum and Outlier.

Plasma lipidome
The genetic data for 179 plasma lipids were obtained from the latest genome-wide association studies (GWAS) based on the GeneRISK cohort. This cohort recruited 7,266 participants from southern Finland between 2015 and 2017, including 2,624 males and 4,642 females aged 45 to 66 years old. Blood samples were collected from fasting participants for serum, plasma, and DNA extraction. The lipidomic analysis based on mass spectrometry was conducted by Linotype GmbH (Dresden, Germany) to analyze the lipids of the participants. Ultimately, this study identified 495 genome-wide associations of plasma lipids at 56 genetic loci [13].
Inflammatory proteins
The genetic data for 91 inflammatory proteins were obtained from the latest GWAS conducted by Zhao et al. This study recruited 11 cohorts comprising a total of 14,824 participants of European ancestry. Protein profiling data for each cohort were generated by the Olink laboratory in Uppsala. The researchers used the Olink Target platform to measure 91 plasma proteins and performed genome-wide protein quantitative trait locus (pQTL) mapping and meta-analysis. A total of 180 pQTLs were identified (59 cis, 121 trans). This GWAS identified genetic determinants of inflammation-related proteins, which can aid in further understanding the etiology of immune-mediated LC [14].
LC and subtypes
The GWAS related to LC was derived from a meta-analysis based on the International Lung Carcinoma Consortium (ILCCO) and the Transdisciplinary Research in Cancer of the Lung (TRICL) consortium. It included a total of 85,716 participants of European ancestry, comprising 29,266 patients and 56,450 controls. Additionally, the histological types were further categorized into LADC, SqCLC, and SCLC. Supplementary Table S2 displayed all genetic data.
Screening genetic instruments
Genetic instruments for MR analysis need to meet three criteria: (1) they should be strongly associated with the exposure; (2) they should be unrelated to potential confounding variables; (3) they should only affect the outcome through the exposure. To obtain enough single nucleotide polymorphisms (SNPs) for MR analysis, for plasma lipidome and inflammatory proteins, we selected SNPs with p<1E-5. This threshold is also used in many MR studies [15, 16]. Then, genetic instruments in linkage disequilibrium (LD) were further excluded. Subsequent SNPs used for MR should meet the criteria of r2<0.001 and distance >10,000kb. Additionally, the F statistic corresponding to each SNP was calculated to exclude weak genetic instrument bias. SNPs with F statistics less than 10 were further excluded. Finally, we excluded palindromic SNPs to ensure consistent effects of genetic instruments on exposure and outcomes. The plasma lipidome, inflammatory proteins, and LC-associated SNPs were presented in Supplementary Tables S3, S4, and S5, respectively.
Forward MR analysis
In the primary analysis, the inverse variance-weighted (IVW) method was employed to assess the causal effects of 179 plasma lipids on LC and subtypes. Using the Wald ratio method, the individual impact of each genetic instrument on the outcome was calculated. The IVW method conducts a meta-analysis of the Wald ratios corresponding to each genetic instrument, and the results are presented as odds ratios (OR) and 95% confidence intervals (CI). The robustness of the IVW method was assessed using the MR-Egger, Weighted Median, Simple Mode, and Weighted Mode methods. These MR methods have been described in our previous study [17]. Due to the high correlation among the characteristics of plasma lipidome and inflammatory proteins involved in this study, which is based on a shared large number of SNPs, to correct for the "measurement pleiotropy" effect, we introduced the MR Bayesian model averaging (MR-BMA) method. The MR-BMA method is more suitable for detecting the effects of related biomarkers acting through the same causal pathway [18]. Statistical significance is considered only when the IVW method is consistent with other MR methods and when both the IVW and MR-BMA methods yield p-values less than 0.05.
Reverse MR analysis
Because mediation MR analysis requires excluding the influence of reverse causality, we tested the impact of LC and subtypes on changes in plasma lipidome. For LC and subtypes, we selected SNPs with p < 5E-8 for MR analysis. As mentioned earlier, we removed SNPs in LD and calculated the F-statistic. The methods used for MR analysis were consistent with those used in forward MR analysis.
Sensitivity analysis
Cochran's Q test was used to assess the heterogeneity among the genetic instruments corresponding to each exposure [19]. MR-Egger regression and MR-PRESSO were employed to detect whether MR analysis was influenced by potential horizontal pleiotropy [20, 21]. Results with heterogeneity or horizontal pleiotropy were further excluded.
Mediation MR analysis
The lipid for mediation analysis needs to meet the following conditions: (1) The lipid has a causal effect on LC or subtypes; (2) LC or subtypes do not have a reverse causal effect on the lipid. For lipids that meet these conditions, we first detect the causal effect of specific lipids on 91 inflammatory proteins. Next, we will determine the causal relationship between inflammatory proteins significantly influenced by specific lipids and LC or subtypes. Once the inflammatory proteins acting as mediators are identified, we will further calculate the direct effect of lipids on LC or subtypes, the indirect effect (mediation effect) mediated by inflammatory proteins, and the proportion of the effect of lipids on LC or subtypes explained by inflammatory proteins. We use the Coefficient product method for mediation analysis [22, 23]. The total effect of lipids on LC, obtained using the IVW method, is denoted as β. The effect of lipids on inflammatory proteins is denoted as β1, and the effect of inflammatory proteins on LC is denoted as β2. Here, the mediation effect exerted by inflammatory proteins is calculated as β1×β2. The direct effect is the effect of exposure on the outcome after excluding the influence of the mediator, calculated as β - β1×β2. The proportion of the mediation effect to the total effect is calculated as (β1×β2)/β. All analyses in this study were conducted in R (version 4.3.2). The R packages used include the "TwoSampleMR" package [24] and the "MR-PRESSO" package [21].
Results
Causal effects of plasma lipidome on LC and subtypes
LC
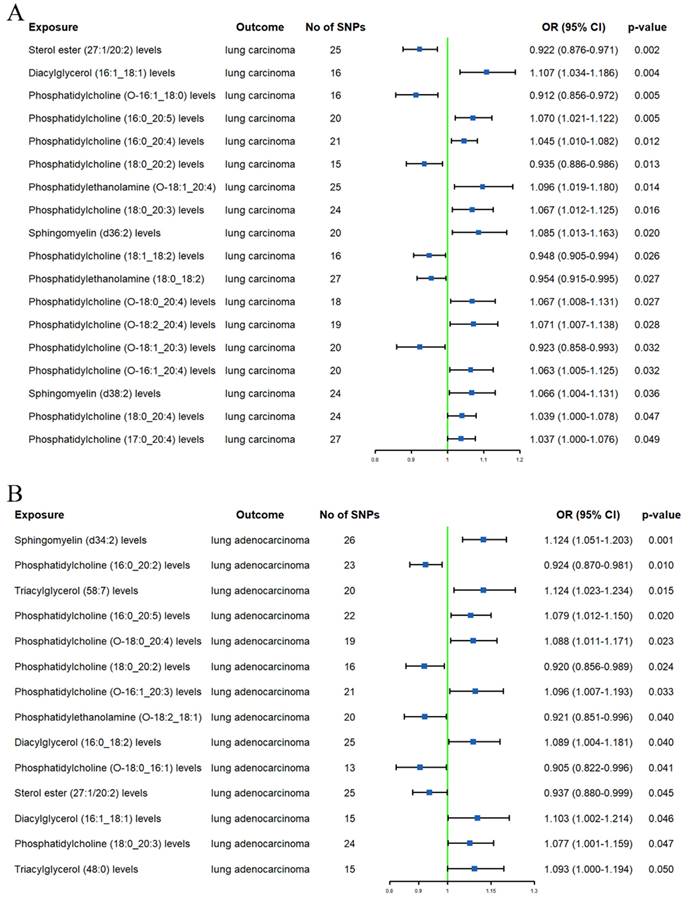
In the initial analysis, a total of 19 lipids were identified to have a causal relationship with LC (Supplementary Table S6). After using the MR-BMA method to remove lipids with non-significant results (p ≥ 0.05) (Supplementary Table S7), there were still 12 lipids associated with LC (Figure 2A). Specifically, an increase in genetically predicted SM (d36:2) levels (OR 1.085, 95% CI 1.013-1.163) and DAG (16:1_18:1) levels (OR 1.107, 95% CI 1.034-1.186) was associated with an increased risk of LC. However, an increase in SE (27:1/20:2) levels (OR 0.922, 95% CI 0.876-0.971) was associated with a decreased risk of LC. Additionally, PC (O-16:1_18:0), PC (18:0_20:2), PC (18:1_18:2), and PC (O-18:1_20:3) levels were found to be associated with a decreased LC risk (ORs = 0.912, 0.935, 0.948, and 0.923, respectively). In contrast, PC (16:0_20:5), PC (O-18:2_20:4), PC (16:0_20:4) levels, PC (O-16:1_20:4), and PC (17:0_20:4) levels increased the risk of LC (ORs = 1.070, 1.071, 1.045, 1.063, and 1.037, respectively). Additional MR methods (MR-Egger, Weighted Median, Simple Mode, and Weighted Mode) yielded consistent results with the IVW method, further confirming the robustness of our findings.
Forest plots of causal effect of plasma lipidome on lung carcinoma (A) and lung adenocarcinoma (B).
LADC
In the preliminary analysis, 14 lipids were found to be associated with LADC (Supplementary Table S6). After excluding lipids that did not pass the MR-BMA test (Supplementary Table S7), we finally identified 10 lipids (Figure 2B). The IVW method revealed that high standard deviation (SD) of TAG (48:0) (OR 1.093, 95% CI 1.000-1.194), TAG (58:7) (OR 1.124, 95% CI 1.023-1.234), DAG (16:0_18:2) (OR 1.089, 95% CI 1.004-1.181), and DAG (16:1_18:1) levels (OR 1.103, 95% CI 1.002-1.214) were associated with higher LADC risk. A high SD of SE (27:1/20:2) levels (OR 0.937, 95% CI 0.880-0.999) was associated with lower LADC risk. We also observed that higher PC (16:0_20:5) and PC (O-16:1_20:3) levels were associated with increased LADC risk (ORs = 1.079 and 1.096, respectively); however, higher PC (18:0_20:2) and PC (16:0_20:2) levels were associated with decreased LADC risk (ORs = 0.920 and 0.924, respectively). Additionally, a high SD of PE (O-18:2_18:1) levels (OR 0.921, 95% CI 0.851-0.996) was also associated with lower LADC risk. Furthermore, four additional MR analyses showed consistent directions with the main method.
SqCLC
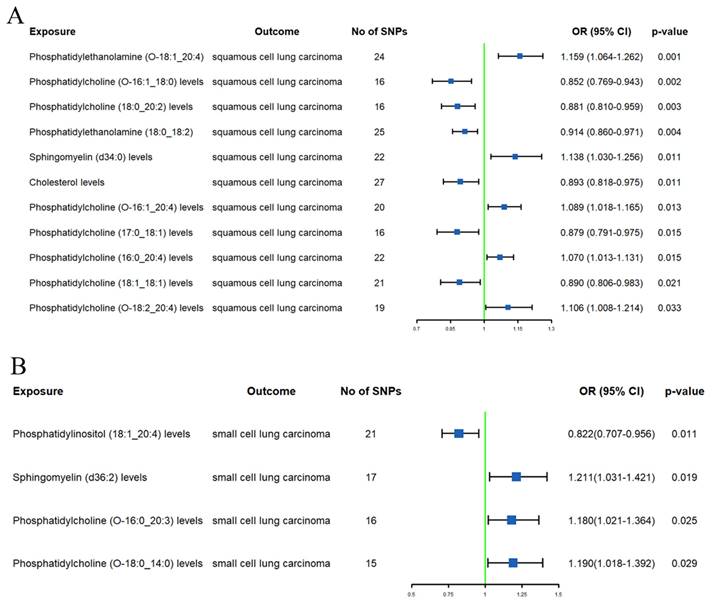
After excluding 1 lipid using MR-BMA (Supplementary Table S7), we identified 14 lipids with a causal relationship with SqCLC (Supplementary Table S6 and Figure 3A). High genetically predicted SM (d34:0) levels (OR 1.138, 95% CI 1.030-1.256) were associated with an increased SqCLC risk. Conversely, a high PE (O-18:1_20:4) (OR 0.822, 95% CI 0.707-0.956) and PE (18:0_18:2) (OR 0.914, 95% CI 0.860-0.971) levels were associated with a low SqCLC risk. Additionally, PC (O-16:1_20:4), PC (16:0_20:4), and PC (O-18:2_20:4) levels were associated with an increased SqCLC risk (ORs = 1.089, 1.070, and 1.106, respectively). A high SD of PC (18:0_20:2), PC (O-16:1_18:0), PC (18:1_18:1), and PC (17:0_18:1) levels were associated with a decreased SqCLC risk (ORs = 0.881, 0.852, 0.890, and 0.879, respectively). It is noteworthy that although the IVW method suggested associations of SM (d38:2), SM (d36:1), PC (O-16:0_18:1), and PC (18:0_20:4) levels with SqCLC risk, additional MR analyses showed directions opposite to the primary method (Supplementary Table S6).
Forest plots of causal effect of plasma lipidome on squamous cell lung carcinoma (A) and small cell lung carcinoma (B).
SCLC
After excluding 4 lipids using the MR-BMA method (Supplementary Table S7), we found only 4 lipids associated with SCLC risk (Supplementary Table S6 and Figure 3B). Specifically, a high SD of SM (d36:2) (OR 1.211, 95% CI 1.031-1.421), PC (O-16:0_20:3) (OR 1.180, 95% CI 1.021-1.364), and PC (O-18:0_14:0) levels (OR 1.190, 95% CI 1.018-1.392) were associated with an increased SCLC risk. PI (18:1_20:4) levels (OR 0.822, 95% CI 0.707-0.956) were associated with lower SCLC risk. The results of additional MR analyses were consistent with those of the IVW method in terms of direction (Supplementary Table S6).
Reverse causal effects of LC and subtypes on plasma lipidome
To assess potential reverse causality effects, we used LC and its subtypes as exposures and plasma lipidome as outcomes. Based on the IVW method analysis, we only found potential causal effects of an increased risk of LADC (OR 0.934, 95% CI 0.873-1.000) with decreased PC (16:0_20:2) levels. No other evidence was found for the causal effects of LC and subtypes on plasma lipidome (Supplementary Table S8).
MR sensitivity analysis
Cochran's Q test revealed heterogeneity among the genetic instruments for PC (O-16:1_20:4), PC (O-18:1_20:3), TAG (58:7), DAG (16:0_18:2), and SM (d36:1) levels (Supplementary Table S9). MR-Egger regression indicated that the causal effect of PC (O-16:1_20:3) on LADC was influenced by horizontal pleiotropy (Supplementary Table S9). Additionally, MR-PRESSO detected outliers among the genetic instruments for PC (O-16:1_20:4) and SM (d36:1) levels (Supplementary Table S9).
Mediation effect of inflammation proteins
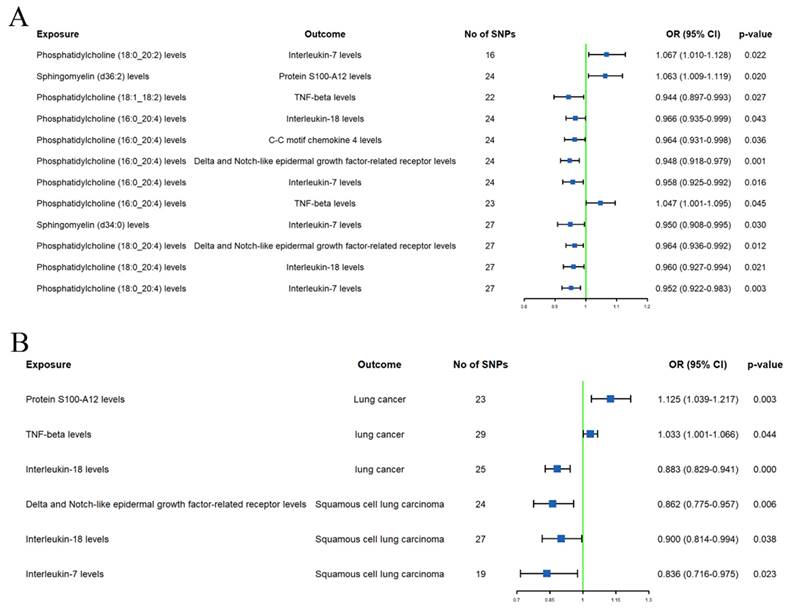
The initial mediation MR analysis identified 8 inflammation proteins that are simultaneously associated with both plasma lipidome and LC or subtypes. However, causal analyses for the effects of plasma lipidome on inflammation proteins (Supplementary Table S10) or inflammation proteins on LC or subtypes (Supplementary Table 11) showed that the additional MR analysis results for protein S100-A12, C-X-C motif chemokine 10, monocyte chemoattractant protein-3, and C-C motif chemokine 4 levels were inconsistent with the main analysis direction. Sensitivity analysis also revealed heterogeneity in the effect of C-X-C motif chemokine 10 levels on LC, and the causal relationship between C-C motif chemokine 4 levels and SqCLC was influenced by horizontal pleiotropy (Supplementary Table S9).
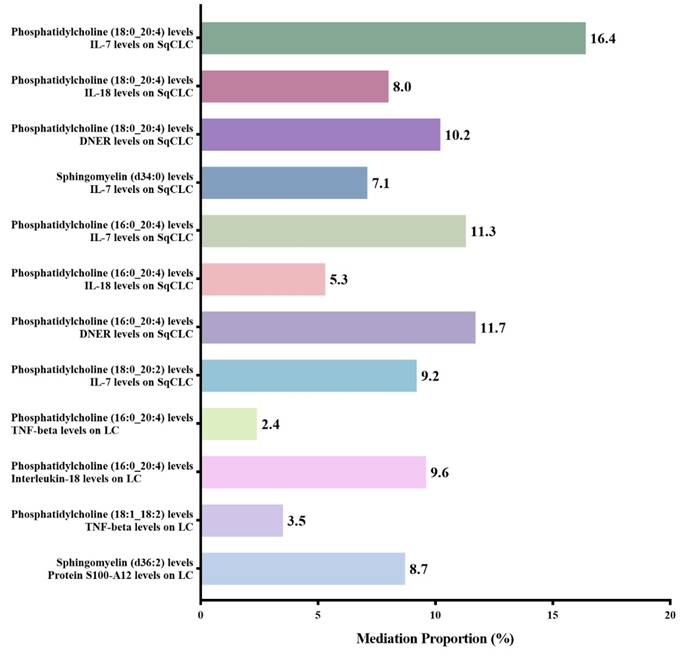
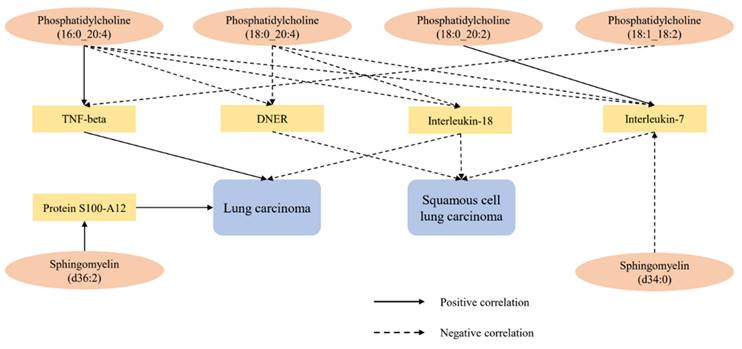
After excluding the above-mentioned inflammatory proteins, we finally identified 5 inflammatory proteins that exerted mediating effects (Supplementary Table S12 and Figure 4). Specifically, protein S100-A12 levels mediate the effect of SM (d36:2) levels on LC, with a mediation effect accounting for 8.7%; TNF-beta levels mediate the effects of PC (18:1_18:2) and PC (16:0_20:4) levels on LC, with mediation effects of 3.5% and 9.6%, respectively; IL-7 levels mediate the effects of PC (18:0_20:2), PC (16:0_20:4), SM (d34:0), and PC (18:0_20:4) levels on SqCLC, with mediation effects of 9.2%, 11.3%, 7.1%, and 16.4%, respectively; IL-18 levels mediate not only the effect of PC (16:0_20:4) levels on LC (mediation effect of 9.6%) but also the effects of PC (16:0_20:4) and PC (18:0_20:4) levels on SqCLC, with mediation effects of 5.3% and 8.0%, respectively. Additionally, delta and Notch-like epidermal growth factor-related receptor (DNER) levels mediate the effects of PC (16:0_20:4) and PC (18:0_20:4) levels on SqCLC, with mediation effects of 11.7% and 10.2%, respectively (Supplementary Table S12 and Figure 5). No evidence was found for other inflammatory proteins mediating the causal effects of plasma lipids on LC and subtypes. Figure 6 provides an intuitive illustration of the regulatory network of plasma lipidome and inflammatory proteins on LC and subtypes.
Forest plots of causal effects of plasma lipidome on inflammatory proteins (A) and causal effects of inflammatory proteins on lung carcinoma or subtypes (B).
Mediation analysis of inflammatory proteins between plasma lipidome and lung carcinoma or subtypes.
Regulatory network of lung carcinoma and subtypes by plasma lipidome and inflammatory proteins.
Discussion
This study employed an MR design to elucidate the causal relationship between plasma lipidome and LC and subtypes. Mediation MR analysis revealed that inflammatory proteins may play a significant role in the impact of plasma lipidome on LC. Our findings highlight the complex interplay between plasma lipidome, inflammatory proteins, and LC, underscoring the potential for devising preventive strategies.
The lipid metabolism network in cancer cells is highly complex. On one hand, lipid metabolism in cancer cells is regulated not only by intracellular oncogenic signals but also by the tumor microenvironment, which comprises various components, including lipids. On the other hand, aberrant lipid metabolism can alter oncogenic signals in cancer cells, affecting nearby non-cancerous cells by modifying the secretion of substances, including lipids, from cancer cells [25]. Excessive lipid levels are also associated with cancer cell proliferation and metastasis. By altering the composition of lipid membranes, cancer cells can invade adjacent tissues and overcome cell death mechanisms. Cancer cells can acquire energy and protect themselves from oxidative stress damage by modulating lipid breakdown and synthesis metabolism. Additionally, cancer cells can exploit lipid metabolism to regulate the activity of immune cells, thereby evading the host's normal immune clearance functions [26]. Key genes/proteins associated with lipid metabolism may serve as potential indicators for predicting the prognosis of various cancers [27].
As the most abundant phospholipid in the body, multiple studies have explored the relationship between PC and LC. One study, based on scRNA-seq and lipidomics, found that five PC subclasses could serve as important detection features for early LC [28]. Another metabolomic analysis indicated that levels of saturated PC and monounsaturated PC significantly increase in NSCLC, whereas levels of polyunsaturated PC decrease significantly in NSCLC patients [3]. A cross-omics analysis focusing on clinical features and lipidomic profiles among different subtypes of LC patients found that PC (19:0-19:0 and 19:0-21:2) is specifically associated with LADC [29]. Our study identified 7 subclasses of PC associated with the risk of LC and subtypes. Specifically, a high PC (16:0_20:2), PC (O-16:1_18:0), PC (17:0_18:1), PC (18:0_20:2), PC (18:1_18:1), PC (18:1_18:2), and PC (O-18:1_20:3) levels were associated with a decreased risk of LC, LADC, and SqCLC. On the contrary, a high PC (16:0_20:4), PC (16:0_20:5), PC (17:0_20:4), PC (O-16:0_20:3), PC (O-16:1_20:3), PC (O-16:1_20:4), PC (O-18:0_14:0), and PC (O-18:2_20:4) levels increased the risk of LC, LADC, SqCLC, and SCLC. It is worth noting that different subclasses of PC exhibited bidirectional effects on the risk of LC or subtypes. This suggests that the spatial structure of lipids, such as carbon atoms, double bonds, or isomerism, may affect the proliferation and differentiation of cancer cells.
PE is the second most abundant phospholipid after PC. PE participates in various cellular functions, including activating oxidative phosphorylation, maintaining mitochondrial morphology, and participating in cell death pathways such as ferroptosis and apoptosis [30, 31]. Mechanistic studies have found that excessive production of mitochondrial PE can inhibit cancer cell proliferation [32]. Evidence from retrospective studies suggests significant differences in the levels of PE among patients with malignant or benign pulmonary nodules [33]. However, cross-omics analysis revealed that specific PE subtype levels are significantly higher in SCLC compared to other LC subtypes or healthy controls [29]. This study found that high PE (O-18:1_20:4), PE (18:0_18:2), and PE (O-18:2_18:1) levels are associated with a low LADC or SqCLC risk. Our results further support the role of PE as a protective factor against LC.
DAG, as a lipid second messenger, is involved in transmitting intracellular signals that influence mammalian cell proliferation, survival, and motility. DAG has been widely implicated in the onset, progression, and metastasis of cancers [34]. Some molecules that indirectly regulate DAG, such as protein kinase C (PKC) isoform PKCε and phosphatidic acid phosphatase Lipin-1, are associated with the progression and adverse outcomes of LC [35, 36]. Our study provided further evidence that elevated DAG (16:1_18:1) and DAG (16:0_18:2) levels are associated with an increased LC and LADC risk.
Our study also demonstrated that elevated SM(d36:2) and SM(d34:0) levels are associated with an increased LC, SqCLC, or SCLC risk. A case-control study conducted in Japan supports our findings [37]. This study found that increased SM levels in primary LC tissue samples are associated with an increased risk of LC recurrence. Furthermore, we also found that elevated genetically predicted TAG (48:0) and TAG (58:7) levels are associated with an increased LADC risk, while increased SE (27:1/20:2) levels are associated with a low LC and LADC risk. High PI (18:1_20:4) levels are associated with a lower SCLC risk. However, there is currently no observational evidence linking these three lipids to LC.
Our mediation MR analysis also revealed the involvement of inflammatory proteins in the causal effect of plasma lipidome on LC or subtypes. Among them, IL-7 and IL-18 are both pro-inflammatory cytokines, and several studies have reported the therapeutic potential of IL-7 and IL-18 in LC [38-42]. Our MR analysis revealed that high IL-7 and IL-18 levels are associated with a low LC or SqCLC risk. PC (18:0_20:2), PC (16:0_20:4), and PC (16:0_20:4) levels may potentially influence the incidence of SqCLC by modulating the levels of IL-7. PC (16:0_20:4) may also increase the risk of LC and SqCLC by reducing the levels of IL-18. Additionally, protein S100-A12 is involved in the regulation of SM (d36:2) for LC. TNF-beta partially mediates the effects of PC (18:1_18:2) and PC (16:0_20:4) on LC. The causal effects of PC (16:0_20:4) and PC (18:0_20:4) on SqCLC are partially mediated by DNER. These three inflammatory proteins have been reported to be associated with prostate cancer, papillary thyroid carcinoma, ovarian cancer, gastric cancer, and breast cancer [43-47]. Our study is the first to identify their involvement in the effects of the lipidome on LC or subtypes. The mechanisms of these inflammatory proteins on LC require further investigation.
This is the first study to systematically explore the causal relationship between plasma lipidome and LC and subtypes using MR design. We also identified 5 inflammatory proteins playing crucial mediating roles in the association between plasma lipidome and LC and subtypes. However, MR methods have limitations that need to be acknowledged. First, despite using SNPs as genetic instruments to mitigate confounding effects, some unmeasured confounders that influence the genetic instruments may still bias the results of MR analysis. In this study, we rigorously employed 4 additional MR methods, the BMA-MR approach, and sensitivity analyses to maximize the reduction of potential biases from unmeasured confounders. Second, to obtain adequate genetic instruments, our study used a threshold of p < 1E-5 for SNP screening. However, this may violate the MR assumption that there is a strong association between genetic instruments and exposure. Therefore, further MR analysis is needed when more appropriate genetic instruments are available. Finally, this MR study was primarily based on European populations. Since the same genetic instruments may have different effects in different ethnic groups, this may limit the generalizability of our results to broader populations.
In conclusion, our findings underscore the intricate network between plasma lipidome and inflammatory proteins in regulating LC. Further research is needed to identify the key lipids and inflammatory proteins that play critical roles in cancer development, along with corresponding functional studies and experimental validations to elucidate the underlying mechanisms involved. Additionally, our study highlights the potential of integrating plasma lipidome into clinical practice. Incorporating plasma lipidome into cancer risk assessment and prevention strategies may pave the way for new approaches in the prevention and management of LC.
Abbreviations
LC: lung carcinoma;
PC: phosphatidylcholines;
PE: phosphatidylethanolamines;
DAG: diacylglycerols;
TAG: triacylglycerols;
SM: sphingomyelins;
PI: phosphatidylinositols;
CE: cholesterol esters;
SE: sterol esters;
NSCLC: non-small cell lung carcinoma;
LADC: lung adenocarcinoma;
SqCLC: squamous cell carcinoma;
SCLC: small cell lung carcinoma;
CRP: C-reactive protein;
MIG: monokine induced by gamma interferon;
SAA: serum amyloid A;
MR: Mendelian Randomization;
GWAS: genome-wide association studies;
pQTL: protein quantitative trait locus;
ILCCO: International Lung Carcinoma Consortium;
TRICL: Transdisciplinary Research in Cancer of the Lung;
SNPs: single nucleotide polymorphisms;
LD: linkage disequilibrium;
IVW: inverse variance-weighted;
OR: odds ratios;
CI: confidence intervals;
MR-BMA: MR Bayesian model averaging;
DNER: delta and Notch-like epidermal growth factor-related receptor;
PKC: protein kinase C.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers: 82070017, 82070014 and 81870009).
Data availability statement
The GWAS data of plasma lipidome, inflammatory proteins and lung carcinoma are accessible under application at https://www.ebi.ac.uk/gwas/ (accessed on 1 March 2024), and exposure data sources and handling of these data are described in Methods.
Author contributions
H.Y. and G.F. conceived and designed the experiment; J.F. and X.J. ran the analysis and verified the underlying data; H.Y. wrote the original manuscript. Y.Z., C.B., and C.Z. were involved in data interpretation; H.Y., J.F., and C.Z. participated in revising the manuscript. All authors have read and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Zhang L, Zhu B, Zeng Y, Shen H, Zhang J, Wang X. Clinical lipidomics in understanding of lung cancer: Opportunity and challenge. Cancer letters. 2020;470:75-83
3. Chen Y, Ma Z, Zhong J, Li L, Min L, Xu L. et al. Simultaneous quantification of serum monounsaturated and polyunsaturated phosphatidylcholines as potential biomarkers for diagnosing non-small cell lung cancer. Scientific reports. 2018;8:7137
4. Lv J, Gao D, Zhang Y, Wu D, Shen L, Wang X. Heterogeneity of lipidomic profiles among lung cancer subtypes of patients. J Cell Mol Med. 2018;22:5155-9
5. Brenner DR, Fanidi A, Grankvist K, Muller DC, Brennan P, Manjer J. et al. Inflammatory Cytokines and Lung Cancer Risk in 3 Prospective Studies. Am J Epidemiol. 2017;185:86-95
6. Suárez GM, Añé-Kourí AL, González A, Lorenzo-Luaces P, Neninger E, Salomón EE. et al. Associations among cytokines, EGF and lymphocyte subpopulations in patients diagnosed with advanced lung cancer. Cancer Immunol Immunother. 2021;70:1735-43
7. Michels N, van Aart C, Morisse J, Mullee A, Huybrechts I. Chronic inflammation towards cancer incidence: A systematic review and meta-analysis of epidemiological studies. Crit Rev Oncol Hematol. 2021;157:103177
8. Shiels MS, Pfeiffer RM, Hildesheim A, Engels EA, Kemp TJ, Park JH. et al. Circulating inflammation markers and prospective risk for lung cancer. J Natl Cancer Inst. 2013;105:1871-80
9. Shiels MS, Katki HA, Hildesheim A, Pfeiffer RM, Engels EA, Williams M. et al. Circulating Inflammation Markers, Risk of Lung Cancer, and Utility for Risk Stratification. J Natl Cancer Inst. 2015;107:djv199
10. Larsson SC. Mendelian randomization as a tool for causal inference in human nutrition and metabolism. Curr Opin Lipidol. 2021;32:1-8
11. Sekula P, Del Greco MF, Pattaro C, Köttgen A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J Am Soc Nephrol. 2016;27:3253-65
12. Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA. et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. Jama. 2021;326:1614-21
13. Ottensmann L, Tabassum R, Ruotsalainen SE, Gerl MJ, Klose C, Widén E. et al. Genome-wide association analysis of plasma lipidome identifies 495 genetic associations. Nat Commun. 2023;14:6934
14. Zhao JH, Stacey D, Eriksson N, Macdonald-Dunlop E, Hedman Å K, Kalnapenkis A. et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24:1540-51
15. Ji D, Chen WZ, Zhang L, Zhang ZH, Chen LJ. Gut microbiota, circulating cytokines and dementia: a Mendelian randomization study. J Neuroinflammation. 2024;21:2
16. Yan H, Jin X, Zhang C, Zhu C, He Y, Du X. et al. Associations between diet and incidence risk of lung cancer: A Mendelian randomization study. Front Nutr. 2023;10:1149317
17. Yan H, Zhu C, Jin X, Feng G. Mendelian randomization reveals no correlations between herpesvirus infection and idiopathic pulmonary fibrosis. PloS one. 2023;18:e0295082
18. Dudbridge F. Polygenic Mendelian Randomization. Cold Spring Harb Perspect Med. 2021;11:a039586
19. Greco MF, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34:2926-40
20. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512-25
21. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693-8
22. Sanderson E. Multivariable Mendelian Randomization and Mediation. Cold Spring Harb Perspect Med. 2021;11:a038984
23. Carter AR, Sanderson E, Hammerton G, Richmond RC, Davey Smith G, Heron J. et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36:465-78
24. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408
25. Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021;218:e20201606
26. Martin-Perez M, Urdiroz-Urricelqui U, Bigas C, Benitah SA. The role of lipids in cancer progression and metastasis. Cell Metab. 2022;34:1675-99
27. Irshad R, Tabassum S, Husain M. Aberrant Lipid Metabolism in Cancer: Current Status and Emerging Therapeutic Perspectives. Curr Top Med Chem. 2023;23:1090-103
28. Wang G, Qiu M, Xing X, Zhou J, Yao H, Li M. et al. Lung cancer scRNA-seq and lipidomics reveal aberrant lipid metabolism for early-stage diagnosis. Sci Transl Med. 2022;14:eabk2756
29. Zhu Z, Zhang L, Lv J, Liu X, Wang X. Trans-omic profiling between clinical phenoms and lipidomes among patients with different subtypes of lung cancer. Clin Transl Med. 2020;10:e151
30. Pohl EE, Jovanovic O. The Role of Phosphatidylethanolamine Adducts in Modification of the Activity of Membrane Proteins under Oxidative Stress. Molecules. 2019;24:4545
31. Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem. 2013;288:4158-73
32. Pandey SK, Paul A, Shteinfer-Kuzmine A, Zalk R, Bunz U, Shoshan-Barmatz V. SMAC/Diablo controls proliferation of cancer cells by regulating phosphatidylethanolamine synthesis. Mol Oncol. 2021;15:3037-61
33. Fahrmann JF, Grapov D, DeFelice BC, Taylor S, Kim K, Kelly K. et al. Serum phosphatidylethanolamine levels distinguish benign from malignant solitary pulmonary nodules and represent a potential diagnostic biomarker for lung cancer. Cancer Biomark. 2016;16:609-17
34. Cooke M, Kazanietz MG. Overarching roles of diacylglycerol signaling in cancer development and antitumor immunity. Sci Signal. 2022;15:eabo0264
35. Casado-Medrano V, Barrio-Real L, Wang A, Cooke M, Lopez-Haber C, Kazanietz MG. Distinctive requirement of PKCε in the control of Rho GTPases in epithelial and mesenchymally transformed lung cancer cells. Oncogene. 2019;38:5396-412
36. Fan X, Weng Y, Bai Y, Wang Z, Wang S, Zhu J. et al. Lipin-1 determines lung cancer cell survival and chemotherapy sensitivity by regulation of endoplasmic reticulum homeostasis and autophagy. Cancer Med. 2018;7:2541-54
37. Takanashi Y, Funai K, Sato S, Kawase A, Tao H, Takahashi Y. et al. Sphingomyelin(d35:1) as a novel predictor for lung adenocarcinoma recurrence after a radical surgery: a case-control study. BMC Cancer. 2020;20:800
38. Andersson A, Yang SC, Huang M, Zhu L, Kar UK, Batra RK. et al. IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J Immunol. 2009;182:6951-8
39. Zhu Y, Jiang X, Ding Z, Ming J. Interleukin 7 inhibit autophagy via P53 regulated AMPK/mTOR signaling pathway in non-small cell lung cancer. Scientific reports. 2022;12:11208
40. Shi L, Xu Z, Yang Q, Huang Y, Gong Y, Wang F. et al. IL-7-Mediated IL-7R-JAK3/STAT5 signalling pathway contributes to chemotherapeutic sensitivity in non-small-cell lung cancer. Cell Prolif. 2019;52:e12699
41. Jaspers JE, Khan JF, Godfrey WD, Lopez AV, Ciampricotti M, Rudin CM. et al. IL-18-secreting CAR T cells targeting DLL3 are highly effective in small cell lung cancer models. J Clin Invest. 2023;133:e166028
42. Timperi E, Focaccetti C, Gallerano D, Panetta M, Spada S, Gallo E. et al. IL-18 receptor marks functional CD8(+) T cells in non-small cell lung cancer. Oncoimmunology. 2017;6:e1328337
43. Åberg AM, Bergström SH, Thysell E, Tjon-Kon-Fat LA, Nilsson JA, Widmark A. et al. High Monocyte Count and Expression of S100A9 and S100A12 in Peripheral Blood Mononuclear Cells Are Associated with Poor Outcome in Patients with Metastatic Prostate Cancer. Cancers. 2021;13:2424
44. Wang X, Sun Z, Tian W, Piao C, Xie X, Zang J. et al. S100A12 is a promising biomarker in papillary thyroid cancer. Scientific reports. 2020;10:1724
45. Buhrmann C, Yazdi M, Popper B, Shayan P, Goel A, Aggarwal BB. et al. Resveratrol Chemosensitizes TNF-β-Induced Survival of 5-FU-Treated Colorectal Cancer Cells. Nutrients. 2018;10:888
46. To HTN, Park JH, Kim JW, Kang D. Delta/Notch-like Epidermal Growth Factor-Related Receptor (DNER), a Potential Prognostic Marker of Gastric Cancer Regulates Cell Survival and Cell Cycle Progression. Int J Mol Sci. 2023;24:10077
47. Wang L, Wu Q, Li Z, Sun S, Yuan J, Li J. et al. Delta/notch-like epidermal growth factor-related receptor promotes stemness to facilitate breast cancer progression. Cell Signal. 2019;63:109389
Author contact
![]() Corresponding authors: Dr. Chenghua Zhu, email: zhuchenghuachengcom; or Dr. Ganzhu Feng, email: fenggzedu.cn.
Corresponding authors: Dr. Chenghua Zhu, email: zhuchenghuachengcom; or Dr. Ganzhu Feng, email: fenggzedu.cn.