Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(4):1281-1295. doi:10.7150/jca.101911 This issue Cite
Research Paper
Exploring breast cancer associated-gene panel for next-generation sequencing and identifying new, pathogenic variants in breast cancer from western China
Jingliang Cheng1,2#, Binghui Song1,2#, Chunli Wei1#, Lianmei Zhang1,3#, Xiaoyan Liu1, Lisha Yang1,2, Singkome Tima2,4, Sawitree Chiampanichayakul2,4, Xiuli Xiao5 ![]() , Songyot Anuchapreeda2,4
, Songyot Anuchapreeda2,4 ![]() , Junjiang Fu1
, Junjiang Fu1 ![]()
1. Key Laboratory of Epigenetics and Oncology, The Research Center for Preclinical Medicine, Southwest Medical University, Luzhou 646000, Sichuan Province, China.
2. Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand.
3. Department of Pathology, The Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University, Huai'an 223300, Jiangsu Province, China.
4. Research Center of Pharmaceutical Nanotechnology, Chiang Mai University, Chiang Mai, 50200, Thailand.
5. Department of Pathology, the Affiliated Hospital, Southwest Medical University, Luzhou 646000, Sichuan Province, China.
# Equal contributions.
Received 2024-8-5; Accepted 2024-12-13; Published 2025-1-13
Abstract

Breast cancer (BC) is the most frequently diagnosed and the leading cause of cancer-related deaths among women worldwide. It is crucial to develop a cost-effective BC genetic panel for detection and diagnosis. In this study, tissue samples from 52 BC patients and peripheral blood samples from 18 healthy volunteers were collected in western China, followed by gDNA extraction. H&E and IHC analysis were employed to detect the expression of invasive BC tissues. We analyzed data using public databases such as COSMIC/ClinVar/HGMD along with our own previously published data and queried commercial BC panels to select high-risk genes. Using Illumina DesignStudio, gene panel primers consisting of 13 genes were designed with 696 primer pairs. The specificity of all primers was validated through common PCR assays. Once the gene panel was completed, multiple polymerase chain reactions (MPCR) were performed using the designed panel primers. The resulting MPCR products were purified to enrich them as library templates. Subsequently, after passing quality tests for library integrity assessment, Next-generation sequencing (NGS) was conducted. Through bioinformatics analysis of the NGS data, 4,571 variants were identified in the annotation files from 52 samples, classified into different types. Among these variants, 358 (approximately 7.8%) were newly discovered and distributed across 11 genes in 52 patients without in the ExAC database. The KMT2C gene exhibited the highest frequency of variants, presenting in 83.0% of 52 patient samples. Variants in BRCA2 (71%), BRCA1 (48%), PALB2 (40%), PIK3CA (23%), and RNF40 (21%) genes were found in over 20% of patients. Additionally, variants were observed in the AKT1 (12%), ERBB2 (10%), ESR1 (8%), TWIST1 (8%), and PIK3R1 (4%) genes. Further analysis using PolyPhen-2, SIFT, CADD, and Mutation Taster tools analysis showed that out of these new variants, 49 (49/358) had potential pathogenic effects on protein functions and structure across 52 patients. Consequently, a high-risk gene panel has been preliminarily established for early detection/diagnosis that will contribute to earlier prevention and treatment strategies for individuals with BC, particularly those residing in developing or underdeveloped countries. The identification of novel pathogenic variants within our cohort not only expands knowledge regarding genetic diagnosis applications for BC patients but also facilitates genetic counseling services for affected individuals and their families.
Keywords: breast cancer, gene panel, next-generation sequencing (NGS), pathogenic variants, gene diagnosis, genetic counseling
1. Introduction
Cancer is a prevailing global healthy issue, particularly in developing or undeveloped countries or regions. According to the GLOBOCAN 2022 data [1, 2], breast cancer (BC) has been reported as the second most frequently diagnosed and the fourth leading cause of death among all cancers worldwide. Especially, BC is the most common cancer and the main cause of cancer death among women worldwide. In China, there were approximately 357,200 new cases of BC reported in females in 2022 [3], accounting for 15.47% of new female cases globally with around 75,000 deaths. The increasing incidence and mortality rates of BC patients have made this disease a significant threat to the health of Chinese women. Advancements in clinical diagnosis and treatment have significantly improved BC survival rates, however, there remains a gap between the five-year survival rate of BC patients in China (82.0%) and that observed in developed countries like the United States (90.9%) [4-6]. The discrepancy can be attributed to delayed diagnoses among many BC patients who are already at intermediate or advanced stages with tumor metastasis upon initial detection. Consequently, surgical interventions may occur too late resulting in high post-operative recurrence rates and shorter survival times. To address the challenge posed by rapidly increasing incidence and mortality rates among BC patients, early detection/diagnosis, prevention strategies, and timely treatment must be prioritized [5].
Precision medicine is an effective approach based on scientific understanding of individual differences arising from genome variation, environment, and lifestyle. It involves precise genetic diagnosis to achieve personalized therapy, and early prevention [7, 8]. In developed Western countries, databases such as ClinVar, the Catalogue of Somatic Mutations in Cancer (COSMIC), next-generation sequencing (NGS) technology, and bioinformatics analysis, are utilized for genetic diagnosis in BC patients. Commercial gene panels have already been developed to cater to their needs [5, 6, 9, 10]. However, in China-a developing country with certain underdeveloped regions like western China-factors such as living habits, educational level, and medical conditions (particularly scientific and technological advancements), contribute to a large number of BC patients being diagnosed at advanced stages with poor prognosis [2, 5, 6, 9-11]. BC has become a prevalent malignant tumor significantly impacting the physical and mental health of women while posing a threat to their lives. The primary cause of death among BC patients lies not in the growth of primary tumors but rather in metastasis and recurrence. Therefore, it is imperative to develop a cost-effective gene panel that can effectively facilitate early prevention, detection, and diagnosis.
The etiology and pathogenesis of BC are multifactorial and complex, with genetic factors playing an influential role in approximately 5%-10% of cases, while the majority of cases (90%-95%) are sporadic and caused by somatic mutations. Somatic mutations are not inherited, and the primary method employed in this study involved in collecting cancer tissue for comparison with normal tissue or blood samples. The COSMIC database serves a comprehensive resource for exploring the impacts of somatic mutations on different cancers. Notably, BRCA1 and BRCA2 genes are the most common disease genes associated with hereditary BC due to their high-frequency germline mutation. Currently, variant interpretation guidelines provided by the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) solely focus on germline variants. However, advancements in DNA sequencing techniques such as NGS have facilitated the identification of other BC susceptibility genes including various variants of TP53, PIK3CA, PALB2, and PTEN genes. Furthermore, rare genes have been reported to increase the risk of developing metastatic BC [10, 12-14]. This study aims to establish a high-risk gene panel for the diagnosis BC in western China to enhance knowledge and information pertaining to the clinical application of genetic diagnosis among BC patients. These findings can provide valuable insights for genetic counseling related to both BC patients and their family pedigrees.
2. Materials and Methods
2.1 Sample collection and DNA extraction
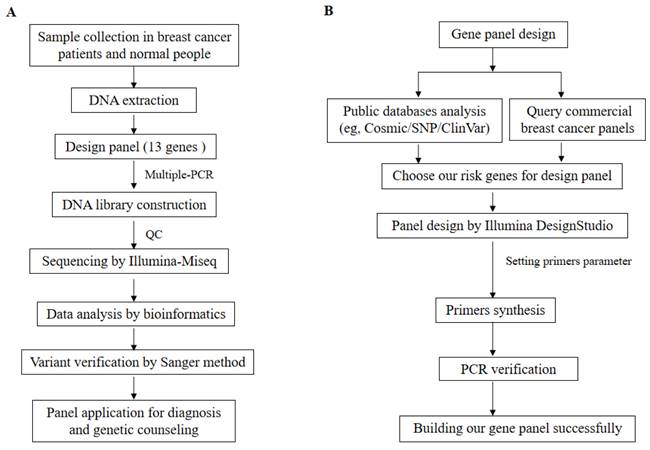
This study was approved in advance by the Ethics Committee of Southwest Medical University and was conducted in accordance with the principles outlined in the Declaration of Helsinki (2013 version). Tissue samples were recruited from 52 patients with BC, along with blood samples from 18 healthy donors from different families. The patient was diagnosed with BC through pathological examination, and the BC tissue of the patients collected through clinical surgery. Normal people have no disease after clinical examination, and blood samples of normal people are collected through clinical physical examination. Clinical examination and family information, including age, sex, tumor sizes, tumor stage, tumor metastasis, pathological diagnosis, and family history were recorded for BC patients. The expression of Her-2/ER/PR patient pathological samples was utilized for BC pathology reporting across various subtypes. Tumor size and metastasis served as grading systems to determine patient status. Subsequently, DNA extraction was performed using a standard phenol/chloroform method [15] on both BC tissues and normal blood samples obtained from patients and healthy donors, respectively. The workflow of this study is illustrated in Figure 1A.
The workflow of our study and gene panel design is as follows. A. Study workflow - firstly, we collected BC tissue from patients and blood samples from normal individuals. Subsequently, DNA was extracted, followed by use of 13 genes in the design panel. Multiple-PCR was completed and the product was purified. Then, a DNA library was constructed, quality control checks were performed using Agilent 5200, and sequencing was carried out using the Illumina Miseq platform. Thirdly, bioinformatics analysis of sequencing data obtained all variants which validated using the Sanger method before applying for diagnosis and genetic counseling through the panel. B. Gene panel design workflow - firstly public databases (e.g., Cosmic/SNP/ClinVar) were analyzed along with commercial BC panels to choose risk genes for designing our gene panel. Secondly, Illumina DesignStudio designed gene panels while setting primer parameters. Thirdly, synthesized primers underwent PCR validation to ensure specificity. Finally, the successful construction of our gene panel.
2.2 Hematoxylin-Eosin staining and immunohistochemistry
Patients with BC tissues were fixed in 10% formalin for one day, embedded in paraffin, and sliced every 5 µm. After dewaxing in xylene and dehydrating, the slides were stained with hematoxylin and eosin (H&E) before being probed by immunohistochemistry (IHC). The H&E and IHC methods were performed as previously described protocols [16-18]. The BRCA1 (cat #: ZM-0347) and TP53 (cat #: ZM-0408) antibodies for IHC were purchased from Beijing Zhong Shan-Golden Bridge Biological Technology Co., Ltd., CN. Slides with a thickness of 5 µm underwent antigen retrieval through incubation in 10 µM sodium citrate buffer at 95°C for three times, 12 min each. Samples were then blocked using a solution containing 5% BSA before primary antibodies for BRCA1 and TP53 (diluted at a ratio of 1:100) were applied overnight. Appropriate biotin-conjugated secondary antibodies (cat #: SP-9000, ZSGB-Bio, CN) were used to incubate the samples at room temperature for an hour before visualization was achieved via streptavidin-conjugated horseradish peroxidase (HRP) and 3,3-diaminobenzidine (DAB) (cat #: ZLI-9017, ZSGB-Bio, CN). Finally, the stained slides underwent retaining with hematoxylin prior to dehydration, mounting, and analysis.
2.3 Design gene panel
To design the gene panel, we initially conducted an analysis using public databases such as COSMIC/ClinVar/HGMD. Specifically, a comprehensive analysis of the COSMIC database was performed as previously described [19]. Subsequently, the top twenty genes were identified with a mutation frequency in BC and searched for specific inhibitors to target these genes through literature research [7, 12, 14, 20-32]. Additionally, considering our previous studies and current investigation on BC genes to select high-risk genes specific to the Chinese population in our region [16, 33-35], they were included in the gene panel. Furthermore, to tailor our panel specifically for Chinese individuals at high risk of BC development, commercially available BC panels were compared from Western developed countries [6, 10, 32, 36-38]. Thirteen genes (AKT1, BRCA1, BRCA2, ESR1, ERBB2, KMT2C, PALB2, PIK3CA, PIK3R1, PTEN, TP53, RNF40, and TWIST1) were selected for primer design to cover all exons and intron junction sequences. The Illumina DesignStudio was employed to design the primers and set their parameters. The primer set underwent multiple assay steps (AmpliSeq for Illumina Gene) to ensure accuracy before displaying all gene primer information on a webpage table that could be downloaded. Consequently, thirteen genes with corresponding primers pairs totaling up to 696 were synthesized into our designed gene panel. Then, the specificity of primers was assessed by PCR. Finally, the successfully implementation of NGS resulted in the completion of our designed gene panel. The workflow illustrating the process is depicted in Figure 1A &B.
2.4 Multiple-PCR and product purified analysis
The multiple-PCR (MPCR) amplification was conducted using genomic DNA (gDNA) as a template in a total volume of 50 µL on a Veriti™ 96-Well Thermal Cycler. The Multiplex PCR Plus Kit for MPCR purchased from QIAGEN, Germany (cat #: 206152) [39] was employed. The components for MPCR were as follows: 25 µL of 2× Multiplex PCR Master Mix*, 5 µL of 10× primer mix, ≤300 ng of template DNA, and RNase-free water to reach a final volume of 50 µL. The cycling protocol for multiplex PCR consisted of an initial denaturation at 95 ºC for 5 min, followed by 35 cycles at 95 ºC for 30 s, then annealing at 60 ºC for 90 s, extension at 72 ºC for 30 s, and final extension at 68 ºC for 10 min. The amplified MPCR products were purified using AMPure XP Beads (Vazyme Biotech Co., Ltd., N411-01).
2.5 Construction of gene panel library and NGS analysis
After the successful design of the gene panel, multiplex PCR (MPCR) was performed to amplify our designed panel primers as the library template. The QIAseq 1-Step Amplicon Library kit (QIAGEN, Germany, Cat #: 180412) was utilized for library construction. Subsequently, each sample library was uniquely identified using different adapter sequences. To assess the quality of the libraries, Agilent Fragment Analyzer Systems (Agilent, USA) were employed for library quality control analysis. Each captured sample achieved a depth exceeding 200 × with over 90% coverage. The raw average data size for each sample amounted to 0.7GB, while the sequencing reads exhibited a quality score value around 20, and a base recognition rate surpassing 99%.
NGS was conducted on the Illumina MiSeq platform (Illumina, USA). For bioinformatics analysis of the NGS data, original reads were converted in FASTQ format and aligned to hg19 using BWA aligner version 0.5.9 [40]. Recalibration and local realignment were performed using Genome Analysis Toolkit (GATK version 1.0.5974) [41], followed by variant calling with Atlas2 toolkit [9]. Common polymorphisms with an allele frequency higher than 0.5% were filtered based on several common variant databases. Variant annotation was carried out using ANNOVAR [42] with RefSeq genes as a reference for coordinate mutations. The variant allele frequency of the variants was calculated from sequencing data. Functional predictions of variants were made using SIFT and PolyPhen-2 tools [43]. Known pathogenic mutations were searched in various databases such as COSMIC/ClinVar/HGMD/dbsnp/ExAC [14, 44-46]. We identified somatic mutations by adding blood samples of healthy individuals to exclude the germline mutations. Loci associated with clinical symptoms underwent screening.
2.6 Variant verification by Sanger method
For the validation of variants identified through NGS analysis, gene panel results were analyzed using a designed primers panel. The patient's gDNA was utilized as a template for PCR amplification on a Life Technology (USA) amplification machine, followed by Sanger sequencing analysis on an ABI-3500DX sequencer [15].
2.7 Variant classification and bioinformatics analysis
We developed an in-house local knowledge base and a proprietary bioinformatics pipeline to automate the construction of classification types for all identified variants and the variant allele frequency was assessed. The clinical significance of all identified variants was assessed following the standards and guidelines of the American College of Medical Genetics and Genomics by ACMG Laboratory Quality Assurance Committee [12, 47]. Minor allele frequencies were analyzed using public databases including the Exome Aggregation Consortium (ExAC) [48], the 5000 Exomes Project [49], the NHLBI Exome Sequencing Project ESP6500 databases [9], etc. Disease-specific variant information was queried from ClinVar [46], Online Mendelian Inheritance in Man (OMIM) [45], COSMIC [44], etc. The variants were analyzed by silico methodology. Variant substitutions' impact on protein structure and function were predicted using SIFT and PolyPhen-2 tools [43]. Nucleotide conservation analysis for all variants was performed by phyloP tool [32, 41]. An in-house local knowledge base was utilized for classifying, storing, organizing, continuously updating, and upgrading variant information along with lines of evidence. This process ensured reproducibility, rigor, and efficiency in reclassification. Novel variants with our cohort were identified through querying the ExAC database [50, 51]. In addition, Combined Annotation Dependent Depletion (CADD) (version 1.7) and Mutation Taster tools were also used to analyze these novel variants [52, 53].
2.8 Statistical analyses
The distribution of gene variants among categories was analyzed by assessing alteration in amino acid function. All statistical analyses were conducted using R software version 3.4.4 (R Foundation for Statistical Computing, Vienna, Austria). A statistically significant difference was considered when the adjusted p-value was less than 0.05 [54].
3. Results
3.1 The breast cancer patient recruitment and clinical findings
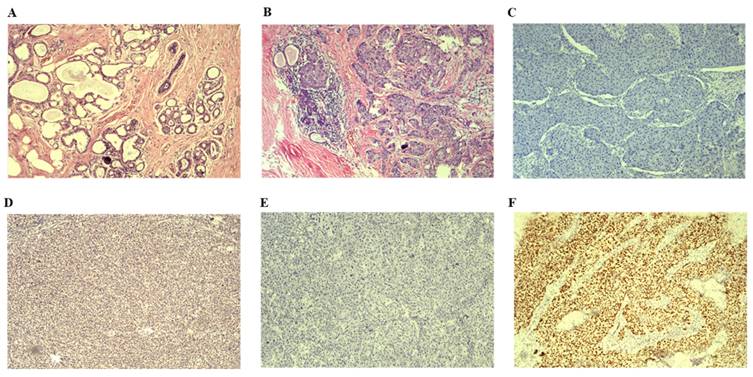
We enrolled 52 patients with BC tissues and 18 healthy blood donors whose DNA passed quality control for the study [Table 1]. Clinical information was collected from all patients, with an average age of diagnosis at 50 years (ranging from 29 to 77 years). The average tumor size was measured at 25 mm (range between 10 and 55 mm). Tumor metastasis was observed in 20 patients (38.5%), predominantly diagnosed as invasive ductal breast carcinomas. Patients were categorized into four subtypes based on the expression of Her-2/ER/PR. Among them, Luminal B subtype had the highest prevalence with 23 patients (44.2%), followed by Luminal A subtype with 14 patients (26.9%). Additionally, 11 patients were classified as Her-2-enriched subtype (21.2% in total), and only four patients were identified as basal-like subtype (7.7%). According to the American Cancer Society (https://www.cancer.org/cancer/types/breast-cancer/about/types-of-breast-cancer.html) and pathological classification of breast cancer (https://mp.weixin.qq.com/s/rP6lUKNQWNP96BuMHeik5A), the breast cancer cases were divided into two types including in situ breast cancer and invasive breast cancer. In addition, we categorized invasive breast cancer into early and late stages [Table 1]. Moreover, all BC samples were malignant based on the pathologic diagnosis by two independent pathologists. The pathological examination involved H&E staining and IHC analysis. Representative H&E staining images of normal breast tissue and invasive BC tissue are shown in Figure 2 A&B. Respective IHC images without any antibodies demonstrated negative expression (Figure 2C&E), while strong positive expressions of BRCA1 and TP53 antibodies were observed in invasive BC tissues (Figure 2D&F).
Demographic summary of the study group
| Number (n) | Percentage (%) | |
|---|---|---|
| Gender | ||
| Female | 65 | 92.8 |
| Male | 5 | 7.2 |
| Cancer | ||
| Yes | 52 | 74.3 |
| No | 18 | 25.7 |
| Age of onset | ||
| <40 years | 13 | 18.6% |
| >40 years | 57 | 82.4% |
| Intrinsic molecular subtype of breast cancer patients | ||
| Luminal A | 14 | 26.9% |
| Luminal B | 23 | 44.2% |
| HER2- enriched | 11 | 21.2% |
| Basal-like | 4 | 7.7% |
| Types of breast cancer | ||
| in situ breast cancer | 5 | 9.6% |
| invasive breast cancer (early-stage) | 30 | 57.7% |
| invasive breast cancer (late-stage) | 17 | 32.7% |
Results of H&E staining images and representative IHC images are presented. A. H&E staining image depicting normal breast tissue is shown. B. H&E staining image illustrating invasive BC tissue is displayed. C. Representative IHC image demonstrating the absence of BRCA1 antibody in invasive BC tissues is provided. D. Representative IHC image displaying the presence of BRCA1 antibody in invasive BC tissues is included. E. Representative IHC image indicating the lack of TP53 antibody in invasive BC tissues is depicted. F Representative IHC image showing the presence of TP53 antibody in invasive BC tissues is exhibited. H&E: Hematoxylin &Eosin; IHC: immunohistochemical staining.
3.2 Gene panel design and NGS analysis results
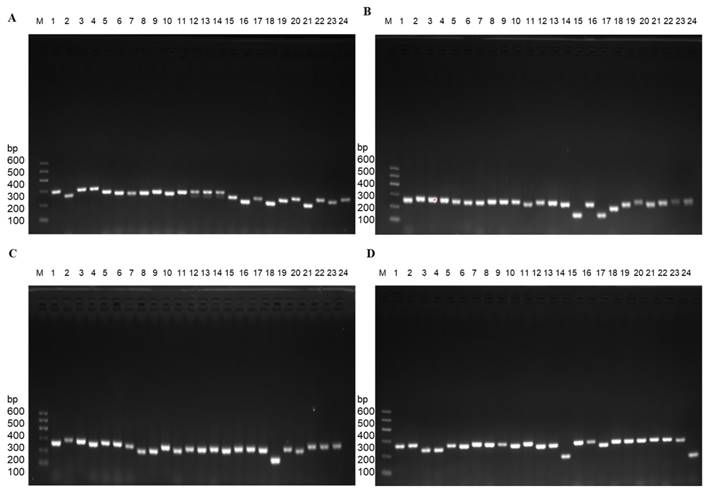
For the design of gene panel primers, Illumina DesignStudio was utilized and incorporated a total of thirteen genes with 696 primer pairs. The selection of these genes was based on comprehensive analysis using public databases (e.g., COSMIC/ClinVar/HGMD), our previous studies [16, 19, 33-35], as well as a relevant investigation conducted on Chinese breast patients [5, 10, 14, 15, 19, 29, 31, 33, 35, 44-46]. Specifically focusing on the PIK3CA, PIK3R1, ESR1, TWIST1, KMT2C, PTEN, BRCA2, AKT1, RNF40, PALB2, ERBB2, TP53, and BRCA1 genes, the information about chromosome, exons, and primer pairs is shown in Table 2. All the genes frequency is about somatic mutations. Germline mutations were excluded. Subsequently, the primers for the panel were synthesized, followed by PCR verification of their specificity. The PCR results for four representative genes (PTEN/TP53/RNF40/KMT2C) are illustrated in Figure 3.
List of genes analyzed and their association with breast cancer
| Gene | Transcript | Chromosome | Num_primers | Num_exons | Alteration | Frequency (%) | Candidate drug | Level of evidence for the target | Gene function/signal pathway | Specific inhibitors |
|---|---|---|---|---|---|---|---|---|---|---|
| PIK3CA | NM_006218.4 | chr3 | 43 | 21 | Amplifications/Mutations | >10 | PI3K inhibitor | 1-2 | PI3K/AKT/mTOR | Alpelisib (FDA-approved), Copanlisib (clinical trial) |
| PIK3R1 | NM_181523.3 | chr5 | 57 | 19 | Mutations | 1-5 | 4 | PI3K/AKT/mTOR | ||
| ESR1 | NM_001122740.2 | chr6 | 48 | 12 | Mutations/Amplifications/Translocations | >10% in metastatic/ER+ MBC resistant to endocrine therapy | 2 | ER signaling | ||
| KMT2C | NM_170606.3 | chr7 | 145 | 59 | Mutations | 5-10 | Drug targeting/epigenetics | 4 | Epigenetics | |
| PTEN | NM_000314.8 | chr10 | 60 | 10 | Mutations/Deletions | 5-10 | AKT inhibitor | 3 | PI3K/AKT/mTOR | Copanlisib (clinical trial) |
| BRCA2 | NM_000059.4 | chr13 | 82 | 27 | Mutations/Deletions | 1-5 | PARP inhibitor | 1 | DNA repair | Rucaparib, Niraparib, Olaparib (FDA-approved) |
| AKT1 | NM_001014431.2 | chr14 | 30 | 15 | Amplifications/Mutations | 1-5 | AKT inhibitor | 2 | PI3K/AKT/mTOR | Capivasertib (AZD-5363, clinical trial) |
| ERBB2 | NM_004448.4 | chr17 | 60 | 32 | Amplifications/Mutations | >10 | HER2 inhibitor | 1/3 | Growth factor receptors | Trastuzumab (FDA-approved) |
| TP53 | NM_000546.6 | chr17 | 24 | 11 | Mutations | >10 | Not known | NA | DNA repair | |
| BRCA1 | NM_007294.4 | chr17 | 56 | 24 | Mutations/Deletions | 1-5 | PARP inhibitor | 1 | DNA repair | Rucaparib, Niraparib, Olaparib (FDA-approved) |
| TWIST1 | NM_000474.4 | chr7 | 11 | 2 | ||||||
| RNF40 | NM_014771.4 | chr16 | 48 | 20 | ||||||
| PALB2 | NM_024675.4 | chr16 | 32 | 13 |
Exemplary PCR results were achieved using gene panel primers. A. PTEN gene with partial primers. B. TP53 gene with total primers. C. RNF40 gene with partial primers. D. KMT2C gene with partial primers.
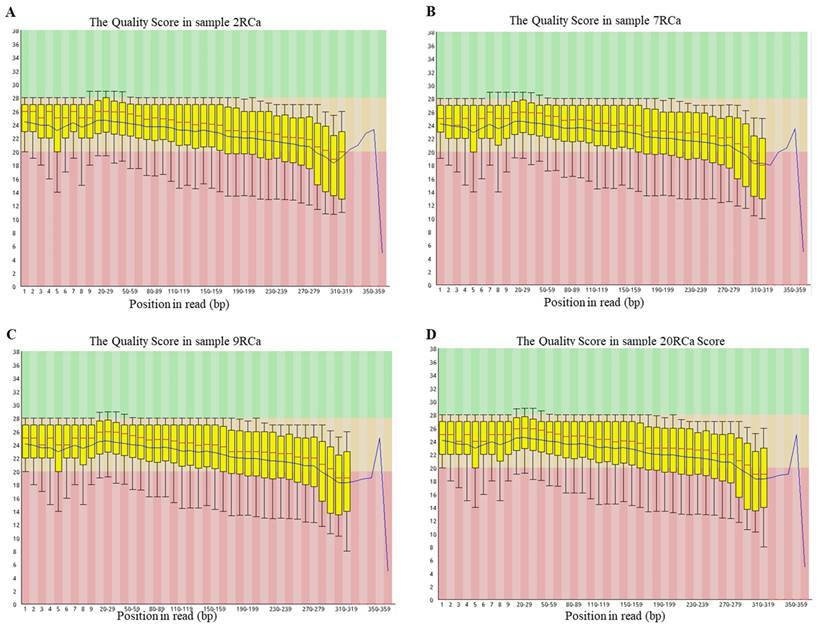
After the successful design of the gene panel primers, MPCR amplification was conducted, and subsequent purification of the products was performed. All samples underwent library construction and successfully passed quality control measures, as depicted in Figure 4, prior to undergoing NGS. The target fragment distribution of the normal library was between 280 bp and 460 bp, and the main peak was about 391 bp. Through bioinformatics analysis of sequenced data, known various types of variants were identified for each sample [Table 3].
Representative quality score profiles for different samples were generated using Agilent technology. A&B&C&D. The quality score was determined for sample 2RCa, sample 7RCa, sample 9RCa, sample 20RCa, respectively.
Known pathogenic variants result
| Sample | Type | Genotype | Gene | Function | Exon | Protein | Coding | ClinVar | Chromosome |
|---|---|---|---|---|---|---|---|---|---|
| 2RCa | SNV | A/T | PIK3CA | missense | 21 | p.His1047Leu | c.3140A>T | Pathogenic | chr3:178952085 |
| 20RCa | SNV | A/G | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952085 |
| 26RCa | SNV | A/G | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952085 |
| 36RCa | SNV | CA/CG | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952084 |
| 40RCa | SNV | ATC/GTC | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952085 |
| 40RCa | SNV | T/C | TP53 | missense | 8 | p.Arg280Gly | c.838A>G | Conflicting Interpretations of pathogenicity | chr17:7577100 |
| 42RCa | SNV | A/G | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952085 |
| TG15 | SNV | ACA/ACG | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952083 |
| TG19 | SNV | A/G | PIK3CA | missense | 21 | p.His1047Arg | c.3140A>G | Pathogenic | chr3:178952085 |
| TG46 | SNV | A/T | PIK3CA | missense | 21 | p.His1047Leu | c.3140A>T | Pathogenic | chr3:178952085 |
| 9RCa | SNV | C/G | PALB2 | missense | 10 | p.Glu1018Asp | c.3054G>C | Conflicting Interpretations of pathogenicity | chr16:23632742 |
| 27RCa | SNV | C/T | TP53 | nonsense | 4 | p.Trp53Ter | c.158G>A | Pathogenic | chr17:7579529 |
| 31RCa | INDEL | GTTTA/G | BRCA2 | frameshiftDeletion | 10 | p.Ile591MetfsTer22 | c.1773_1776delTTAT | Pathogenic | chr13:32907382 |
| 50RCa | SNV | T/A | PIK3CA | missense | 5 | p.Asn345Lys | c.1035T>A | Pathogenic | chr3:178921553 |
| TG02 | SNV | G/C | TP53 | nonsense | 5 | p.Ser166Ter | c.497C>G | Pathogenic | chr17:7578433 |
| TG02 | SNV | T/G | BRCA1 | unknown | 14 | p.? | c.4485-2A>C | Likely pathogenic | chr17:41226540 |
| TG04 | SNV | C/T | TP53 | nonsense | 4 | p.Trp91Ter | c.273G>A | Pathogenic | chr17:7579414 |
| TG34 | INDEL | CAAAAC/CAAAACA | BRCA2 | frameshiftInsertion | 11 | p.Ala1996SerfsTer7 | c.5985_5986insA | Pathogenic | chr13:32914472 |
| TG38 | SNV | T/C | TP53 | missense | 5 | p.Tyr163Cys | c.488A>G | Pathogenic | chr17:7578442 |
| TG39 | SNV | G/A | PIK3CA | missense | 2 | p.Arg38His | c.113G>A | Likely pathogenic | chr3:178916726 |
| TG40 | SNV | C/T | TP53 | missense | 7 | p.Gly245Ser | c.733G>A | Pathogenic | chr17:7577548 |
| TG52 | SNV | G/C | TP53 | missense | 6 | p.His193Asp | c.577C>G | Likely pathogenic | chr17:7578272 |
3.3 Variant verification results
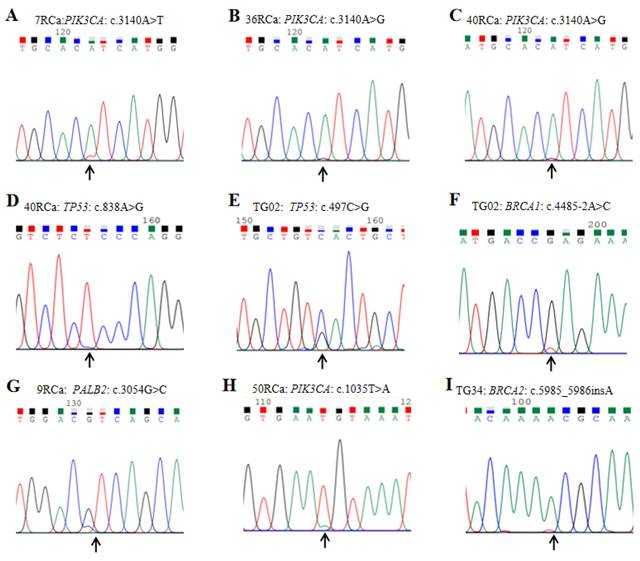
To validate the genetic variations identified by the gene panel, the Sanger sequencing method was employed to confirm partial variants, and representative outcomes are presented in Figure 5. All detected variants were successfully validated through Sanger sequencing. It is worth noting that certain patients exhibited pathogenic variants with SNV peak values less than one-fourth of the allele nucleotide value. Notably, different samples displayed identical variants at the same position with distinct nucleotide types, for instance, samples 7RCa and 36RCa showed PIK3CA variant c.3140A>T and c.3140A>G, respectively (Figure 5 A&B). Furthermore, the sequencing results revealed that a single sample harbored multiple variants in different genes; for example, sample 40RCa exhibited PIK3CA and TP53 variants c.3140A>T and c.838A>G, respectively (Figure 5 C&D), while in sample TG02 demonstrated TP53 and BRCA1 variant c.497C>G and c.4485-2A>C, respectively (Figure 5 E&F). Additionally, we observed PALB2 variant c.3054G>C in sample 9RCa, PIK3CA variant c.1035T>A in sample 50RCa, as well as BRCA2 variant c.5985_5986insA in sample TG34, respectively (Figure 5 G&H&I).
Representative program profiles for variant verification by Sanger sequencing are presented as follows: A. The Sanger sequencing results of sample 7RCa reveal the presence of PIK3CA variant c.3140A>T. B. In sample 36RCa, the Sanger sequencing results indicate the occurrence of PIK3CA variant c.3140A>G. C&D. Sample 40RCa exhibits two variants, namely PIK3CA variant c.3140A>T and TP53 variant c.838A>G, as confirmed by Sanger sequencing. E&F. The Sanger sequencing analysis of sample TG02 identifies TP53 variant c.497C>G and BRCA1 variant c.4485-2A>C. G. The PALB2 variant c.3054G>C is detected in sample 9RCa through Sanger sequencing. H. In sample 50RCa, a PIK3CA variant c.1035T>A is identified using Sanger sequencing. I. Sample TG34 shows an insertion mutation (BRCA2 variant c.5985_5986insA) according to the results obtained from Sanger sequencing.
3.4 Results of different variants' distribution
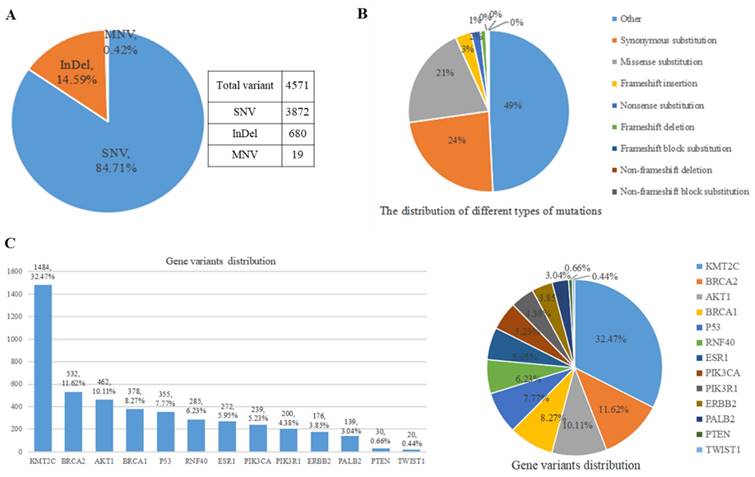
By conducting bioinformatics analysis, 4,571 variants were identified in the annotations files from 70 samples and classified into different types (Figure 6). All the variants were somatic changes. The predominant type of variants was single nucleotide variation (SNV), accounting for approximately 84.71% (3,872 variants) of the total. The second most common type was insertion and deletion (InDel), comprising about 14.59% (680 variants) of all variants. Additionally, multiple nucleotide variants (MNV) were also observed (Figure 6A). Depending on the specific nucleotide alterations, amino acid changes may occur which can potentially impact protein function. These genetic variations were further categorized into various types, including synonymous mutations, missense mutations, frameshift insertion/deletion/block substitution, non-frameshift deletion/block substitution, and other types (Figure 6B). Figure 6B illustrates that the three most prevalent types of amino acid changes were synonymous mutations, missense mutations, and frameshift insertion with frequencies of 23.54% (1076 variants), 20.46% (935 variants), and 3.24% (148 variants), respectively. Through analyzing the distribution patterns of these variants across different genes and their positions within them (Figure 6C), it was found that KMT2C gene exhibited the highest number of variant occurrences with a count of around 1484 representing approximately 32.47% of all detected variants. Following this, the gene BRCA2 displayed the second-highest frequency with 531 variants accounting for 11.62% of all detected variants. The gene TWIST1 had the lowest number of variants with only 20 around representing approximately 0.44% of all detected variants.
Results of different variants of the panel encompassing 13 genes examined in a cohort comprising 52 distinct breast cancer samples and individuals without the disease. A. Distribution analysis was conducted for missense, nonsense, synonymous, and intron mutation. B. The SNV, InDel, and MNV distribution was also investigated. C. Comprehensive assessment of all variant types within our gene panel was performed. Abbreviations: SNV: Single nucleotide variant; InDel: Insertion-Deletion; MNV: Multiple Nucleotide Variant.
3.5 New variants found in our cohort
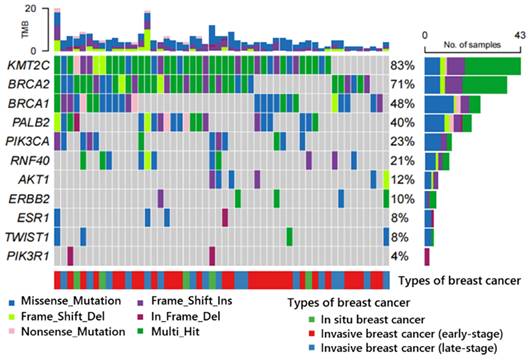
The majority of the variants were identified through comprehensive databases such as ClinVar, OMIM, COSMIC dbsnp, ExAC, etc. Variants that could not be retrieved from these databases were meticulously analyzed and considered for the first time within our cohort. The 358 novel variants (approximately 7.8% of the overall count) were discovered from 52 patients. Subsequently, bioinformatics analysis was conducted to obtain annotation files and classify these variants into distinct types (Figure 7). Figure 7 illustrates that these newly identified variants are distributed among eleven genes, with KMT2C gene exhibiting the highest prevalence in 83.0% of samples. Additionally, BRCA2 (71%), BRCA1 (48%), PALB2 (40%), PIK3CA (23%), and RNF40 (21%) genes displayed variant frequencies exceeding 20% of samples. Furthermore, AKT1 (12%), ERBB2 (10%), ESR1 (8%), TWIST1 (8%), and PIK3R1 (4%) genes exhibited variant occurrences; however, no new variants were detected in TP53 and PTEN genes.
The distribution of all new variants in 11 genes (KMT2C (83.0%), BRCA2(71%), BRCA1 (48%), PALB2(40%), PIK3CA (23%), RNF40 (21%), AKT1 (12%), ERBB2 (10%), ESR1 (8%), TWIST1 (8%) and PIK3R1 (4%)) was observed in a cohort of 52 patients. Notably, no new variant was identified in TP53 and PTEN genes. These variants were classified into different types, including Missense_mutation, Frame_Shift_Ins, Frame_Shift_Del, Nonsense_mutation, and In_Frame_Del. Additionally, the samples were categorized into three groups: in situ breast cancer with 5 patients, invasive breast cancer (early-stage) with 30 patients, and invasive breast cancer (late-stage) with 17 patients. Abbreviations: Frame_Shift_Ins: Frameshift insertion; Frame_Shift_Del: Frame shift deletion; In_Frame_Del: Inframe deletion; Multi_Hit: Multiple Hit.
Depending on the type of function, missense, frameshift insertion, frameshift deletion, nonsense, and in-frame deletion are observed. Missense variants constitute the predominant type with a total frequency of 40%, followed by frameshift insertion at 23%. The remaining variants collectively account for less than 40% of the occurrences. Notably, certain genes (KMT2C and BRCA2) exhibit multiple hits as previously mentioned.
We identified somatic mutations by adding blood samples of healthy individuals to exclude the germline mutations. Based on the sequencing data, the variant allele frequencies were low and all were greater than 2%, which also showed these variants were somatic mutations. PolyPhen-2 and SIFT are commonly employed in-silico tools for missense variant interpretation. A PolyPhen-2 prediction score above 0.3 indicates a probable damaging effect of the variant, resulting in a total of 89 variants (24.9%) out of 358 (52 patients) with a score exceeding this threshold value. Disease-causing (DC) variants exhibit a SIFT value below 0.05, leading to the identification of 101 variants with scores lower than this cutoff among the total number analyzed. Notably, when considering both PolyPhen-2 scores above 0.3 and the SIFT scores below 0.05, a subset of 68 pathogenic variants was found within our cohort consisting of 358 variants across 52 patients [Table 4]. At the same time, CADD and Mutation Taster tools were also used to analyze these variants. The prediction of the Mutation Taster showed that 74.24% of these variants are disease-causing (exclude 17 polymorphisms) and their corresponding probability value is shown in Table 4. In addition, 78.79% of the novel variants had a PHRED score greater than 20 in the CADD tool. Taken together, a combination of PolyPhen-2, SIFT, CADD, and Mutation Taster tools' analysis, 49 variants (49/358) had potential pathogenic effects on protein functions and structures across 52 patients. According to the silico prediction, the variants were conserved across different species and not in the germline.
Results of new probable damaging variants found in our cohort
| Sample | Gene | Chromosome | Function | Exon | Protein | Coding | Variant Allele Frequency | Sift | Polyphen | MutationTaster | PHRED |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2RCa | KMT2C | chr7:151949631 | Missense_Mutation | 10 | p.Arg490Gly | c.1468A>G | 2.94% | 0 | 0.999 | 1 | 19.31 |
| 2RCa | BRCA2 | chr13:32914067 | Non_Synonymous_Mutation | 11 | p.Lys1861=;Val1862Met | c.5583_5584delAGinsGA | 3.64% | 0.04 | 0.937 | poly | - |
| 7RCa | BRCA2 | chr13:32906985 | Missense_Mutation | 10 | p.Lys457Arg | c.1370A>G | 6.58% | 0.01 | 0.949 | poly | 17.19 |
| 9RCa-1 | BRCA2 | chr13:32906459 | Missense_Mutation | 10 | p.His282Asn | c.844C>A | 4.17% | 0.05 | 0.723 | poly | 6.942 |
| 10RCa | KMT2C | chr7:151960147 | Missense_Mutation | 9 | p.Leu418Pro | c.1253T>C | 3.51% | 0 | 1 | 0.999 | 28.6 |
| 11RCa | KMT2C | chr7:151880075 | Missense_Mutation | 35 | p.Glu1750Gly | c.5249A>G | 3.57% | 0 | 0.999 | 0.999 | 26.5 |
| 11RCa | BRCA2 | chr13:32893237 | Missense_Mutation | 3 | p.Trp31Arg | c.91T>A | 22.49% | 0 | 1 | 0.999 | 25.0 |
| 11RCa | PALB2 | chr16:23619317 | Missense_Mutation | 12 | p.Val1073Ala | c.3218T>C | 3.45% | 0 | 0.994 | poly | 24.2 |
| 12RCa | KMT2C | chr7:151853395 | Missense_Mutation | 45 | p.Ser3903Pro | c.11707T>C | 2.56% | 0.02 | 1 | 0.999 | 26.8 |
| 12RCa | KMT2C | chr7:151873368 | Missense_Mutation | 38 | p.Asp3057Gly | c.9170A>G | 2.9% | 0 | 1 | 0.999 | 26.0 |
| 12RCa | KMT2C | chr7:151874022 | Missense_Mutation | 38 | p.Glu2838Val | c.8513A>T | 3.13% | 0 | 0.895 | 0.994 | 21.7 |
| 13RCa | PALB2 | chr16:23614913 | Missense_Mutation | 13 | p.Leu1143Pro | c.3428T>C | 2.67% | 0.02 | 0.562 | 0.999 | 24.7 |
| 16RCa | KMT2C | chr7:151833939 | Missense_Mutation | 59 | p.Asn4905Ser | c.14714A>G | 2.94% | 0 | 0.997 | 0.999 | 25.9 |
| 17RCa | TWIST1 | chr7:19156497 | Missense_Mutation | 1 | p.Lys150Glu | c.448A>G | 5.36% | 0 | 0.993 | 0.999 | 32 |
| 17RCa | ERBB2 | chr17:37871589 | Missense_Mutation | 10 | p.Phe400Ser | c.1199T>C | 2.7% | 0 | 1 | 0.999 | 27.1 |
| 18RCa | PIK3CA | chr3:178919182 | Missense_Mutation | 4 | p.Glu223Lys | c.667G>A | 2.94% | 0 | 0.834 | 0.999 | 25.0 |
| 18RCa | TWIST1 | chr7:19156473 | Missense_Mutation | 1 | p.Phe158Leu | c.472T>C | 3.77% | 0 | 1 | 0.999 | 32 |
| 18RCa | BRCA2 | chr13:32907192 | Missense_Mutation | 10 | p.Asp526Gly | c.1577A>G | 2.88% | 0.01 | 0.354 | poly | 11.21 |
| 18RCa | RNF40 | chr16:30774446 | Missense_Mutation | 3 | p.Met47Thr | c.140T>C | 7.02% | 0 | 0.774 | 0.999 | 25.7 |
| 25RCa | KMT2C | chr7:151879465 | Missense_Mutation | 36 | p.Lys1827Arg | c.5480A>G | 4.4% | 0 | 0.999 | 0.868 | 24.3 |
| 26RCa | KMT2C | chr7:151873278 | Missense_Mutation | 38 | p.Ile3087Thr | c.9260T>C | 3.41% | 0 | 0.985 | 0.999 | 29.8 |
| 26RCa | BRCA2 | chr13:32906459 | Missense_Mutation | 10 | p.His282Asn | c.844C>A | 4.17% | 0.05 | 0.723 | poly | 6.942 |
| 26RCa | RNF40 | chr16:30774446 | Missense_Mutation | 3 | p.Met47Thr | c.140T>C | 7.02% | 0 | 0.774 | 0.999 | 25.7 |
| 27RCa | BRCA2 | chr13:32907224 | Missense_Mutation | 10 | p.Glu537Gly | c.1610A>G | 3.47% | 0.01 | 0.84 | poly | 13.90 |
| 29RCa | TWIST1 | chr7:19156511 | Missense_Mutation | 1 | p.Lys145Arg | c.434A>G | 2.74% | 0 | 0.969 | 0.999 | 33 |
| 29RCa | TWIST1 | chr7:19156527 | Missense_Mutation | 1 | p.Ser140Thr | c.418T>A | 2.74% | 0 | 1 | 0.999 | 31 |
| 29RCa | TWIST1 | chr7:19156545 | Missense_Mutation | 1 | p.Ile134Val | c.400A>G | 2.67% | 0 | 0.542 | 0.999 | 30 |
| 31RCa | BRCA1 | chr17:41245607 | Missense_Mutation | 10 | p.Ser647Arg | c.1941T>A | 3.64% | 0 | 0.991 | 0.891 | 22.4 |
| 33RCa | KMT2C | chr7:151879333 | Missense_Mutation | 36 | p.Ala1871Val | c.5612C>T | 3.45% | 0 | 0.361 | poly | 22.0 |
| 34RCa | PALB2 | chr16:23614949 | Missense_Mutation | 13 | p.Ile1131Thr | c.3392T>C | 2.6% | 0 | 0.603 | 0.712 | 25.5 |
| 36RCa | AKT1 | chr14:105236736 | Missense_Mutation | 14 | p.Asp462Gly | c.1385A>G | 2.99% | 0 | 0.346 | 0.999 | 19.87 |
| 36RCa | BRCA1 | chr17:41245353 | Missense_Mutation | 10 | p.Glu732Gly | c.2195A>G | 2.67% | 0 | 0.868 | poly | 13.11 |
| 40RCa | PIK3CA | chr3:178952118 | Missense_Mutation | 21 | p.Ile1058Thr | c.3173T>C | 2.73% | 0 | 0.717 | 0.999 | 24.6 |
| 42RCa | KMT2C | chr7:151860736 | Missense_Mutation | 43 | p.Gln3309Arg | c.9926A>G | 2.74% | 0.05 | 0.671 | poly | 14.94 |
| 42RCa | PALB2 | chr16:23614925 | Missense_Mutation | 13 | p.Ile1139Thr | c.3416T>C | 2.53% | 0 | 0.946 | poly | 25.1 |
| 42RCa | PALB2 | chr16:23614931 | Missense_Mutation | 13 | p.Ile1137Thr | c.3410T>C | 3.8% | 0 | 0.998 | 0.994 | 25.9 |
| 43RCa | KMT2C | chr7:151833981 | Missense_Mutation | 59 | p.Glu4891Gly | c.14672A>G | 2.7% | 0 | 0.999 | 0.999 | 32.0 |
| 43RCa | KMT2C | chr7:151833997 | Missense_Mutation | 59 | p.Tyr4886His | c.14656T>C | 2.67% | 0 | 1 | 0.999 | 29.9 |
| 43RCa | KMT2C | chr7:151876970 | Missense_Mutation | 37 | p.Gly2464Glu | c.7391G>A | 2.82% | 0.01 | 0.998 | 0.999 | 24.8 |
| 43RCa | BRCA2 | chr13:32910755 | Missense_Mutation | 11 | p.Ser755Pro | c.2263T>C | 2.96% | 0.03 | 0.469 | poly | 11.00 |
| 43RCa | RNF40 | chr16:30780523 | Missense_Mutation | 16 | p.Leu755Pro | c.2264T>C | 2.52% | 0 | 1 | 0.999 | 29.6 |
| 46RCa | BRCA2 | chr13:32912838 | Missense_Mutation | 11 | p.Phe1449Ser | c.4346T>C | 2.56% | 0 | 0.861 | poly | 22.7 |
| 46RCa | PALB2 | chr16:23619314 | Missense_Mutation | 12 | p.Leu1074Arg | c.3221T>G | 4.11% | 0 | 1 | 0.986 | 27.1 |
| 46RCa | PALB2 | chr16:23619329 | Missense_Mutation | 12 | p.Leu1069Arg | c.3206T>G | 2.78% | 0 | 1 | 0.999 | 26.1 |
| 55RCa | RNF40 | chr16:30776578 | Missense_Mutation | 7 | p.Gln283Arg | c.848A>G | 4.84% | 0 | 0.999 | 0.999 | 24.0 |
| TG23 | ERBB2 | chr17:37873643 | Missense_Mutation | 15 | p.Gly603Asp | c.1808G>A | 2.7% | 0 | 1 | 0.999 | 28.9 |
| TG30 | KMT2C | chr7:151849828 | Missense_Mutation | 49 | p.Leu4163Pro | c.12488T>C | 3.85% | 0 | 0.998 | 0.999 | 24.6 |
| TG30 | BRCA2 | chr13:32911820 | Missense_Mutation | 11 | p.Glu1110Lys | c.3328G>A | 5.11% | 0 | 1 | 0.999 | 27.4 |
| TG30 | PALB2 | chr16:23625332 | Missense_Mutation | 11 | p.Ser1065Phe | c.3194C>T | 3.57% | 0 | 1 | 0.999 | 28.9 |
| TG01 | BRCA2 | chr13:32937375 | Missense_Mutation | 18 | p.Asp2679Gly | c.8036A>G | 2.9% | 0 | 0.997 | 0.999 | 26.0 |
| TG02 | BRCA1 | chr17:41245161 | Missense_Mutation | 10 | p.Thr796Ile | c.2387C>T | 15.76% | 0.01 | 0.707 | poly | 15.36 |
| TG04 | BRCA2 | chr13:32971060 | Missense_Mutation | 26 | p.Ala3176Val | c.9527C>T | 3.7% | 0 | 0.989 | 0.995 | 23.0 |
| TG15 | BRCA2 | chr13:32906526 | Missense_Mutation | 10 | p.Glu304Gly | c.911A>G | 3.33% | 0.05 | 0.996 | poly | 22.9 |
| TG19 | KMT2C | chr7:152008963 | Missense_Mutation | 5 | p.Gln220Arg | c.659A>G | 3.7% | 0 | 0.534 | poly | 18.14 |
| TG26 | PALB2 | chr16:23614919 | Missense_Mutation | 13 | p.Asp1141Gly | c.3422A>G | 3.08% | 0.01 | 1 | 0.999 | 26.7 |
| TG26 | BRCA1 | chr17:41243655 | Missense_Mutation | 10 | p.Ser1298Pro | c.3892T>C | 2.67% | 0 | 0.998 | 0.975 | 23.7 |
| TG38 | AKT1 | chr14:105239701 | Missense_Mutation | 10 | p.Leu282Val | c.844C>G | 26.9% | 0 | 0.464 | 0.999 | 23.2 |
| TG38 | RNF40 | chr16:30779233 | Missense_Mutation | 12 | p.Met483Thr | c.1448T>C | 3.77% | 0 | 0.999 | 0.999 | 27.8 |
| TG39 | PIK3CA | chr3:178938941 | Missense_Mutation | 14 | p.Gln728Arg | c.2183A>G | 3.31 | 0 | 0.985 | 0.999 | 23.6 |
| TG40 | PALB2 | chr16:23614982 | Missense_Mutation | 13 | p.Glu1120Gly | c.3359A>G | 3.66% | 0 | 1 | 0.999 | 25.8 |
| TG40 | RNF40 | chr16:30774446 | Missense_Mutation | 3 | p.Met47Thr | c.140T>C | 7.02% | 0 | 0.774 | 0.999 | 25.7 |
| TG52 | KMT2C | chr7:151949652 | Missense_Mutation | 10 | p.Leu483Pro | c.1448T>C | 2.94% | 0 | 1 | 0.999 | 27.0 |
| 60RCa | PIK3CA | chr3:178928058 | Missense_Mutation | 8 | p.Trp446Arg | c.1336T>A | 3.17% | 0 | 1 | 0.999 | 28.2 |
| 60RCa | KMT2C | chr7:151845181 | Missense_Mutation | 52 | p.Phe4611Leu | c.13831T>C | 3.09% | 0 | 0.999 | 0.999 | 23.7 |
| 60RCa | KMT2C | chr7:151845729 | Missense_Mutation | 52 | p.Val4428Ala | c.13283T>C | 2.55% | 0.01 | 0.944 | 0.999 | 26.6 |
| 60RCa | BRCA1 | chr17:41244886 | Missense_Mutation | 10 | p.His888Tyr | c.2662C>T | 3.13% | 0.02 | 0.767 | poly | 12.67 |
Note: Here, 49 variants had potential pathogenic effects on protein functions and structures across 52 patients. poly: polymorphism
4. Discussion
The technology developed for NGS has revolutionized the clinical approach to genetic testing in the field of cancer. Multi-gene panels have demonstrated powerful capabilities in identifying pathogenic variants (PVs) within known BC-related genes, as well as novel variants potentially associated with the disease. Recently, a combination of hereditary gene panels targeting hot mutations such as BRCA1/2 and PALB2, along with multiple genes with various cancers, applied for gene diagnosis [6, 8, 12, 14, 38].
A few of these gene variants were not found in ClinVar, OMIM, and COSMIC databases within the gene panel of variants distribution. Novel mutations were initially discovered in our cohort and confirmed as novel through the ExAC database. Specifically, 68 new variants from PolyPhen-2 and SIFT analysis exhibited potential pathogenicity, providing evidence for ACMG that supports the need for more specific pathogenicity classification when combined with other evidence to enhance clinical application [47]. These pathogenic variants increase the risk of BC and offer guidance for early detection/diagnosis. All newly identified variants in the KMT2C gene ranked the highest within our cohort, which has been recently reported as a biomarker for chemotherapy response [12, 29]. The distribution pattern of new variants in BRCA1/2 genes is as follows: BRCA1/2 genes are associated with BC pathogenesis and account for approximately 5%-10% of known pathogenic variations among all BC-related genes; they also act as modifiers of hereditary BC risk. Analysis conducted on a large number of Chinese hereditary BC patients revealed that germline variations in the BRCA1/2 genes exhibit high ethnic specificity [7, 14, 15, 32, 36, 37]. The PALB2 (partner and localizer of BRCA2) gene ranks after KMT2C and BRCA1/2 genes among new variants, it encodes a protein that interacts with BRCA2, and its mutations are associated with significantly increased female BC risk [11]. PIK3CA (23%) and RNF40 (21%) variants were detected in over 20% of samples. The mutation rate of PIK3CA is particularly high in male breast cancer (MBC), ranking first among known BC-associated variations recorded in the COSMIC database. Among our patients' cases involving this gene mutation type p. H1047R was predominant [12, 19, 31, 51]. In our novel variants of the RNF40 gene, 19 additional variants were identified, out of which six were classified as DC mutation based on PolyPhen-2 scores greater than 0.3 and SIFT scores less than 0.05. RNF40 has been reported to exhibit both tumor-suppressive and oncogenic roles in BC cells. The occurrence of these gene variants in BC patients has been relatively infrequent [33-35]. Similarly, the distribution of new variants in AKT1 (12%), ERBB2 (10%), ESR1(8%), TWIST1(8%), and PIK3R1(4%) genes was found to be less than 15% among all patients [31]. These genetic variations across multiple genes pose an increased risk of BC development in humans; however, early detection can facilitate timely diagnosis, prevention, and treatment.
Variants of the TP53 and PTEN genes were identified in our patient cohort. Additionally, these variants have been associated with an increased susceptibility to cancer development. Tumor protein p53 (TP53), a transcription factor, is a well-known tumor suppressor gene that exhibits high mutation rates across various cancer types, including BC. Although germline in the TP53 gene is rare and occurs in only approximately 1% of all BC cases, carriers of such mutation face a significantly higher lifetime risk of developing BC, estimated at around 80%-90%, compared to non-carriers [14, 30]. A recent study has also demonstrated that pathogenic variants in the PTEN gene are linked to earlier disease onset and an elevated risk for female BC [13, 14].
Despite extensive research on clinical inhibitors for most high-risk genes with hotspot variants in this gene panel [6, 10, 19-23, 25-28, 50, 55], our study acknowledges certain limitations. The majority of these variants are somatic mutations found in sporadic cases of BC. However, the resistance mechanisms to novel pathogenic variants remain elusive. Therefore, identifying and targeting these pathogenic variants for early BC treatment still presents a significant challenge.
In our study of the patient tissue with clinical information, we consistently observed a large tumor size, with a maximum diameter of 55 mm, several times larger than that seen in BC patients from Western countries. The average age at diagnosis patients was around 50 years, contrasting with the late age at diagnosis typically observed in developed Western countries. Consequently, patients with tumors were more susceptible to metastasis and recurrence, leading to a decrease in their five-year survival rate [1-3, 10]. Early detection, diagnosis, and treatment of BC can improve the survival rate of patients. It is one of the few malignant tumors that can reduce the mortality rate through early diagnosis. Compared with conventional imaging examination, genetic detection has obvious advantages, which can diagnose BC earlier before the appearance of typical clinical manifestations and improve the level of cancer prevention and treatment. Therefore, the novel gene panel based on the high-risk genes may have a high value for the early detection of BC. Currently available data on germline and somatic mutations in BC patients are expanding. Our findings suggest that this novel variant holds significance for early diagnosis/prevention, and treatment strategies specifically tailored for Chinese individuals. Furthermore, it contributes to the overall knowledge base and facilitates clinical applications for diagnosing BC patients as well as providing genetic counseling services for affected families. Notably, utilizing a gene panel for patient gene diagnosis proves cost-effective compared to whole exome sequencing (WES). Compared with general NGS, the gene panel is based on high-risk genes of BC, which has fewer genes and high sequencing depth, easier interpretation, and more efficient detection. This approach not only benefits undeveloped regions of China like some areas in southwest China but also holds potential value for undeveloped countries across Asia or Africa, such as Thailand [4, 7, 10, 31, 38].
5. Conclusions
In conclusion, we have preliminarily established a cost-effective and high-risk gene panel for early detection of BC. We found a subset of 49 new pathogenic variants within our cohort consisting of 358 variants across 52 patients on protein functions and structures, and the non-pathogenic variants that may also play a risk factor in BC patients. This breakthrough will contribute to the timely identification, effective prevention, and preventative treatment of BC patients, particularly those residing in developing or undeveloped regions. Moreover, our cohort's discovery of novel pathogenic variants not only enriches the existing knowledge base for clinical application of genetic diagnosis in BC patients but also enhances genetic counseling services pertaining to both BC patients and their family pedigrees.
Acknowledgements
The authors thank the people from Chiang Mai University, the Center of Excellence in Pharmaceutical Nanotechnology, Chiang Mai University, and The Research Center for Preclinical Medicine, Southwest Medical University.
Funding
This study was supported by the Special Training Program for Young Science and Technology Talents from Southwest Medical University (No. 00031726), the Key Project of Applied Basic Research of Southwest Medical University (No. 2023ZD010), and the Project Fund for Young Innovative Talents of Huai'an No. 1 Hospital Affiliated to Nanjing Medical University (No. QC202209), and was partially funded by the Sichuan Science and Technology Program (No. 2022YFS0623), National Natural Science Foundation of Sichuan Province (No. 2025ZNSFSC0986), and the National Natural Science Foundation of China (Nos. 82073263 and 31701087).
Ethics approval and consent to participate
The study was approved by the Ethical Committee of Southwest Medical University and the Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University, and in accordance with the recommendations of the Helsinki Declaration to ensure compliance with ethical standards.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Author contributions
J.F. and S. A. were in charge of the idea, and project design. X. X., L.Y., and L.Z. recruited samples. J. C, C. W., L. Y., X.L., and B.S. performed DNA extraction, MPCR amplification and library construction, and sequencing. J. C, and B.S., performed bioinformatics analysis. S.T. and S.C. performed project design, review, and editing. J.C., J. F., and S. A. wrote and revised the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63
2. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17-48
3. Han B, Zheng R, Zeng H, Wang S, Sun K, Chen R. et al. Cancer incidence and mortality in China, 2022. J Natl Cancer Cent. 2024;4:47-53
4. Zeng H, Zheng R, Zhang S, Zou X, Chen W. Female breast cancer statistics of 2010 in China: estimates based on data from 145 population-based cancer registries. J Thorac Dis. 2014;6:466-70
5. Wang F, Yu ZG. Current status of breast cancer prevention in China. Chronic Dis Transl Med. 2015;1:2-8
6. Nagini S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer Agents Med Chem. 2017;17:152-63
7. Breast Cancer Association C, Dorling L, Carvalho S, Allen J, Gonzalez-Neira A, Luccarini C, et al. Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med. 2021;384:428-39
8. Precision medicine improves outcomes in metastatic breast cancer. Nature. 2022
9. Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C. et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8
10. Im SA, Gennari A, Park YH, Kim JH, Jiang ZF, Gupta S. et al. Pan-Asian adapted ESMO Clinical Practice Guidelines for the diagnosis, staging and treatment of patients with metastatic breast cancer. ESMO Open. 2023;8:101541
11. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A. et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165-7
12. Heng YJ, Hankinson SE, Wang J, Alexandrov LB, Ambrosone CB, de Andrade VP. et al. The Association of Modifiable Breast Cancer Risk Factors and Somatic Genomic Alterations in Breast Tumors: The Cancer Genome Atlas Network. Cancer Epidemiol Biomarkers Prev. 2020;29:599-605
13. Cummings S, Alfonso A, Hughes E, Kucera M, Mabey B, Singh N. et al. Cancer Risk Associated With PTEN Pathogenic Variants Identified Using Multigene Hereditary Cancer Panel Testing. JCO Precis Oncol. 2023;7:e2200415
14. Subasioglu A, Guc ZG, Gur EO, Tekindal MA, Atahan MK. Genetic, Surgical and Oncological Approach to Breast Cancer, with BRCA1, BRCA2, CDH1, PALB2, PTEN and TP53 Variants. Eur J Breast Health. 2023;19:55-69
15. Cheng J, Peng J, Fu J, Khan MA, Tan P, Wei C. et al. Identification of a novel germline BRCA2 variant in a Chinese breast cancer family. J Cell Mol Med. 2020;24:1676-83
16. Fu J, Qin L, He T, Qin J, Hong J, Wong J. et al. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275-89
17. Li D, Liu X, Zhang L, He J, Chen X, Liu S. et al. COVID-19 disease and malignant cancers: The impact for the furin gene expression in susceptibility to SARS-CoV-2. Int J Biol Sci. 2021;17:3954-67
18. Li D, Cheng J, Zhang W, Zhang L, Maghsoudloo M, Fu J. et al. Tripartite motif-containing 28 (TRIM28) expression and cordycepin inhibition in progression, prognosis, and therapeutics of patients with breast invasive carcinoma. J Cancer. 2024;15:4374-85
19. Zhou J, Imani S, Shasaltaneh MD, Liu S, Lu T, Fu J. PIK3CA hotspot mutations p. H1047R and p. H1047L sensitize breast cancer cells to thymoquinone treatment by regulating the PI3K/Akt1 pathway. Mol Biol Rep. 2022;49:1799-816
20. Jin RY, Tang T, Zhou S, Long X, Guo H, Zhou J. et al. Design, synthesis, antitumor activity and theoretical calculation of novel PI3Ka inhibitors. Bioorg Chem. 2020;98:103737
21. Tan ES, Cao B, Kim J, Al-Toubah TE, Mehta R, Centeno BA. et al. Phase 2 study of copanlisib in combination with gemcitabine and cisplatin in advanced biliary tract cancers. Cancer. 2021;127:1293-300
22. Cortesi L, Rugo HS, Jackisch C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. 2021;16:255-82
23. Yu D. Mechanisms of ErbB2-mediated paclitaxel resistance and trastuzumab-mediated paclitaxel sensitization in ErbB2-overexpressing breast cancers. Semin Oncol. 2001;28:12-7
24. Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977-84
25. Andrikopoulou A, Chatzinikolaou S, Panourgias E, Kaparelou M, Liontos M, Dimopoulos MA. et al. "The emerging role of capivasertib in breast cancer". Breast. 2022;63:157-67
26. Tharin Z, Richard C, Derangere V, Ilie A, Arnould L, Ghiringhelli F. et al. PIK3CA and PIK3R1 tumor mutational landscape in a pan-cancer patient cohort and its association with pathway activation and treatment efficacy. Sci Rep. 2023;13:4467
27. Brett JO, Spring LM, Bardia A, Wander SA. ESR1 mutation as an emerging clinical biomarker in metastatic hormone receptor-positive breast cancer. Breast Cancer Res. 2021;23:85
28. Berger F, Marce M, Delaloge S, Hardy-Bessard AC, Bachelot T, Bieche I. et al. Randomised, open-label, multicentric phase III trial to evaluate the safety and efficacy of palbociclib in combination with endocrine therapy, guided by ESR1 mutation monitoring in oestrogen receptor-positive, HER2-negative metastatic breast cancer patients: study design of PADA-1. BMJ Open. 2022;12:e055821
29. Liu X, Qiu R, Xu M, Meng M, Zhao S, Ji J. et al. KMT2C is a potential biomarker of prognosis and chemotherapy sensitivity in breast cancer. Breast Cancer Res Treat. 2021;189:347-61
30. Kwong A, Shin VY, Ho CYS, Au CH, Slavin TP, Weitzel JN. et al. Mutation screening of germline TP53 mutations in high-risk Chinese breast cancer patients. BMC Cancer. 2020;20:1053
31. Xiao W, Zhang G, Chen B, Chen X, Wen L, Lai J. et al. Characterization of Frequently Mutated Cancer Genes and Tumor Mutation Burden in Chinese Breast Cancer. Front Oncol. 2021;11:618767
32. Guindalini RSC, Viana DV, Kitajima J, Rocha VM, Lopez RVM, Zheng Y. et al. Detection of germline variants in Brazilian breast cancer patients using multigene panel testing. Sci Rep. 2022;12:4190
33. Fu S, Cheng J, Wei C, Yang L, Xiao X, Zhang D. et al. Development of diagnostic SCAR markers for genomic DNA amplifications in breast carcinoma by DNA cloning of high-GC RAMP-PCR fragments. Oncotarget. 2017;8:43866-77
34. Tarcic O, Granit RZ, Pateras IS, Masury H, Maly B, Zwang Y. et al. RNF20 and histone H2B ubiquitylation exert opposing effects in Basal-Like versus luminal breast cancer. Cell Death Differ. 2017;24:694-704
35. Fu J, Liao L, Balaji KS, Wei C, Kim J, Peng J. Epigenetic modification and a role for the E3 ligase RNF40 in cancer development and metastasis. Oncogene. 2021;40:465-74
36. Slavin TP, Niell-Swiller M, Solomon I, Nehoray B, Rybak C, Blazer KR. et al. Corrigendum: Clinical Application of Multigene Panels: Challenges of Next-Generation Counseling and Cancer Risk Management. Front Oncol. 2015;5:271
37. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K. et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25-33
38. Gardner SA, Weymouth KS, Kelly WS, Bogdanova E, Chen W, Lupu D. et al. Evaluation of a 27-gene inherited cancer panel across 630 consecutive patients referred for testing in a clinical diagnostic laboratory. Hered Cancer Clin Pract. 2018;16:1
39. Watahiki M, Kawahara R, Suzuki M, Aoki M, Uchida K, Matsumoto Y. et al. Single-Tube Multiplex Polymerase Chain Reaction for the Detection of Genes Encoding Enterobacteriaceae Carbapenemase. Jpn J Infect Dis. 2020;73:166-72
40. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
41. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297-303
42. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164
43. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812-4
44. Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A. et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355-8
45. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789-98
46. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062-D7
47. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24
48. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285-91
49. Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM. et al. A global reference for human genetic variation. Nature. 2015;526:68-74
50. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110-21
51. Hutchinson KE, Chen JW, Savage HM, Stout TJ, Schimmoller F, Cortes J. et al. Multiple PIK3CA mutation clonality correlates with outcomes in taselisib + fulvestrant-treated ER+/HER2-, PIK3CA-mutated breast cancers. Genome Med. 2023;15:28
52. Schubach M, Maass T, Nazaretyan L, Roner S, Kircher M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024;52:D1143-D54
53. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361-2
54. R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for statistical Computing. 2018
55. Fu J, Li D, Zhang L, Maghsoudloo M, Cheng J, Fu J. Comprehensive analysis, diagnosis, prognosis, and cordycepin (CD) regulations for GSDME expressions in pan-cancers. Cancer Cell Int. 2024;24:279
Author contact
![]() Corresponding authors: Dr. Junjiang Fu, Key Laboratory of Epigenetics and Oncology, the Research Center for Preclinical Medicine, Southwest Medical University, Luzhou, Sichuan 646000, P R China. Tel/Fax: +86-830-3160283; E-mail: fujunjiangedu.cn. Dr. Songyot Anuchapreeda, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand. E-mail: songyot.anuchac.th. Dr. Xiuli Xiao, E-mail: xiaoxiuliedu.cn.
Corresponding authors: Dr. Junjiang Fu, Key Laboratory of Epigenetics and Oncology, the Research Center for Preclinical Medicine, Southwest Medical University, Luzhou, Sichuan 646000, P R China. Tel/Fax: +86-830-3160283; E-mail: fujunjiangedu.cn. Dr. Songyot Anuchapreeda, Department of Medical Technology, Faculty of Associated Medical Sciences, Chiang Mai University, Chiang Mai 50200, Thailand. E-mail: songyot.anuchac.th. Dr. Xiuli Xiao, E-mail: xiaoxiuliedu.cn.