Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(8):2466-2475. doi:10.7150/jca.98431 This issue Cite
Research Paper
High MIG-6 expression promotes tumor proliferation and metastasis of gastric cancer
Wenqiu Zhao1,2*, Tao Jin3*, Yun Liu4, Shihe Shao3,4 ![]() , Feilun Cui2
, Feilun Cui2 ![]()
1. Department of Clinical Laboratory, Xuzhou Traditional Chinese Medicine Hospital Affiliated to Nanjing University of Chinese Medicine, China.
2. The Affiliated Taizhou Second People's Hospital of Yangzhou University, China.
3. Yixing People's Hospital, China.
4. School of Medicine, Jiangsu University, China.
* Wenqiu Zhao and Tao Jin contributed equally to this manuscript.
Received 2024-5-14; Accepted 2025-3-31; Published 2025-4-21
Abstract

Background: Mitogen-inducible gene-6 (MIG-6) is a feedback inhibitor that targets activated epidermal growth factor receptor (EGFR) and suppresses tumor growth fueled by constitutively activated EGFR. Nevertheless, the action mechanism of MIG-6 in gastric cancer (GC) remains to be elucidated.
Methods: Western blotting, fluorescence quantitative PCR, and immunohistochemistry were performed to detect the expression of MIG-6 in GC cell lines and tissues. Public databases were used to analyze MIG-6 in patients with GC. Furthermore, the GC cell lines were selected for the knockdown and overexpression of MIG-6.
Results: Bioinformatics and histological analyses showed that MIG-6 was elevated in human GC tissues and cells. The Kaplan-Meier plotter showed that patients with elevated MIG-6 expression had significantly shorter survival. Furthermore, small interference RNA-mediated reduction of MIG-6 expression decreased EGFR/AKT signaling, as well as the proliferation and metastasis of human GC cells in vitro, whereas its overexpression increased these actions. Also, MIG-6 overexpression promoted the epithelial-mesenchymal transition of GC cells as well as the expression of the migration-associated protein matrix metalloproteinase-9 in vitro.
Conclusion: These results suggest that MIG-6 can serve as a new prognostic biomarker or potential therapeutic target for the identification of patients with poor survival.
Keywords: Gastric cancer, EGFR, MIG-6, metastasis, proliferation, survival.
1. Introduction
The global incidence of gastric cancer (GC), a malignant tumor of the gastric mucosa, and the mortality associated with the disease rank fifth and fourth in human cancers, respectively [1]. High salt intake, a diet poor in fruits and vegetables, ethnicity, heredity, and Helicobacter pylori infection are the risk factors for the illness [2]. Unfortunately, the early stages of the disease are often asymptomatic, and at the time of surgery, approximately 60% of the patients with GC have locally progressed and metastatic illness. Surgical resection has a very low therapeutic efficacy [3, 4]. Therefore, identifying new biomarkers and investigating the molecular mechanisms of metastasis and proliferation for the diagnosis and treatment of GC require urgent attention.
Mitogen-inducible gene-6 (MIG-6), also referred to as ERBB receptor feedback inhibitor 1 (ERRFI1), receptor-associated late transducer (RALT), or gene 33, is an immediate early response gene that can be directly activated by several mitogens and common chronic stress stimuli, such as hypoxia, hormones, and growth factors (such as epidermal growth factor) [5]. MIG-6 encodes a scaffolding adaptor protein that interacts with other signal molecules to influence signal transduction. MIG-6 can selectively target activated epidermal growth factor receptor (EGFR) and act as an EGFR feedback inhibitor, which indicates its critical role in human cancers. Additionally, studies have revealed that MIG-6 inhibits EGFR signaling on two different levels: by binding to the kinase domain and decreasing EGFR catalytic activity and by causing EGFR molecules to be internalized and eventually degraded [6, 7]. For instance, Maity and Reschke [8, 9] observed that suppressing the expression of MIG-6 prolongs the activation of the EGFR signaling pathway and promotes the genesis and development of malignancies, including lung and liver tumors. According to research by Ying et al. [10], MIG-6 alleviates the malignant potential of glioblastoma multiforme cells by promoting EGFR trafficking into late endosomes/lysosomes and encouraging its destruction. Furthermore, in lung cancer and glioblastoma in humans, MIG-6 is genetically altered and transcriptionally repressed but not in other malignancies [9, 10]. Although several studies have shown that MIG-6 is a tumor suppressor gene in human malignancies, mounting evidence suggests that MIG-6 plays contradictory roles in growth regulation and tumor progression. He et al. [11], for instance, noted that MIG-6 controls an EGFR-independent pathway to stimulate the proliferation of triple-negative breast cancer cells. Additionally, Kang et al. [12] claimed that MIG-6 overexpression accelerates cell proliferation, invasion, and epithelial-mesenchymal transition (EMT) in EGFR-directed tyrosine kinase inhibitor (TKI)-resistant lung cancer cells compared with EGFR-TKI sensitive cells. These findings allude that MIG-6 is a context-dependent regulator in malignancies and that the connection between MIG-6 and EGFR is extremely complex. Thus, the role of MIG-6 in GC remains unclear.
Here, our bioinformatics and histological analyses uncover that MIG-6 is upregulated in GC and that MIG-6 upregulation is positively correlated with poorer clinical outcomes in GC. Moreover, MIG-6 overexpression increased EGFR/AKT signaling and promoted tumor proliferation and metastasis of GC cells in vitro. Also, MIG-6 overexpression induces EMT in GC cells. These results imply a hitherto unreported tumor-promoting role of MIG-6 in GC. Overall, our results showed that MIG-6 could regulate GC and may serve as a potential target for antineoplastic therapies.
2. Materials and Methods
2.1 Patients and samples
This study was approved by the Ethics Committee of the School of Medicine, Jiangsu University, and informed consent was obtained from all patients. A total of 53 surgical specimens of GC were randomly selected between 2015 and 2016 from the General Surgery Department of the First People's Hospital of Zhenjiang, Jiangsu, China. Inclusion criteria: patients who had not received any form of anti-tumor therapy, such as chemoradiotherapy before surgery, and were diagnosed with GC after a pathological diagnosis. Exclusion criteria: patients with a family history of GC and those who had received radiotherapy or chemotherapy. The samples were acquired from the tumor tissue (T, avoiding tumor necrotic region) and the matching paracancerous tissue (P, distance from tumor focus >5 cm). The Cancer Staging Manual, seventh edition, of the American Joint Committee on Cancer was used to determine the participants' tumor stages.
2.2 Immunohistochemistry
The gastric tumor tissues were serially sectioned at a thickness of 4 μm and embedded in paraffin. Hydrogen peroxide (3%) was used to block endogenous peroxidase activity for 10 min, and bovine serum albumin (5%, Boster Bioengineering, Wuhan, China) was used to block the slides. The tissue sections were incubated with primary antibodies MIG-6 (Cell Signaling Technology, 1:300) overnight at 4°C. The sections were subsequently incubated overnight in a humidified chamber at 4°C, visualized with 3,3 diaminobenzidine, and counterstained with hematoxylin and eosin for microscopic examination.
MIG-6 protein expression in the cytoplasm of tumor cells was evaluated by measuring the intensity of staining and the proportion of positively stained cells. The scoring system was as follows: For positive cells that stained yellowish, light-brown, or dark-brown, a score of 1-3 was given; otherwise, a score of 0 was awarded.
2.3 Cell culture
Human gastric epithelial cells GES-1 and the four human GC cell lines (BGC-823, SGC-7901, MGC-803, and AGS) were sourced from our laboratory. Fetal bovine serum (FBS, Gibco, Grand Island, NY, USA)-supplemented F12 medium (Gibco) was used to culture the AGS cells, and RPMI-1640 medium (Gibco) supplemented with 10% FBS was used to culture the other cancer cell lines. All cells were cultured in a humid incubator at 37°C with 5% CO2.
2.4 Plasmid construction and small interference RNA (siRNA) and cell transfection
The pcDNA-MIG-6 plasmids and vector plasmids were purchased from Bioworld Company (Nanjing, China). Lipofectamine 2000 (Invitrogen, Shanghai, China) was used to transfect the BGC-823 cells with the plasmids. Western blotting was performed to evaluate the efficiency of the overexpression. The siRNA-targeted MIG-6 procured from GenePharma Company (Shanghai, China) was transfected into SGC-7901 cells for downregulating the expression of MIG-6 in GC cells.
2.5 RNA extraction and real-time PCR analysis
Total RNA was extracted from paraffin sections of GC tissues using TRIzol reagent according to the manufacturer's instructions (Invitrogen, Life Technologies Corporation, CA, USA), and cDNA was synthesized using the HiScript® QRT SuperMix in the qPCR Kit (Vazyme, Nanjing, China). The mRNA levels of MIG-6 were evaluated using the Applied Biosystems Real-Time PCR System. The sequences of the qRT-PCR primers were as follows: MIG-6: 5′-ATAGAAGATGGTCAGCAGAAG-3′, MIG-6-reverse: 5′-CATTGAGGTAAGACGGAAGG-3′. The 2-∆∆CT relative quantitative approach was used to quantify the data, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was employed as the internal control.
2.6 Western blotting
Phenylmethanesulfonyl fluoride and phosphatase inhibitors were added to radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology, Shanghai, China), and the mixture was used to lyse cell proteins. An equal amount of proteins (100 μg) was then separated using SDS-PAGE (12%) and transferred to polyvinylidene fluoride membranes. Before being incubated with primary antibodies overnight at 4°C, the membranes were blocked with 5% nonfat dry milk powder for 2 h at room temperature. The enhanced chemiluminescence system (Image Quant LAS 4000 mini, USA) was used to view the membranes after applying the appropriate secondary antibody at room temperature for 2 h. The relative densities of the bands were measured using ImageJ (http://rsb.info.nih.gov/ij/). The relative protein levels were calculated using the loading control GAPDH. Each test was repeated at least three times.
2.7 Transwell migration assay
CoStar transwell chambers (8 m pore size, Corning, NY, USA) were used for the transwell migration tests, which were performed with 1 × 105 cells per well. Cells were seeded in the top chambers of the wells using 300 μL of serum-free medium to promote cell migration, whereas the lower chambers were filled with 600 μL of medium containing 10% FBS. A cotton swab was used to scrape out the cells on the upper side of the membrane during 24 h of incubation at 37°C and 5% CO2. The cells that had moved to the bottom surface of the membrane were stained with crystal violet after being fixed with 4% paraformaldehyde. The cells were photographed and counted in five arbitrary fields.
2.8 Cell proliferation assay
Following the manufacturer's instructions, the cell counting kit-8 (CCK-8 kit, Tongren, Shanghai, China) was used to assess the proliferative ability of BGC-823 and SGC-7901 cells. In 96-well plates, 1 × 103 transfected cells were planted and cultured. CCK-8 solution (10 μL) was then added to each well before being incubated for 1 h. At different time intervals (24 h, 48 h, and 72 h), the absorbance at 450 nm was measured using a microplate reader.
2.9 Colony-formation assay
Colony-formation experiments were performed in triplicate on six-well plates with 1,000 cells per well. The cells were cultivated in a humidified incubator with 5% CO2 at 37°C. When discernible clones developed in the wells after 14 d, culturing was stopped, and the cells were fixed with 4% paraformaldehyde, and stained with crystal violet.
2.10 Statistical analyses
GraphPad 8.0 was used for all statistical analyses. The student's t-test was used to identify whether significant differences were present between groups. Fisher's exact test was used to determine the relationship between MIG-6 expression and clinicopathological variables. Overall survival curves were produced using the Kaplan-Meier technique, and the log-rank test was used for comparison and Fisher's exact test for estimation. Statistical significance was considered as a P value of <0.05. All tests were conducted at least three times.
3. Results
3.1 MIG-6 expression in GC tissues is positively correlated with disease progression and poor prognosis
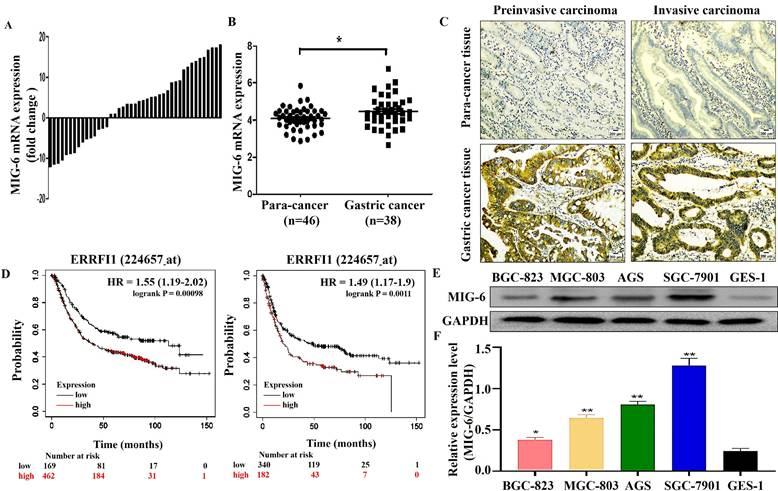
To investigate MIG-6 expression in GC, MIG-6 mRNA levels in GC tissues and paracancerous tissues were determined using qRT-PCR. The findings indicated that among the 43 paraffin sections of the GC tissues, the MIG-6 mRNA level was elevated in 65.1% (28/43) of the tissues (Figure 1A), which agrees with the analysis of the ONCOMINE dataset (Figure 1B) (https://www.oncomine.org). Immuno-histochemical examination of MIG-6 protein expression in 53 pairs of tumor tissues and paracancerous tissues revealed that the expression was positive in GC tissues (Figure 1C). In addition, MIG-6 expression was considerably higher in the cancer tissues of 64.2% (34/53) patients than in matching paracancerous tissues. Moreover, high MIG-6 expression was positively correlated with higher differentiation (P = 0.02) and advanced clinical stage (P = 0.02) in patients with GC (Table 1). Furthermore, the Kaplan-Meier plotter signified that MIG-6 protein expression was inversely correlated with overall survival and progression-free survival in patients with GC (Figure 1D), indicating that MIG-6 may be a predictive biomarker for poor prognosis. As this result differs from those from previous publications on MIG-6, the following functional and molecular testing studies were performed.
The expression of MIG-6 in the GC tissues and cell lines. (A) The mRNA level of MIG-6 in 43 pairs of GC tissues and paracancerous tissues was detected by qRT-PCR. (B) The mRNA level of MIG-6 in the paracancerous tissues (n = 38) and GC tissues (n = 46) from the ONCOMINE database. (C) Immunohistochemistry detected the protein level of MIG-6 in the paracancerous tissues (left panel) and GC tissues (right panel) (Original magnification: ×100). (D) The effect of MIG-6 mRNA expression on the overall survival and progression-free survival in GC patients was analyzed by Kaplan-Meier Plotter (http://www.kmplot.com). (E) and (F) The expression of MIG-6 in human gastric epithelial cells GES-1 and GC cell lines were detected by western blotting. *P < 0.05, **P < 0.01
The correlation of MIG-6 expression with the clinical features of gastric cancer (GC)
| Characteristics | n | High MIG-6 (n = 34) | Low MIG-6 (n = 19) | P-value |
|---|---|---|---|---|
| Gender | NS | |||
| Male | 43 | 28 | 15 | |
| Female | 10 | 6 | 4 | |
| Age (years) | 0.20 | |||
| ≥60 | 38 | 22 | 16 | |
| <60 | 15 | 12 | 3 | |
| Tumor differentiation | 0.02 | |||
| Poor or moderate | 41 | 30 | 11 | |
| Well | 12 | 4 | 8 | |
| Primary tumor size (cm) | 0.13 | |||
| ≥5 | 24 | 18 | 6 | |
| <5 | 29 | 16 | 13 | |
| Clinical stage | 0.02 | |||
| I or II | 30 | 15 | 15 | |
| III or IV | 23 | 19 | 4 | |
| Invasion depth | 0.37 | |||
| T1-2 | 13 | 7 | 6 | |
| T3-4 | 40 | 27 | 13 | |
| Lymph node metastasis (N factor) | 0.40 | |||
| Absent (N0) | 21 | 15 | 6 | |
| Present (N1-N3) | 32 | 19 | 13 |
P < 0.05 are indicated in bold
3.2 The expression of MIG-6 differs in normal gastric mucosal epithelial cells and human GC cell lines
MIG-6 was expressed in both GES-1 and GC cells. However, compared with GES-1 cells, the expression was higher in GC cells. The expression was the lowest in BGC-823 cells and the highest in SGC-7901 cells (Figure 1E).
3.3 MIG-6 regulates EGFR activation and signaling in GC cells
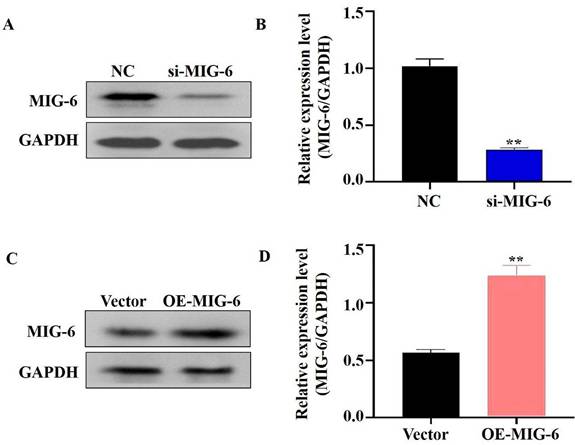
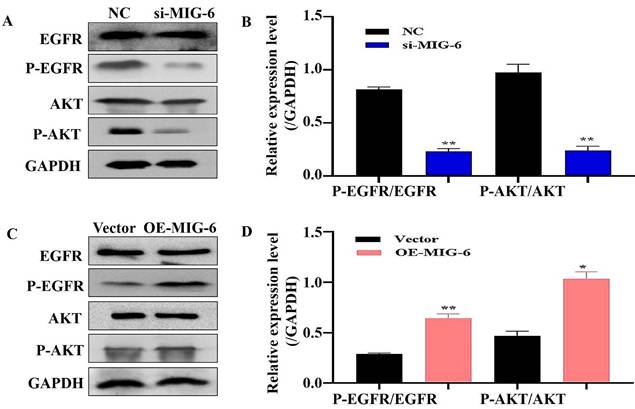
EGFR plays a critical role in cancer inhibition and growth [13]. MIG-6 suppresses tumor growth by blocking EGFR and its downstream signaling pathways [14]. The findings indicated that MIG-6 expression was knocked down in SGC-7901 cells (Figure 2A and B) and that the expressions of P-EGFR and P-AKT were decreased in SGC-7901 cells (Figure 3A and B). MIG-6 was subsequently overexpressed in BGC-823 cells (Figure 2C and D). Furthermore, MIG-6 overexpression induced the expressions of P-EGFR and P-AKT in BGC-823 cells (Figure 3C and D). These data demonstrate that MIG-6 regulates EGFR activation and signaling in GC cells.
The expression of MIG-6 was enhanced in BGC-823 cells and knocked down in SGC-7901 cells. (A) and (B) The expression of MIG-6 in SGC-7901 cells transfected with si-MIG-6 decreased significantly. **P < 0.01 versus SGC-7901 cells transfected with NC SGC-7901 cells. (C) and (D) pcDNA3.0-MIG-6 could efficiently increase the expression of MIG-6 in BGC-823 cells. **P < 0.01 versus blank BGC-823 cells.
The regulation of MIG-6 in the EGFR/AKT pathway. (A) and (B) The knockdown of MIG-6 inhibits EGFR and AKT phosphorylation expression in SGC-7901 cells. (C) and (D) The overexpression of MIG-6 promotes EGFR and AKT phosphorylation expression in BGC-823 cells, *P < 0.05, **P < 0.01.
3.4 Aberrant MIG-6 expression influences the proliferation of GC cells
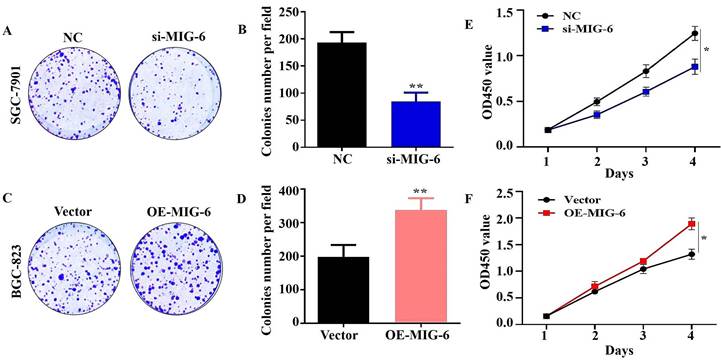
The results of the CCK-8 assay showed that the knockdown of MIG-6 drastically reduced GC cell growth and colony formation (Figure 4A and E). Compared with the vector group, the proliferation of BGC-823 cells was enhanced in the OE-MIG-6 group (Fig. 4F) and it formed larger and a greater number of clones (Figure 4C). Overall, these findings illustrate that MIG-6 expression could promote GC cell proliferation in vitro.
The knockdown of MIG-6 reduced the ability of proliferation of SGC-7901 cells (A) and (B) Colony formation assay was used to detect the ability of proliferation in SGC-7901 cells after MIG-6 knockdown. (C) and (D) Colony formation assay was performed to detect the ability of proliferation in BGC-823 cells with MIG-6 overexpression. (E) and (F) CCK-8 assay was performed in BGC-823 and SGC-7901 cells to examine the cell proliferation ability (Scale bar = 100 μm), *P < 0.05, **P < 0.01.
3.5 MIG-6 overexpression induces EMT in GC cells
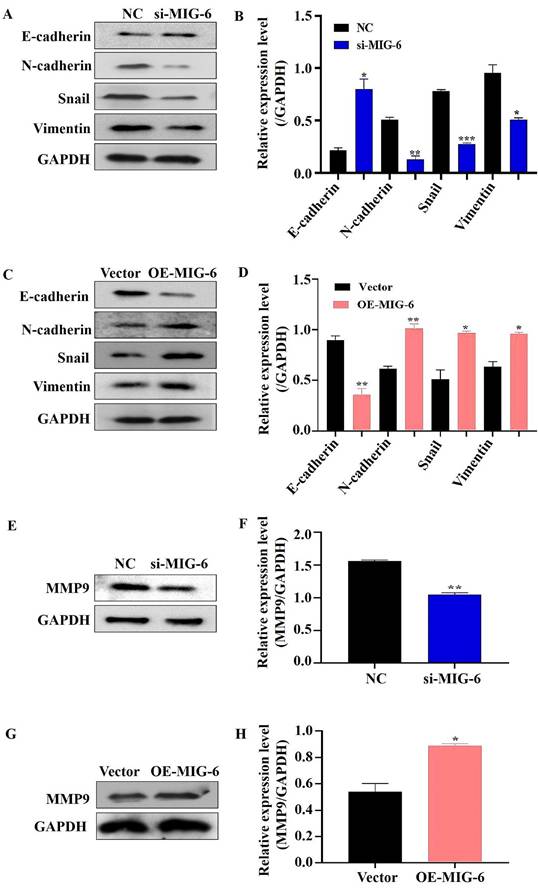
A developmental and physical transformation known as EMT occurs before epithelial cell metastasis and invasion [15]. The phenotype, shape, and polarity of epithelial cells change, and stromal cell features are simultaneously acquired [16]. MIG-6 downregulation decreased the expressions of N-cadherin, snail, and vimentin in SGC-7901 cells, but the epithelial marker E-cadherin increased drastically (Figure 5A and B). The overexpression of MIG-6, on the contrary, led to a modest decrease in E-cadherin and an increase in N-cadherin, snail, and vimentin expressions in BGC-823 cells (Figure 5C and D). Overall, these data suggest that aberrant MIG-6 expression modulates EMT, which in turn modifies GC cell migratory capacity in vitro.
The expression of MIG-6 affected EMT-associated proteins in BGC-823 and SGC-7901 cells. (A) and (B) Western blotting revealed EMT phenotype with reduced Snail, Vimentin, and N-cadherin and boosted the E-cadherin expression in SGC-7901 cells transfected with si-MIG-6. (C) and (D) EMT phenotype displayed that high MIG-6 expression could induce Snail, Vimentin, and N-cadherin expression and reduce the E-cadherin expression. (E) and (F) The expression of MMP9 was decreased by western blotting in SGC-7901 cells transfected with si-MIG-6. (G) and (H) The expression of MMP9 was increased by western blotting in BGC-823 cells transfected with pcDNA3.0-MIG-6. *P < 0.05, **P < 0.01, ***P < 0.001.
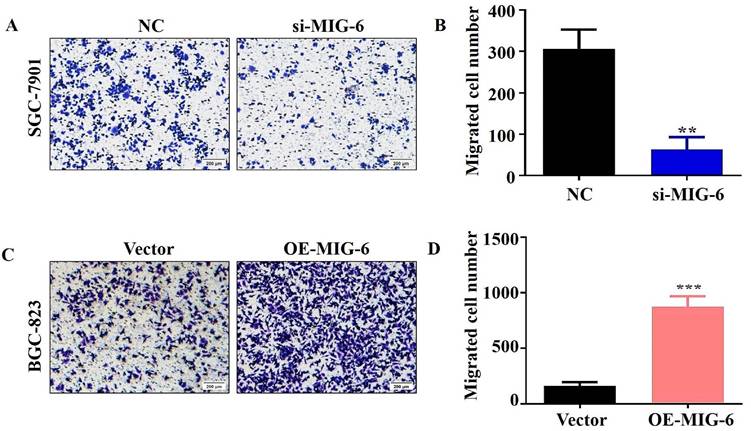
3.6 Aberrant MIG-6 expression influences cell metastasis in vitro
The transwell migration experiment showed that compared with the NC group, the migration of the SGC-7901 cells was dramatically decreased in the si-MIG-6 group (Figure 6A and B). MIG-6 overexpression greatly increased the number of migratory BGC-823 cells (Figure 6C and D). Owing to their proclivity to degrade the extracellular matrix, matrix metalloproteinases (MMPs) are assumed to play an important role in tumor invasion and metastasis in several cancers [17]. Therefore, the expression of the migratory marker MMP9 was identified using western blotting. MMP9 expression was significantly reduced in SGC-7901 cells transfected with si-MIG-6 (Figure 5E and F). MMP9 was significantly higher in MIG-6-overexpressing cells than in vector groups (Figure 5G and H). Collectively, our findings suggest that MIG-6 regulates GC cell motility in vitro.
The migratory ability was evaluated by using transwell migration assay in BGC-823 and SGC-7901 cells. (A) and (B) The knockdown of MIG-6 reduced the migratory ability of SGC-7901 cells. (C) and (D) The overexpression of MIG-6 enhanced the migratory ability of BGC-823 cells. (200× magnification) **P < 0.01.
4. Discussion
MIG-6 is a multiadapter protein known for its interaction with and negative regulation of EGFR. MIG-6-deficient mice exhibit hyperactivation of EGFR signaling and skin hyperplasia, and are highly prone to tumor formation in skin, lung, and other tissues. Chang et al. [18] compared multiple erlotinib-resistant cancer cell lines with their paired erlotinib-sensitive lines and showed that MIG-6 expression is significantly increased in erlotinib-resistant cell lines. The observed MIG-6 overexpression in erlotinib-resistant cells was linked to a decrease in EGFR activity and activation of AKT. These findings imply that MIG-6 plays contradictory roles in different tumor environments. MIG-6 acts as a tumor suppressor and is genetically altered or transcriptionally inhibited in lung cancer and glioblastoma. In these instances, constitutively active forms of EGFR promote tumor growth [9,10]. Nonetheless, MIG-6 deletions are rare in GC; hence, more investigations into MIG-6 are warranted. This study observed that the expression of MIG-6 was significantly increased in GC tissues and cells compared with normal controls. In addition, histological studies revealed that MIG-6 expression in GC tissues contributes to disease progression and poor prognosis, uncovering a previously under-recognized tumor-promoting role of MIG-6 in cancer.
The expression of MIG-6 has been demonstrated to be suppressed in nonsmall-cell lung cancer tissues and liver cancer, leading to increased EGFR/AKT signaling and enhanced cell proliferation and metastasis [8,19]. However, this study found that MIG-6 upregulation in GC tissue and cells could regulate the EGFR/AKT pathway and promote proliferation and metastasis, thereby resulting in unanticipated protumor outcomes. MIG-6 interacts with EGFR via an ErbB-binding region in its C terminus. Biochemical studies on MIG-6 uncovered that a 77 amino acid-region (336-412) is responsible for EGFR inhibition [20]. Functional investigation of this area revealed the presence of an X-ray crystal structure (MIG-6 residues 336-364, known as segment 1) that facilitates EGFR binding and partially suppresses EGFR by preventing the formation of asymmetric dimers. However, for EGFR suppression to be entirely effective in vitro and cells, 50 additional residues (residues 365-412, known as segment 2) must be present [7, 21]. The inhibitory effect of MIG-6 is controlled by EGFR-mediated Y394 phosphorylation, which is preceded by Src-mediated Y395 [22]. A study has reported that MIG-6 is an activity-based EGFR inhibitor and that its selectivity for active EGFR is determined by EGFR-mediated MIG-6 phosphorylation at Tyr394 [22]. Hence, we hypothesized that Y394 phosphorylation of MIG-6 was defective in GC cells, which affected the capacity of MIG-6 to decrease EGFR. However, additional studies are required to test this notion.
The EMT is a biological process in which cells lose their epithelial properties and instead acquire mesenchymal characteristics. Tumor initiation, malignant growth, tumor stemness, tumor cell motility, blood intravasation, metastasis, and treatment resistance have been documented to be related to EMT [15, 23]. The activity of adherens junctions is dramatically altered during EMT, which could be mainly attributed to the replacement of E-cadherin by N-cadherin, a process known as “cadherin switching” [24]. According to Chen et al. [25], vimentin expression is directly connected to poor clinical outcomes in breast cancer. Our study found that E-cadherin expression was reduced in BGC-823 cells overexpressing MIG-6 although MMP9, N-cadherin, snail, and vimentin expressions were elevated. When siRNA was applied to SGC-7901 cells, E-cadherin increased but MMP9, N-cadherin, snail, and vimentin decreased. This result suggests that the expression of MIG-6 may induce EMT and facilitate the development of GC and its metastasis. Nevertheless, the underlying mechanisms are yet to be elucidated. Further studies could focus on the precise mechanism by which MIG-6 governs GC cell metastasis and invasion during tumor growth, particularly in vivo.
In conclusion, this study identified that enhanced MIG-6 expression drives GC cell metastasis, proliferation, and EMT and contributes to disease progression in terms of tumor clinical stage and differentiation. The Kaplan-Meier plotter demonstrated that patients with GC exhibiting high MIG-6 expression had a significantly shorter survival. Furthermore, our findings indicated that MIG-6 may regulate the EGFR/AKT pathway and promote tumor growth and metastasis. In addition, our data showed that reducing MIG-6 inhibits tumor development in GC, thus revealing a hitherto unknown prosurvival role of MIG-6 in GC. These findings are encouraging as they suggest that MIG-6 has the potential to be used as a diagnostic and prognostic biomarker in GC.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (81772157), Science and Technology Project of Jiangsu Provincial Xuzhou Municipal Health Commission (XWKYHT20240076), Jiangsu Funding Program for Excellent Postdoctoral Talent (2023ZB180) and Postdoctoral Fellowship Program of CPSF (GZC20230992).
Author contributions
Wenqiu Zhao: Conceptualization, Methodology, Data curation, Formal analysis, Investigation, Validation, Writing - original draft, Writing -Review & editing, Funding acquisition. Tao Jin: Formal analysis, Investigation, Validation. Yun Liu: Data curation, Formal analysis, Validation. Shihe Shao: Conceptualization, Resources, Funding acquisition, Project administration, Supervision, Writing - review & editing Feilun Cui: Conceptualization, Project administration, Supervision, Writing - review & editing.
Data availability
All data generated or analysed during this study are included in this published article.
Ethical statement
All patients obtain the informed consent and it is approved by the Ethics Committee of the School of Medicine, Jiangsu University.
Consent for publication
All authors approved the final manuscript and the submission to this journal.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635-48
3. Lordick F, Carneiro F, Cascinu S, Fleitas T, Haustermans K, Piessen G. et al. Gastric cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2022;33:1005-20
4. Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J. et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193-205
5. Kim TH, Lee DK, Cho SN, Orvis GD, Behringer RR, Lydon JP. et al. Critical tumor suppressor function mediated by epithelial Mig-6 in endometrial cancer. Cancer Res. 2013;73:5090-9
6. Frosi Y, Anastasi S, Ballarò C, Varsano G, Castellani L, Maspero E. et al. A two-tiered mechanism of EGFR inhibition by RALT/MIG-6 via kinase suppression and receptor degradation. J Cell Biol. 2010;189:557-71
7. Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG-6 to an activating kinase domain interface. Nature. 2007;450:741-4
8. Reschke M, Ferby I, Stepniak E, Seitzer N, Horst D, Wagner EF. et al. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology. 2010;51:1383-90
9. Maity TK, Venugopalan A, Linnoila I, Cultraro CM, Giannakou A, Nemati R. et al. Loss of MIG-6 Accelerates Initiation and Progression of Mutant Epidermal Growth Factor Receptor-Driven Lung Adenocarcinoma. Cancer Discov. 2015;5:534-49
10. Ying H, Zheng H, Scott K, Wiedemeyer R, Yan H, Lim C. et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A. 2010;107:6912-7
11. He J, Li CF, Lee HJ, Shin DH, Chern YJ, Pereira De Carvalho B. et al. MIG-6 is essential for promoting glucose metabolic reprogramming and tumor growth in triple-negative breast cancer. EMBO Rep. 2021;22:e50781
12. Kang DH, Jung SS, Yeo MK, Lee DH, Yoo G, Cho SY. et al. Suppression of Mig-6 overcomes the acquired EGFR-TKI resistance of lung adenocarcinoma. BMC Cancer. 2020;20:571
13. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol. 2020;61:167-79
14. Cheng WL, Feng PH, Lee KY, Chen KY, Sun WL, Van Hiep N. et al. The Role of EREG/EGFR Pathway in Tumor Progression. Int J Mol Sci. 2021;22:12828
15. Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796:75-90
16. Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333-9
17. Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S. et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. 2017;16:124
18. Chang X, Izumchenko E, Solis LM, Kim MS, Chatterjee A, Ling S. et al. The relative expression of Mig-6 and EGFR is associated with resistance to EGFR kinase inhibitors. PloS one. 2013;8:e68966
19. Li Z, Dong Q Fau - Wang Y, Wang Y Fau - Qu L, Qu L Fau - Qiu X, Qiu X Fau - Wang E, Wang E. Downregulation of Mig-6 in nonsmall-cell lung cancer is associated with EGFR signaling. Mol Carcinog. 2012;51:522-34
20. Wang Z, Raines LL, Hooy RM, Roberson H, Leahy DJ, Cole PA. Tyrosine phosphorylation of mig6 reduces its inhibition of the epidermal growth factor receptor. ACS Chem Biol. 2013;8:2372-6
21. Wang Z, Longo PA, Tarrant MK, Kim K, Head S, Leahy DJ. et al. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat Struct Mol Biol. 2011;18:1388-93
22. Park E, Kim N, Ficarro SB, Zhang Y, Lee BI, Cho A. et al. Structure and mechanism of activity-based inhibition of the EGF receptor by Mig6. Nat Struct Mol Biol. 2015;22:703-11
23. Pastushenko I, Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019;29:212-26
24. Cavallaro U, Schaffhauser B, Christofori G. Cadherins and the tumour progression: is it all in a switch? Cancer Lett. 2002;176:123-8
25. Chen Z, Fang Z, Ma J. Regulatory mechanisms and clinical significance of vimentin in breast cancer. Biomed Pharmacother. 2021;133:111068
Author contact
![]() Corresponding author: Shihe Shao: School of Medicine, Jiangsu University, 301 Xuefu Road, Zhenjiang, 212013, Jiangsu, China; E-Mail: shaoshihe2006com; Feilun Cui: The Affiliated Taizhou Second People's Hospital of Yangzhou University, 225500 Taizhou, Jiangsu, China; E-Mail: pdcuifeiluncom.
Corresponding author: Shihe Shao: School of Medicine, Jiangsu University, 301 Xuefu Road, Zhenjiang, 212013, Jiangsu, China; E-Mail: shaoshihe2006com; Feilun Cui: The Affiliated Taizhou Second People's Hospital of Yangzhou University, 225500 Taizhou, Jiangsu, China; E-Mail: pdcuifeiluncom.