Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2012; 3:67-82. doi:10.7150/jca.3965 This volume Cite
Review
Telomerase-Independent Paths to Immortality in Predictable Cancer Subtypes
Stephen T Durant ![]()
AstraZeneca - DNA Damage Response, Bioscience, Oncology iMed, Alderley Park, Cheshire, SK10 4TG, England, UK.
Received 2011-12-16; Accepted 2012-1-28; Published 2012-1-31
Abstract
The vast majority of cancers commandeer the activity of telomerase - the remarkable enzyme responsible for prolonging cellular lifespan by maintaining the length of telomeres at the ends of chromosomes. Telomerase is only normally active in embryonic and highly proliferative somatic cells. Thus, targeting telomerase is an attractive anti-cancer therapeutic rationale currently under investigation in various phases of clinical development. However, previous reports suggest that an average of 10-15% of all cancers lose the functional activity of telomerase and most of these turn to an Alternative Lengthening of Telomeres pathway (ALT). ALT-positive tumours will therefore not respond to anti-telomerase therapies and there is a real possibility that such drugs would be toxic to normal telomerase-utilising cells and ultimately select for resistant cells that activate an ALT mechanism. ALT exploits certain DNA damage response (DDR) components to counteract telomere shortening and rapid trimming. ALT has been reported in many cancer subtypes including sarcoma, gastric carcinoma, central nervous system malignancies, subtypes of kidney (Wilm's Tumour) and bladder carcinoma, mesothelioma, malignant melanoma and germ cell testicular cancers to name but a few. A recent heroic study that analysed ALT in over six thousand tumour samples supports this historical spread, although only reporting an approximate 4% prevalence. This review highlights the various methods of ALT detection, unravels several molecular ALT models thought to promote telomere maintenance and elongation, spotlights the DDR components known to facilitate these and explores why certain tissues are more likely to subvert DDR away from its usually protective functions, resulting in a predictive pattern of prevalence in specific cancer subsets.
Keywords: telomerase, cancer, Alternative Lengthening of Telomeres, DNA damage response, immortality
Introduction
Human telomeres are regions of 4-15 kilobases of repetitive hexameric (TTAGGGn) DNA sequences at the ends of each chromosome (1). They end with a 3'-overhang that most likely folds back and invades its complementary strand to form a t-loop (2). A complex of telomere-specific shelterin proteins bind and cap telomeres, further preventing chromosomal ends from being recognised as DNA double strand breaks (DSB) by the DNA damage response (DDR) machinery (3,4). As the lagging strands of telomeres are incapable of being fully replicated during each round of cell division, telomeres undergo progressive shortening during normal cellular proliferation. Eventually, they become so short that they trigger the DDR, termed crisis (3,5,6). This normally results in replicative senescence and eventually checkpoint-driven cell death, defining cellular lifespan and safeguarding an organism against unlimited cellular proliferation and cancer (4,7). The vast majority of tumours initially lose the function of important cell cycle checkpoints such as p53 and Retinoblastoma (Rb) proteins and eventually activate a telomere maintenance mechanism (TMM). TMMs use either the telomerase enzyme - the holoenzyme remarkably consisting of its own ribosomal RNA sequence (hTERC) and a catalytic enzyme (hTERT) that synthesises new telomeric DNA from its own template (8), or telomerase-independent Alternative Lengthening of Telomere (ALT) pathways (9)(10).
During the early stages of human development, telomerase is needed to compensate for the huge amount of cell divisions needed to complete embryogenesis and its expression is robustly switched off at a later stage (11,12). At birth through to adulthood, the only cells that continue the requirement for telomerase activity are dividing male germ cell lineages, specific bone marrow stem cells, activated lymphocytes, and proliferative skin and gastrointestinal cells (12). However, 60-70% of immortalised cell lines grown in tissue culture and an estimated 85-90% of cancer tissue overcome this limited proliferative capacity by up-regulating telomerase activity (4,10,13-16). Thus, a pharmaceutical interest in targeting telomerase as an anti-cancer therapeutic rationale is well established. Geron Corporation's intravenous GRN163L (Imetelstat) oligonucleotide-based therapy hybridises to the template region of hTERC and has completed a Phase I clinical trial in patients with chronic lymphocytic leukaemia, multiple myeloma, breast cancer and non-small cell lung cancer (NSCLC) (17). Geron's most advanced hTERT-based GV1001 (GemVax) vaccine designed to raise immune cytotoxic T cell responses against a 16-mer peptide from the active site of human hTERT is in Phase I & II clinical trials (in NSCLC, hepatocellular carcinoma and non-resectable pancreatic carcinoma) and in a randomized Phase III clinical trial in patients with locally advanced or metastatic pancreatic cancer (ClinicalTrials.gov Identifier: NCT00425360) (18,19).
However, it is important to note that i) telomerase-deficient ALT tumours will not respond to these therapies, ii) toxic effects in blood and other telomerase-utilising regenerative tissue may be limiting (Phase I trials of Imeteltstat noted reversible anaemia, thrombocytopenia and neutropenia - although it is possible that these could be due to off-target effects or the concurrent standard chemotherapy used (20,21)) iii) long treatment regimes would be required to gradually erode telomeres of varying lengths in particular tumours (22), iv) telomerase inhibition is known to have TMM-independent effects on cell growth and telomere length depending on whether hTERT or hTERC is targeted (23). Lastly, v) there is a significant possibility that such drugs would ultimately select for resistant cells that activate an ALT mechanism (24).
ALT is therefore also a potentially attractive drug target since repression of ALT in ALT-dependent immortal cell lines results in selective senescence and cell death (25). In particular, ALT inhibition, by siRNA-targeting of ALT components, appears to result in a more rapid telomere dysfunction (26)(10,27-29) which may increase therapeutic efficacy. Furthermore, in the equally established pursuit of targeting telomere DNA itself in cancer cells, early data from studies using the macrocyclic compound Telomestatin, which binds to G4-quadruplexes commonly formed in G-rich regions of DNA such as promoters and telomeres, show effective killing of telomerase-positive and ALT cell lines (30). However, regardless of issues surrounding overcoming drug-like physical properties of G4-quadruplex binders, it is not known if their mode of action is specific to ALT or TMMs in general since approximately half of all gene promoters in the human genome and many oncogenic promoters e.g. c-myc, VEGF, HIF-1a, Bcl-2, Ret, c-kit and KRAS also contain G-rich sequences and are known to form G-quadruplexes in vitro (31,32) (for detailed reviews on G-quadruplexes see (33,34)).
Several caveats to specifically targeting ALT must be considered. It is possible that ALT-positive primary tumours could give rise to telomerase reactivated secondary tumours and vice versa especially if each TMM is suppressed epigenetically rather than by non-functional mutation/s. It has also been shown that telomerase can be transfected into an ALT cell line and appear active alongside ALT (35). Lastly, there are documented cases of tumors that appear to be both telomerase- and ALT-negative, although it is possible that these may just be early tumours that have not yet experienced sufficient telomere shortening to need a TMM and may simply end up spontaneously regressing due to an inability to engage any TMM. Spontaneously regressing telomerase-deficient Type IVS neuroblastoma may be a classic example of this (36,37). This review explores i) how ALT is detected in cells and tissues, ii) unravels the escape pathways that ALT-positive cells are thought to use, iii) identifies the main components of ALT as potentially attractive drug targets and iv) speculates why certain malignancies, recently highlighted in a comprehensive survey of ALT across >6000 samples confirming previously predicted patterns of ALT prevalence across cancers, choose ALT over telomerase reactivation to achieve immortality.
The ALT phenotype and ALT detection in cell lines and tissue
ALT was first deduced in human cell lines from the fact that some telomerase-deficient lines were able to be maintained in culture for many hundreds of population doubling times (38). Later phenotypic studies revealed that, unlike telomerase-positive cells, ALT-dependent cells almost always contain heterogeneous telomere length distribution (38) and form ALT-associated 'promyelocytic leukemia (PML) bodies' or 'APBs' (13,39). These phenotypes are either undetectable or have very low levels of activity in normal somatic cells, therefore providing valuable biomarkers for ALT. Here's how these attributes are tested in cell lines and tissue microarrays (TMAs):
- Measuring telomere length is the definitive method for identifying ALT. One of the hallmarks of ALT is telomere length heterogeneity, ranging from very long (up to 20kb - twice as long as those in non-ALT cells) to very short telomeres. A gradual erosion of telomere length in ALT+ cells is usually at the 50kb per cell division rate and critically short telomeres at a cell's crisis point are then thought to re-stimulate ALT mechanism/s. These varied and fluctuating lengths of telomeres can be measured by terminal restriction fragment Southern blotting, fluorescence in-situ hybridisation (FISH) and single telomere length analysis (STELA) on chromosomal metaphase spreads. These labour intensive methods can be applied to cell lines and formalin fixed paraffin-embedded (FFPE) human tissue biopsies or TMAs tissues only with well-preserved genomic material but are low throughput for screening large numbers of compounds for finding ALT inhibitors. Furthermore, in order to demonstrate cells can maintain telomeres for sufficient periods of time, at least 20 doubling times are needed in tissue culture, dramatically reducing practicality and are not that sensitive to subtle TMM activities (40).
- The TRAP (telomere repeat amplification protocol) assay has been a long-established PCR-based tool for measuring telomerase activity (8,41-45). This is a quantitative assay but only amenable to detection in fresh or frozen cell/tissue samples, and since telomerase requires its RNA template component, the assay is subject to degradation due to RNA instability and to tissue-derived PCR inhibitors. This may have accounted for false-positive summaries of over-all ALT status across tissues that has been estimated to be up to 15% of all cancers. Finally, the requirement for a positive readout for ALT, rather than a positive readout for telomerase activity also renders this assay not as suitable as those directly measuring ALT activity.
- APBs are subsets of Promyelocytic leukaemia (PML) bodies that are punctate regions within the nucleus where PML protein and proteins involved in DNA repair and replication concentrate. These bodies are thought to be sites of storage, macromolecular processing and degradation and can be statically attached to the nuclear matrix or mobile with varying sizes. APBs are defined by containing PML proteins, telomeric DNA sequences and telomere-protecting proteins (termed Shelterins e.g. TRF1 and TRF2) within them. Detecting APBs by IHC and FISH is a simple and robust measure of ALT which can be used to score cell lines and TMAs. APBs vary in size with larger foci usually increasing during conditions of cellular stress and cell cycle arrest and may signal ALT cells with critically short telomeres. DNA damage response (DDR) proteins are known to be present at APBs alongside the histone marker of DSBs - γH2AX. Indeed the APB foci that form using γH2AX and TRF2 antibodies have been termed Telomere dysfunctional foci (TIFs) to reflect the fact that these are recognised as perturbed DNA damaged sites (Cesare and Reddel, 2010; Saharia et al 2010; Saharia et al, 2009).
- Finally, a recent report by Heaphy et al identified a 100% (19/19) perfect correlation between ALT+ status and occurrence of mutations in ATRX and DAXX genes as judged by telomere FISH in pancreatic neuroendocrine tumour samples (46). Out of 439 samples across other cancers, 8/24 ALT+ cases were ATRX mutated and the other 16 ALT+ samples with no mutations detected did not have detectable levels of protein. Thus, screening for ATRX/DAXX mutations/expression may therefore be a surrogate ALT score in patient tissues and may represent a useful prognostic and predictive biomarker for aligning drugs that inhibit ALT to specific patient cohorts likely to respond. Indeed, the John's Hopkins University from where the authors published this work has filed a patent application relating to the use of DAXX and ATRX mutations as diagnostic markers (46).
Modelling the mechanisms of ALT
Sufficiently long telomeres serve to protect the ends of chromosomes from DDR-mediated signalling whilst also providing most normal cells with a defined lifespan once they reach a critically short length. However, their heterochromatic nature and the interference of DNA-protein complexes and higher-order DNA secondary structures (telomeres are G-rich repetitive sequences that are prone to form G-quadruplexes) can induce frequent replication pausing and so demand the activity of certain DDR pathways under normal conditions. Furthermore, a fork stalled in telomeric repeats cannot be rescued by a converging fork since telomeres are replicated in a unidirectional way. Therefore, telomerase-proficient cells utilise one or more forms of homologous recombination (HR)-mediated replication fork resolution. In ALT cells without functioning telomerase, HR in various forms (template-driven recombination-mediated DNA replication, and/or break-induced DNA replication (BIR) - pathways commonly used in DDR to resolve stalled and collapsed replication forks) - has been modelled to principally operate not only to resolve paused replication forks but to maintain and elongate telomeres. HR has only been studied in ALT cells under abnormal conditions in which yeast, mouse and human cell lines have been genetically modified so it is difficult to assess how ALT is switched on pathologically. However, much has been learned over the past decade about the roles of the components of DDR that seem to be utilised in telomerase-positive and telomerase-negative cell lines. Without telomerase or other telomere-capping/protecting (shelterin) components present, ALT is likely an unbalanced process of unregulated homologous telomere recombination events counteracting telomere shortening by using templates from the same telomere, a sister chromatid telomere or other chromosome, or extrachromosomal t-circle DNA common in ALT cells (10) to sustain cellular survival indefinitely.
The first evidence that ALT mechanisms involve recombination processes came from studies of Saccharomyces cerevisiae strains deficient in components of telomerase (TLC1 or EST1) (47,48). Most of these cells died but some clones emerged and were called type 1 or type 2 survivors. Both were found to depend on RAD52 since double knock-outs were unviable. RAD52 is essential for double strand break repair by homologous recombination. Interestingly, rare colonies from the double-knockout (tlc1/rad52) strain are rescued by also knocking out EXO1 (Maringele and Lydall 2004) revealing a possible inhibitory function of EXO1 in these ALT strains. Further and stronger evidence for a recombination-based mechanism for ALT came from studies using plasmid integration into one telomeric region. The tagged DNA was seen to transfer from one telomere and increased in number to other chromosomal ends after a number of cell divisions (49).
A variety of recombination-based models have now been proposed to describe how telomeres can be maintained and extended without functional telomerase activity (Figure 1 A and B). Unequal telomeric sister chromatid exchange (T-SCE) has been suggested to facilitate ALT (Figure 1B). This is supported by the fact that T-SCE is elevated in ALT+ cells as measured by CO-FISH (50). However, this model has come under scrutiny due to the fact that there would not be any net gain in telomere length since one sister would elongate at the expense of the other. It is has therefore been suggested that perhaps if all the elongated telomeres were to be segregated into the daughter cell at the expense of the parental cell, unequal recombination might produce a net gain for the next generation. Another model has been suggested to get round this issue which proposes that break-induce replication (BIR) could do the job (Figure 1A) (51). BIR is a repair mechanism that synthesises DNA up to many kilobases away from a break site using a homologous donor template (in this case telomeric DNA) (see Figure 1A). As is seen in normal proliferative somatic cells, ALT cells are also characterized by the presence of extrachromosomal linear and circular telomere DNA molecules. The extrachromosomal t-circles can in principle be utilized as homologous templates for HR-driven telomere elongation or simply undergo rolling-circle-replication, both mechanisms envisaged to facilitate ALT (Figure 1C).
Proposed mechanisms of ALT. A) Break-induced replication. a and b models differ in timing of the lagging strand synthesis but both result in newly synthesized G and C rich strands on the recipient telomere without loss from the donor telomere. c shows a unidirectional replication fork establishing and following Holliday Junction resolution both donor and recipient telomeres experience semi-conservative replication. B) Telomeric-Sister Chromatid Exchange and C) Rolling circle and t-circle formation after t-loop resolution providing a linear DSB for subsequent HR-mediated invasion into homologous templates.
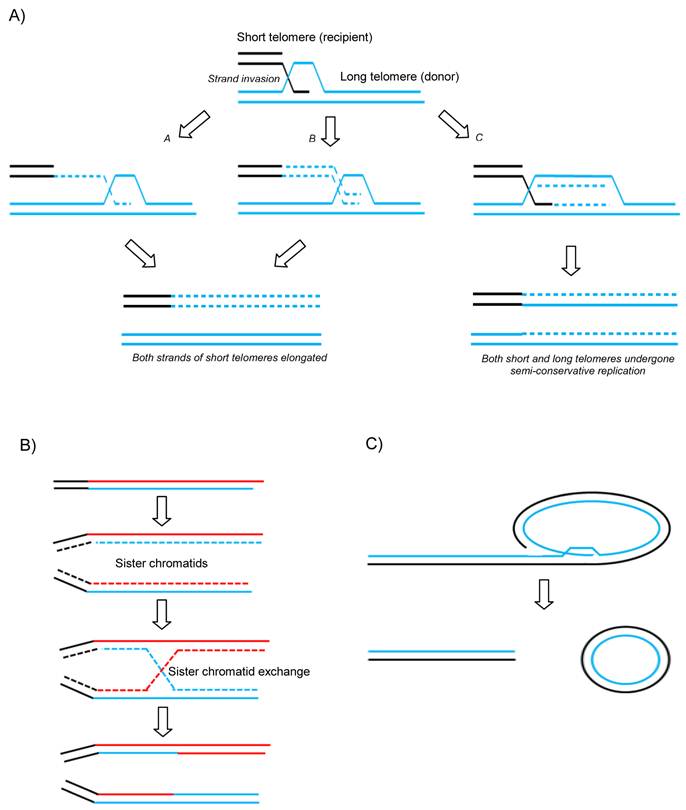
Recent evidence suggests that the latter mechanism of ALT is used in normal somatic cells in a balanced and controlled manner (52). Telomere shortening in normal cells not only occurs by gradual attrition but also by a faster form of telomere trimming (TT, also known as telomere rapid deletion - TRD) (52) and this occurs when HR resolves t-loops into t-circles and much shortened linear telomeres. It has been proposed that this level of activity is increased in ALT cells in which TT is counteracted by elevated levels of HR-mediated telomere elongation. This would explain the drastically varied length of telomeres and increased detection of t-circles in ALT cells and suggests that subtle imbalances occurring in the regulation of TT occurs between mortal and immortal states.
DNA damage response (DDR) components - roles in telomerase-positive and negative cells
Many proteins with specific roles in signalling global DNA damage and DNA repair have been shown to locate and function at telomeres in telomerase positive and negative cell lines (53) and implicated in telomere length homeostasis and chromosome end protection (for a detailed review see (54). Since it is known that persistent DNA damage can elicit enhanced and deregulated DNA repair activity in cancer cells (55), it seems likely that recruitment of the DDR machinery in ALT cells represents a more subversive and unregulated form of activity at telomeres. However it must be stressed that, in fact, the roles of many of the components of DDR during normal TMMs still remain poorly understood.
DNA damage signalling
In telomerase-positive cells, experimental suppression of the proteins known to facilitate telomere capping and mask the presence of a DSB at telomeric ends (termed Shelterins e.g. TRF2 and in some studies POT1) triggers a DDR. This is typified by ataxia telangiectasia mutant (ATM) kinase auto-phosphorylation at S1981 - the master signaller for DSB repair; phosphorylation of H2AX (γ-H2AX) - the amplification signal for DSBs on chromatin and the recruitment of DNA repair factors including 53BP1, MDC1, Rad17, MRN complex (MRE11, RAD50, NBS1) and Rif1 (56,57). These telomeric sites are known as telomere dysfunction-induced foci (TIFs) and signal the presence of DSBs at chromosomal ends. Shortly afterwards cellular senescence and apoptosis ensues in a p53 and Rb-dependent manner. These experimentally forced observations suggest that this occurs naturally in somatic and cancer cells that approach stages of critically shortened telomeres. It is not surprising therefore that the majority of ALT cell lines and tumours lack normal p53 and Rb tumour suppressor functions that would normally trigger cell death in response to persistent DSBs (58-60).
Studies show that ATM and PARP1-mediated DNA damage signalling is critical to the induction of cell death in cells with critically shortened telomeres and inhibition of PARP activity leads to the extension of cellular lifespan (61). In telomerase-positive cells with shortened telomeres or in ALT cells, the phosphorylated form of TRF2 - one of the telomere shelterin proteins - concentrates on telomeres and it is known that ATM catalyses this (56,57). However, since TRF2 is known to suppress the function of ATM at telomeres, the precise role of ATM is currently unclear. The individual roles of PARP1 and PARP2 at telomeres, which normally have roles in signalling the presence of single strand breaks in the genome, is also unclear. PARP1 and PARP2 are sporadically detected at normal telomeres and shortened ones and can poly-ADP-ribosylate TRF2 which also removes TRF2 from telomeres, possibly allowing access to DNA repair proteins. PARP1 has been reported to colocalise with TRF2 in a telomerase-positive cell line, whereas PARP2 was shown to colocalise with TRF2 in an ALT cell line (62).
MRE11, RAD50, NBS1 (MRN complex)
Recruitment of MRN is an early DNA repair event at shortened telomeres in telomerase-positive and negative cell lines and these proteins were the first to be identified as necessary for ALT-mediated telomere elongation (10,63). This complex tethers DNA ends, facilitates 5' to 3' resection of the DNA ends to create 3' overhangs for strand invasion necessary for HR (64) and it has been suggested that this activity stabilises telomere loop formation (54). In ALT cell lines, MRN locates in APBs and in turn recruits BRCA1. MRN is also required for ATM-mediated phosphorylation of TRF1 and its dissociation from telomeres in ALT and non-ALT cells, indicating MRN's involvement in facilitating HR events at shortened telomeres (65). Inhibiting MRN function by transient or long-term repeated siRNA transfection against either protein or over-expression of SP100 (a PML protein), results in telomere shortening but only to a certain length after prolonged inhibition when telomeres remained stable (63,66). This resulted in viable cell line progeny thereafter and may reflect either redundant pathways stepping in or incomplete MRN depletion.
Non-homologous end-joining (NHEJ)
DNA-PK, which consists of DNA-PKcs and Ku70/80 subunits, interacts with telomerase, catalyses NHEJ and represses normal telomeric HR activity - the predominant pathway suggested to be involved in ALT as previously discussed (67). Celli et al conclude that mammalian chromosome ends are highly susceptible to HR and NHEJ together with TRF1/TRF2 functions to repress HR-mediated sister chromatid exchanges operating in ALT. Likewise, defects in HR facilitators like BRCA1 and BRCA2 have been reported to up-regulate NHEJ and this seems to be true at telomeres too as BRCA1 and BRCA2 deficient cells display gross chromosomal end-end fusions and instability (68). BRCA1 is known to regulate telomerase via its transcriptional regulation activity and it colocalises with TRF2 in telomerase-positive cell lines and within APBs of ALT cells, although its role in ALT is unclear since expression of a dominant negative mutant of BRCA1 results in increased telomere length in telomerase-positive cells but not in ALT cells (69).
The formation of t-circles in ALT cells depends on recombination proteins X-ray repair cross-complementing 3 (XRCC3), NBS1 and Ku70/80 and down-regulation of either one of these factors causes a decrease in the levels of t-circles and growth suppression in these cells (70). Since XRCC3 is a protein that forms a complex with the Rad51 paralogue Rad51C in promoting HR (71)and the Ku proteins facilitate NHEJ, it seems that a subtle and complex system involving both HR and NHEJ DSB pathways operate in t-circle formation. This complexity is highlighted by the fact that although Ku70/80 and NHEJ is known to be involved in telomerase-positive TMM, Li et al from Reddel's lab showed that Ku70/80 depleted human SAOS2 cells did not display the telomere deletions observed in telomerase positive human cells lacking Ku70/80 and did not display any significant difference in the overall distribution of telomere signals, nor any increase in chromosomal instability compared to control ALT cells (70). Only t-circle depletion was noted, highlighting the predominant role of NHEJ in this particular mechanism of ALT. As mentioned earlier, recent evidence from Reddel's lab suggests that t-circle formation results from the rapid telomere trimming (TT) process associated with telomere shortening in telomerase -positive but more starkly in telomerase-negative ALT cells (52). In agreement with studies showing a role for XRCC3 in t-circle formation, Pickett et al also noted that XRCC3 depletion resulted in an almost complete loss of t-circles as measured by 2D-gel electrophoresis. However, GEN1 depletion did not cause a reduction, the putative Holliday Junction resolving HR protein, further supporting a model that describes only the involvement NHEJ and separate activities of certain HR components/subpathways in facilitating ALT.
Homologous recombination (HR)
Many DNA repair proteins involved in HR are found at normal and dysfunctional telomeres but are particularly active in ALT, as previously discussed. In addition to HR components like Rad52 and MRN highlighted already, BLM is one of five known ATPase-driven RecQ family helicases that possesses 3-5' DNA unwinding activity, Holliday Junction branch migration and single stranded DNA annealing activity. These activities are required for resolving stalled and collapsed replication forks and this may be operative in advance of or behind a telomeric replication fork. In ALT cells, BLM is known to colocalise with TRF1 and TRF2 at telomeres in S-phase which supports such a function. WRN is another RecQ helicase which possesses exonuclease activity and can interact with DNA-PKcs, RPA, MRN and Rad51 in response to DSBs. WRN normally functions to repress inappropriate recombination such as T-SCE implicated in ALT and is thought to unwind DNA to influence telomerase access to the 3' overhang at the end of telomeres. However, WRN is also found in APBs in ALT cells alongside TRF1 and TRF2 in S-phase where it is thought T-loops require resolution to promote telomere elongation. Knocking down HR components including Rad51D, MUS81, BLM or FANCA/D2 by RNAi dramatically shortens telomeres in ALT+ cells and is associated with reduced cell survival (26)(10,27-29). RTEL is a DNA helicase thought to operate with BLM and Mus81 in resolving recombination intermediates and was first cloned by Ding et al in 2004 and named Regulator of Telomere length since Rtel(-/-) embryonic stem cells showed telomere loss and displayed many chromosome breaks and fusions upon differentiation in vitro (72). It is currently unknown if RTEL functions in telomerase-deficient cells but this would not be surprising considering the fact that other RecQ helicases have associated functions in ALT.
Nucleases involved in telomere maintenance and ALT
Since MRE11 is intricately involved in ALT mechanisms and is known to regulate NHEJ and BRCA1 activity during HR at stalled replication forks (HR) (73-75), it would be interesting to know if other DSB repair components that are particularly associated with resolving stalled and collapsed DNA replication forks are associated with telomeric DNA. For instance, Metnase and FAN1 are nucleases that facilitate these replication processes (76-79) and it is conceivable that such activities could promote ALT-dependent TMMs. Other DNA repair nucleases such as ERCC1/XPF, Mus81 and FEN1 are known to operate at telomeres. ERCC1 and its partner nuclease XPF is responsible for cleaving the 5' end of bubble structures present after nucleotide excision repair initiates DNA unwinding of damaged DNA. ERCC1/XPF also cleaves the G-strand overhangs present at uncapped telomeres which generate substrates for NHEJ in telomerase-positive cells and it is thought that ERCC1/XPF prevents incorrect recombination during chromosome division. This is supported by studies showing dividing ERCC1/XPF deficient cells accumulating small pieces of telomeric DNA called telomeric DNA-containing double minutes (TDMs) which result from recombination between telomeres and similar sequences elsewhere on a chromosome (80).
Mus81, FEN1 and WRN (a RecQ helicase which also possesses nuclease activity) are structure-specific nucleases known to function in ALT and function in lagging-strand DNA replication, HR and the restart of stalled replication forks. Both FEN1 and MUS81 have been found to localize to telomeres in ALT cells during the G2 phase when HR of telomeric DNA is more likely to occur and are known to be required for telomere stability in ALT cells (27). Knocking down MUS81 or FEN1 by RNAi dramatically shortens telomeres in ALT+ cells and is associated with reduced cell survival (26)(10,27-29). Since MUS81 is an endonuclease that cleaves various DNA substrates during HR and is required for the survival of cells undergoing aberrant replication and recombination (81-83), it is not surprising that studies show a role for MUS81 in ALT. FEN1 is another structure-specific endo- and exonuclease that preferentially cleaves 5'-flap DNA substrates and is therefore required for the final steps of long-patch base excision repair which resolves single strand breaks and base lesions (84). It is also active in normal DNA replication where it processes Okazaki fragments on the lagging DNA strand (85). Since we know telomeres can be dramatically shortened by trimming, loss of Mus81, FEN1 or WRN may accelerate this process as cells lose the ability to resolve difficulties encountered during DNA replication. This may be particularly pertinent at telomeres of telomerase-deficient cells in which G-rich quadruplexes are especially difficult to replicate. FEN1 and WRN directly interact in lagging strand replication, HR and the restart of stalled replication forks (86) and the FEN1/WRN telomere-loss phenotype can be rescued by exogenously expressing telomerase (87). However, WRN promotes t-circle formation in TRF2-positive ALT cells and associates with other telomere shelterin proteins and in APBs (88-90), but WRN deficiency leads, in fact, to stimulation of ALT in telomerase-negative mouse cells (91). This suggests that WRN functions in telomerase-positive cells to control resolution of perturbed replication forks but restricts HR-mediated processes required for ALT-mediated telomere elongation.
Structural maintenance of chromosome proteins in ALT
Inter-twinings between sister chromatids are produced by the DNA replication process and since replication forks connect sister chromatids, incomplete DNA replication impairs chromosome segregation. The SMC5/6 SUMO E3 ligase complex is one of three structural maintenance of chromosome (SMC) complexes within eukaryotic cells which function in relieving these torsional stresses by post translational modification of proteins including those involved in telomere capping (92). Since replication fork stalling is common at telomeres, a number of studies associate Smc5/6 with repair of stalled/collapsed replication forks especially in telomerase-defective ALT cells (92). Potts and Yu reported a role for the SMC5/6 complex in the maintenance of telomeres in human ALT cells (93). Smc5/6 depletion in ALT cells inhibits telomere recombination, causing telomere shortening and cell senescence. These studies uncovered a role for the complex in sumoylating components of the telomere shelterin complex, including TRF1, TRF2, RAP1, and TIN1, and facilitating telomere recruitment or retention within APBs. More recently, Chavez et al utilized S. cerevisiae to determine the roles of sumoylation in telomerase-deficient mutants during senescence and demonstrated that sumoylation-deficient telomerase-null cells senesce at an elevated rate (94). Senescent cells deficient in SMC5/6-mediated sumoylation exhibited elevated levels of recombination intermediates selectively at their telomere ends and the authors conclude that this may serve to ensure the faithful completion of template switch recombination. Thus, Smc5/6 appears to counteract accumulation of HR structures at telomeres in senescing telomerase negative yeast cells (92).
Epigenetic regulation at telomeres and in ALT
Telomeres are generally tightly packed heterochromatic regions of the genome, and are less active in transcription than the more open and transcriptionally active euchromatic regions that usually replicate first in S-phase of the cell cycle (95). Thus, telomeres are enriched with histone modifications (e.g. methylation of H3K9, K27, K20) that remodel chromatin to promote tighter conformations that are more silent and protected. Specifically, tri-methylation of histone H3K9 (H3K9me3) by Suv39H1/2 methyltransferases facilitates heterochromatin Protein -1 (HP1) to promote transcriptional silencing. Murine SUV39H1/2 deficiency and subsequent loss of H3K9me3 at telomeres respectively, results in telomere elongation (96). Likewise, over-expression of Dot1 (Disruptor of Telomeres-1 discovered in yeast with known homolog in humans), results in hypermethylated H3K79 at telomeres and also results in telomere elongation (95-97).
DNA itself can be methylated in heterochromatic regions which tend to be rich in CpG dinucleotide sequences and DNA methyltransferases (DNMT3A and B) perform these functions following semi-conservative DNA replication. Mouse deficient in DNMT1 or 3A/B show loss of telomere CpG methylation and also display elongated telomeres. However unlike in humans, only mouse telomeric DNA has been shown to be rich in CpGs (98). SIRT1 is the human homolog of yeast Sir2 deacetylase (type III histone deacetylase), a component of the silent information regulator (SIR) complex encompassing Sir2/Sir3/Sir4. Increasing evidence suggests a major role for SIRT1 in DDR. SIRT1 is recruited to the chromatin upon different DNA damage insults, where it promotes efficient repair of DSBs by HR (99). SIRT1 deacetylates WRN (100)(101,102) and Nbs1 (103,104) and in so doing inhibits Nbs1 phosphorylation and modulates its S-phase checkpoint activity (105). Yeast Sir2 is recruited to telomeres through Rap1 and this complex spreads into subtelomeric DNA via histone deacetylation. Human SIRT1 has been shown to be a positive regulator of telomere length in vivo and attenuates telomere shortening in telomerase-positive cells (106). It is currently unknown if Sirtuins are involved in ALT, however, due to its promotery roles in HR, it would not at all be surprising.
As mentioned earlier, Heaphy et al identified a 100% (19/19) correlation between ALT+ status and occurrence of mutations in ATRX and DAXX genes in pancreatic neuroendocrine tumour samples (46). Out of 439 samples across other cancers 8/8 ALT+ cases were ATRX mutated and a further 16 ALT+ samples had no detectable ATRX/DAXX protein. Exons 2-19 of ATRX were also homozygously deleted in the prototypical ALT+ cell line U2OS and this produced no detectable ATRX protein by immunolabeling. ATRX is a member of the SWI/SNF family of chromatin remodelers and possesses ATPase-driven DNA helicase activity. This unwinding function may be particular active at telomeres since recent studies show a cell cycle regulated role for ATRX in heterochromatin assembly at repetitive G-rich regions. These studies revealed that ATRX and DAXX associated with the histone variant H3.3 and shown to be required for the localization of H3.3 at telomeres and for the repression of telomeric RNA in mouse embryonic stem cells (107-109). Precise roles of ATRX and DAXX in ALT however are currently unknown.
The choice between ALT and telomerase reactivation in cancer
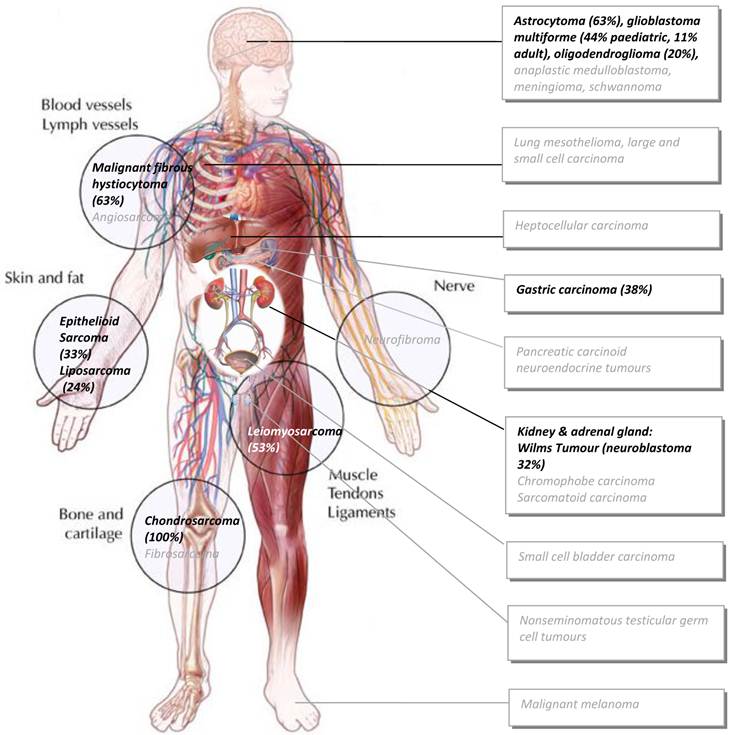
Since ALT is well known to be enriched in sarcomas, it has been suggested that ALT tends to occur in cells of mesenchymal origin in which perhaps telomerase may be more tightly controlled (110). Recent experimental evidence supports this notion. Dominant negative mutant telomerase transfection in bladder T24 cells resulted in a switch towards ALT (as measured by APB foci) after the 27th passage in culture and this was associated with a change in gene expression and morphology concomitant with epithelial to mesenchymal transition (EMT) (111), a transition common during disease progression. Sarcomas are cancerous tumours generally of mesenchymal origin and form in the connective tissues of muscle, tendon, fat, blood vessels, or other soft tissues of the body (lymph nodes, nerves, and tissues around joints) with the legs, stomach, arms and trunk being the most common sarcoma sites (See Figure 2). However, ALT is also found commonly in certain types of CNS tumors, neuroendocrine tumors and testicular germ cell tumors. Thus, it may not be so much that ALT is a mesenchymal trait per se, rather it may just be difficult for most cancers of epithelial tissues to engage ALT (10). It is tempting to speculate that as mesenchymal connective tissue originates from the highest embryonic potential of stem cells (112) and that telomerase activity is highly active in early mammalian embryogenesis (11,12), the robust silencing of telomerase that follows upon completion of embryogenesis may persist much more strongly in mesenchymal tissue through to adulthood than other tissues that never required such strong silencing.
Malignancies prevalent in ALT. Black text in circles denote sarcomas most prevalent in ALT (>20% of those tested on average). Black text in boxes denote the main tumours in which ALT has been detected to a significant degree of frequency (>20% of those tested on average). Grey text in boxes indicate subsets where ALT has been detected but to a lesser degree (<20% and >4% and these make up the rest of the top-10 most ALT prevalent cancer cases. See text for detailed breakdown of ALT in each subtype. (Adapted from the Massachussets Ganeral Hospital sarcoma website: http://www2.massgeneral.org/cancerresourceroom/types/pedi/illustrations/sarcoma.asp)
It was reported that telomerase is actively repressed at the chromatin level in ALT cell lines (113). Later, the same group uncovered a hiearchical gene cluster of 297 up and down regulated genes from a global gene expression profile of mesenchymal liposarcoma cell lines and tumour tissue that may function to repress telomerase activity and activate an ALT pathway (114). Lower c-Myc activity in ALT-positive cells was one gene highlighted, which is consistent with the fact that c-Myc is a known hTERT transcriptional activator (115). It would be particularly interesting to know if a common ALT gene signature, if one exists across all ALT tumours, together with certain epigenetic imbalances that might be prevalent in ALT tumours, may commonly enhance or bias towards HR-mediated responses to stalled replication forks or DSBs.
Recent evidence suggests that cancer cells just entering the point of crisis with some remaining long telomeres present on their chromosomes are more likely to undergo ALT than telomerase activation (116). This is based on the observation that longer telomeres in yeast type II survivors (good models for human ALT sharing similar characteristics) are preferential substrates for recombination. The authors speculate that mutations in cancer cells that cause crisis with remaining long telomeres will drive an immortal route via ALT. It would be interesting to see if ATRX/DAXX mutations, recently identified to track 100% with ALT (46,117), are indeed associated with longer telomeres at the ends of some of the chromosomes in these cells.
Disease linkage
Many studies across various tumour biopsies over the past two decades have culminated in the estimation that 85-90% of all human tumours utilise telomerase activity (10,14,40,118-123). However, the presence of ALT in tumours has not been extensively examined across a large tissue array under the same assay conditions. Recently, an ambitious survey analysed over 6000 tissue samples scoring for ALT using FISH to probe for APB foci (124,125). Overall, 3.7% of tissues in that study were determined to be ALT-positive which is lower than previously estimated. However, despite this large study, ALT has not been screened within large enough cohorts within individual cancer subtypes to give an overall accurate estimation of ALT across disease and this will require further screening at centres where sufficient biopsies are collected and scored under the same conditions, especially in large cohorts of previously identified enriched ALT segments. That aside, the overall spread of ALT across the board (see Table 1) was in agreement with previous findings (25,126-135).
Tumour types in which ALT was detected in this survey, in descending order of prevalence (124,125) (see also Figure 2).
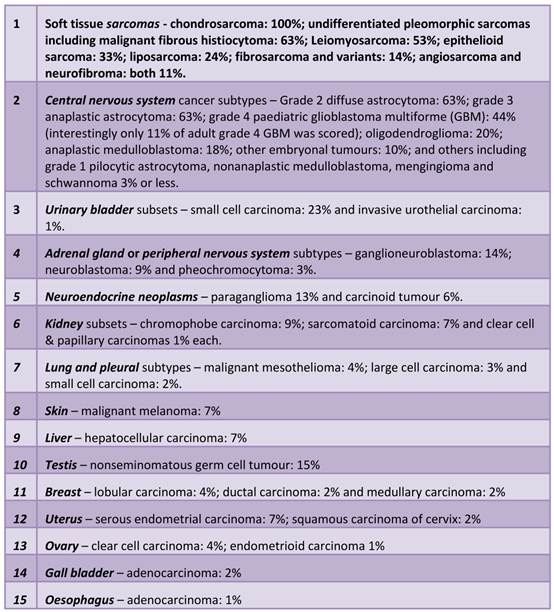
The accumulated evidence from previous studies that have focussed on one particular cancer subtype using limited numbers of TMA samples have reported similar prevalences of ALT as has been documented in this recent survey. However, a particular study highlighted ALT prevelance in 38% (17/42) of cases of gastric carcinoma tissues analysed (136). This does not seem to be in concordance with Heaphy et al's survey in which no occurences of ALT in 155 samples of gastric adenocarcinoma were detected. This may reflect the fact most samples were adenocarcinomas of the stomach and these might not utilise ALT as commonly as other gastric carcinomas. Omori et al also saw ALT frequency to be even higher in tissues scored as having microsattelite instability (mismatch repair deficient) although the mechanism connecting ALT with MSI is currently unknown.
Perhaps surprisingly, another recent study has identified a particularly high prevalence of ALT (32%) in Wilm's Tumour (WT) samples (paediatric nephroblastoma of the kidney) - one of the most common solid tumours of childhood (137). Many paediatric tumours over-express telomerase. Thus, many studies of WT and other paediatric malignancies have focused on telomerase over-activation (138-140). This paediatric tumour seems to activate ALT quite frequently. However, since these tumours contain a mix of epithelial and mesenchymal tissue - its gene expression profile resembling the earliest epithelial to mesenchymal transitory (EMT) stage - this prompted the authors to investigate ALT in WT which to their knowledge had not been done previously. Therefore the mesenchymal status of WT may provide the driver for ALT in this disease as previously discussed. Alternatively, a pattern may be emerging in which paediatric malignancies may be particular prevalent in ALT, recently highlighted in paediatric verses adult glioblastoma multiforme (see Table 1). One possible reason for this could be that a recent and robust silencing of telomerase activity following embryogenesis may provide a vulnerable time frame for ALT to take over under such telomerase repressed conditions. Accordingly, late onset telomerase-positive tumours might take advantage of gradually more relaxed telomerase-silencing mechanisms, perhaps after an accumulation of precluding mutations that contribute to telomerase reactivation. It would be interesting to know the ALT prevalence in other early-onset paediatric malignancies.
Figure 2 illustrates the most frequent ALT-bearing tumour types currently documented in the literature and incorporates previously documented highly prevalent subsets into the findings of the most recent and comprehensive survey published this year. It represents a large collection of broad tumour types, many of which are rare niche segments and others more common, but most tend to have particularly high unmet medical needs. Therefore, drug discovery programs aimed at targeting specific ALT components will be of significant clinical value in the pursuit of developing personalized healthcare medicines in particularly difficult to treat cancer subsets.
Concluding remarks
The pathways and specific components of ALT represent extremely attractive targets for therapeutic intervention in cancers that thwart telomere erosion independently of telomerase activity. To this end, factors such as redundancy and telomerase reactivation must be considered as possible challenges in the future. However, the rapid shortening of telomeres seen when ALT is inhibited in ALT-positive tumour cell lines and the clear steer towards specific tumour types prevalent in ALT provide exciting therapeutic opportunities with defined lines of site to the clinic. As more studies that focus on the specific mechanisms involved in ALT help us to understand exactly why certain tumours become dependent on it for survival, we will move ever closer to developing effective therapies designed to deliver mortal blows to these specific cancers subtypes.
Acknowledgements
I would like to thank Nicol Keith, Alan Meeker, Roger Reddel and Sheila Stewart for helpful insights and regret not being able to include all studies pertinent to this broad and important area of research.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD. et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc.Natl.Acad.Sci.U.S.A. 1988Sep;85(18):6622-6626
2. Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H. et al. Mammalian telomeres end in a large duplex loop. Cell. 1999May14;97(4):503-514
3. Vaziri H, Benchimol S. From telomere loss to p53 induction and activation of a DNA-damage pathway at senescence: the telomere loss/DNA damage model of cell aging. Exp.Gerontol. 1996;31(1-2):295-301
4. Shay JW, Wright WE. Telomerase activity in human cancer. Curr.Opin.Oncol. 1996Jan;8(1):66-71
5. Harley CB. Telomere loss: mitotic clock or genetic time bomb? Mutat.Res. 1991;256(2-6):271-282
6. Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Lansdorp PM. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc.Natl.Acad.Sci.U.S.A. 1994Oct11;91(21):9857-9860
7. d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T. et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003Nov13;426(6963):194-198
8. Blackburn EH, Greider CW, Henderson E, Lee MS, Shampay J, Shippen-Lentz D. Recognition and elongation of telomeres by telomerase. Genome. 1989;31(2):553-560
9. Cesare AJ, Reddel RR. Telomere uncapping and alternative lengthening of telomeres. Mech. Ageing Dev. 2008;129(1-2):99-108
10. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat.Rev.Genet. 2010May;11(5):319-330
11. Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev.Genet. 1996;18(2):173-179
12. Ulaner GA, Giudice LC. Developmental regulation of telomerase activity in human fetal tissues during gestation. Mol.Hum.Reprod. 1997Sep;3(9):769-773
13. Reddel RR, Bryan TM, Colgin LM, Perrem KT, Yeager TR. Alternative lengthening of telomeres in human cells. Radiat.Res. 2001Jan;155(1 Pt 2):194-200
14. Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA. et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin.Cancer Res. 2005Jan1;11(1):217-225
15. Shay JW, Wright WE. The reactivation of telomerase activity in cancer progression. Trends Genet. 1996Apr;12(4):129-131
16. Holt SE, Wright WE, Shay JW. Regulation of telomerase activity in immortal cell lines. Mol.Cell.Biol. 1996Jun;16(6):2932-2939
17. Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J.Hematol.Oncol. 2008Oct29;1:20
18. Roth A, Harley CB, Baerlocher GM. Imetelstat (GRN163L)--telomerase-based cancer therapy. Recent Results Cancer Res. 2010;184:221-234
19. Ouellette MM, Wright WE, Shay JW. Targeting telomerase-expressing cancer cells. J.Cell.Mol.Med. 2011Jul;15(7):1433-1442
20. Buseman CM, Wright WE, Shay JW. Is telomerase a viable target in cancer? Mutat.Res. 2012Feb1;730(1-2):90-7
21. Ouellette MM, Wright WE, Shay JW. Targeting telomerase-expressing cancer cells. J.Cell.Mol.Med. 2011Jul;15(7):1433-1442
22. Sidorov IA, Hirsch KS, Harley CB, Dimitrov DS. Cancer treatment by telomerase inhibitors: predictions by a kinetic model. Math.Biosci. 2003Feb;181(2):209-221
23. Folini M, Gandellini P, Zaffaroni N. Targeting the telosome: therapeutic implications. Biochim.Biophys.Acta. 2009Apr;1792(4):309-316
24. Siddiqa A, Cavazos DA, Marciniak RA. Targeting telomerase. Rejuvenation Res. 2006;9(3):378-390
25. Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat.Rev.Genet. 2010May;11(5):319-330
26. Royle NJ, Mendez-Bermudez A, Gravani A, Novo C, Foxon J, Williams J. et al. The role of recombination in telomere length maintenance. Biochem.Soc.Trans. 2009Jun;37(Pt 3):589-595
27. Saharia A, Stewart SA. FEN1 contributes to telomere stability in ALT-positive tumor cells. Oncogene. 2009Feb26;28(8):1162-1167
28. Zeng S, Yang Q. The MUS81 endonuclease is essential for telomerase negative cell proliferation. Cell.Cycle. 2009Jul15;8(14):2157-2160
29. Zeng S, Xiang T, Pandita TK, Gonzalez-Suarez I, Gonzalo S, Harris CC. et al. Telomere recombination requires the MUS81 endonuclease. Nat.Cell Biol. 2009May;11(5):616-623
30. Temime-Smaali N, Guittat L, Sidibe A, Shin-ya K, Trentesaux C, Riou JF. The G-quadruplex ligand telomestatin impairs binding of topoisomerase IIIalpha to G-quadruplex-forming oligonucleotides and uncaps telomeres in ALT cells. PLoS One. 2009Sep9;4(9):e6919
31. Patel DJ, Phan AT, Kuryavyi V. Human telomere, oncogenic promoter and 5'-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007;35(22):7429-7455
32. Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35(2):406-413
33. Huppert JL. Four-stranded nucleic acids: structure, function and targeting of G-quadruplexes. Chem.Soc.Rev. 2008Jul;37(7):1375-1384
34. Huppert JL. Structure, location and interactions of G-quadruplexes. FEBS J. 2010Sep;277(17):3452-3458
35. Cerone MA, Londono-Vallejo JA, Bacchetti S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum.Mol.Genet. 2001Sep1;10(18):1945-1952
36. Hiyama E, Hiyama K, Yokoyama T, Matsuura Y, Piatyszek MA, Shay JW. Correlating telomerase activity levels with human neuroblastoma outcomes. Nat.Med. 1995Mar;1(3):249-255
37. Hiyama E, Hiyama K, Nishiyama M, Reynolds CP, Shay JW, Yokoyama T. Differential gene expression profiles between neuroblastomas with high telomerase activity and low telomerase activity. J.Pediatr.Surg. 2003Dec;38(12):1730-1734
38. Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995Sep1;14(17):4240-4248
39. Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999Sep1;59(17):4175-4179
40. Henson JD, Reddel RR. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010Sep10;584(17):3800-3811
41. Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985Dec;43(2 Pt 1):405-413
42. Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987Dec24;51(6):887-898
43. Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989Jan26;337(6205):331-337
44. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL. et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994Dec23;266(5193):2011-2015
45. Xin H. Telomeric repeat amplification protocol: measuring the activity of the telomerase. Methods Mol.Biol. 2011;735:107-111
46. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C. et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011Jul22;333(6041):425
47. Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 1993Apr23;73(2):347-360
48. Teng SC, Zakian VA. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol.Cell.Biol. 1999Dec;19(12):8083-8093
49. Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat.Genet. 2000Dec;26(4):447-450
50. Bailey SM, Brenneman MA, Goodwin EH. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004Jul16;32(12):3743-3751
51. Royle NJ, Foxon J, Jeyapalan JN, Mendez-Bermudez A, Novo CL, Williams J. et al. Telomere length maintenance--an ALTernative mechanism. Cytogenet.Genome Res. 2008;122(3-4):281-291
52. Pickett HA, Henson JD, Au AY, Neumann AA, Reddel RR. Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum.Mol.Genet. 2011Dec1;20(23):4684-4692
53. Verdun RE, Crabbe L, Haggblom C, Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol.Cell. 2005Nov23;20(4):551-561
54. De Boeck G, Forsyth RG, Praet M, Hogendoorn PC. Telomere-associated proteins: cross-talk between telomere maintenance and telomere-lengthening mechanisms. J.Pathol. 2009Feb;217(3):327-344
55. Durant ST, Paffett KS, Shrivastav M, Timmins GS, Morgan WF, Nickoloff JA. UV radiation induces delayed hyperrecombination associated with hypermutation in human cells. Mol.Cell.Biol. 2006Aug;26(16):6047-6055
56. Tanaka H, Mendonca MS, Bradshaw PS, Hoelz DJ, Malkas LH, Meyn MS. et al. DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc.Natl.Acad.Sci.U.S.A. 2005Oct25;102(43):15539-15544
57. Huda N, Tanaka H, Mendonca MS, Gilley D. DNA damage-induced phosphorylation of TRF2 is required for the fast pathway of DNA double-strand break repair. Mol.Cell.Biol. 2009Jul;29(13):3597-3604
58. Vousden KH, Lane DP. P53 in Health and Disease. Nat.Rev.Mol.Cell Biol. 2007Apr;8(4):275-283
59. Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B. et al. Arginine methylation regulates the p53 response. Nat.Cell Biol. 2008Dec;10(12):1431-1439
60. Durant ST, Cho EC, La Thangue NB. p53 methylation--the Arg-ument is clear. Cell.Cycle. 2009Mar15;8(6):801-802
61. Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH. et al. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. 1997Oct1;16(19):6018-6033
62. Dantzer F, Giraud-Panis MJ, Jaco I, Ame JC, Schultz I, Blasco M. et al. Functional interaction between poly(ADP-Ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol.Cell.Biol. 2004Feb;24(4):1595-1607
63. Jiang WQ, Zhong ZH, Henson JD, Neumann AA, Chang AC, Reddel RR. Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol.Cell.Biol. 2005Apr;25(7):2708-2721
64. Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007Dec10;26(56):7741-7748
65. Wu Y, Xiao S, Zhu XD. MRE11-RAD50-NBS1 and ATM function as co-mediators of TRF1 in telomere length control. Nat.Struct.Mol.Biol. 2007Sep;14(9):832-840
66. Zhong ZH, Jiang WQ, Cesare AJ, Neumann AA, Wadhwa R, Reddel RR. Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J.Biol.Chem. 2007Oct5;282(40):29314-29322
67. Celli GB, Denchi EL, de Lange T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat.Cell Biol. 2006Aug;8(8):885-890
68. Bodvarsdottir SK, Steinarsdottir M, Bjarnason H, Eyfjord JE. Dysfunctional telomeres in human BRCA2 mutated breast tumors and cell lines. Mutat.Res. 2012Jan3;729(1-2):90-9
69. French JD, Dunn J, Smart CE, Manning N, Brown MA. Disruption of BRCA1 function results in telomere lengthening and increased anaphase bridge formation in immortalized cell lines. Genes Chromosomes Cancer. 2006Mar;45(3):277-289
70. Li B, Reddy S, Comai L. Depletion of Ku70/80 reduces the levels of extrachromosomal telomeric circles and inhibits proliferation of ALT cells. Aging (Albany NY). 2011Apr;3(4):395-406
71. Liu Y, Tarsounas M, O'regan P, West SC. Role of RAD51C and XRCC3 in genetic recombination and DNA repair. J.Biol.Chem. 2007Jan19;282(3):1973-1979
72. Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M. et al. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004Jun25;117(7):873-886
73. Durant ST, Nickoloff JA. Good timing in the cell cycle for precise DNA repair by BRCA1. Cell.Cycle. 2005Sep;4(9):1216-1222
74. Durant ST, Paffett KS, Shrivastav M, Timmins GS, Morgan WF, Nickoloff JA. UV radiation induces delayed hyperrecombination associated with hypermutation in human cells. Mol.Cell.Biol. 2006Aug;26(16):6047-6055
75. Shrivastav M, Miller CA, De Haro LP, Durant ST, Chen BP, Chen DJ. et al. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair (Amst). 2009Aug6;8(8):920-929
76. Lee SH, Oshige M, Durant ST, Rasila KK, Williamson EA, Ramsey H. et al. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc.Natl.Acad.Sci.U.S.A. 2005Dec13;102(50):18075-18080
77. De Haro LP, Wray J, Williamson EA, Durant ST, Corwin L, Gentry AC. et al. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010Sep;38(17):5681-5691
78. MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ. et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. 2010Jul9;142(1):65-76
79. Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K. et al. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc.Natl.Acad.Sci.U.S.A. 2011Apr19;108(16):6492-6496
80. Zhu XD, Niedernhofer L, Kuster B, Mann M, Hoeijmakers JH, de Lange T. ERCC1/XPF removes the 3' overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol.Cell. 2003Dec;12(6):1489-1498
81. Chen XB, Melchionna R, Denis CM, Gaillard PH, Blasina A, Van de Weyer I. et al. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol.Cell. 2001Nov;8(5):1117-1127
82. Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR 3rd, Russell P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001Nov16;107(4):537-548
83. Osman F, Whitby MC. Exploring the roles of Mus81-Eme1/Mms4 at perturbed replication forks. DNA Repair (Amst). 2007Jul1;6(7):1004-1017
84. Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G. et al. Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011Apr15;145(2):198-211
85. Zheng L, Shen B. Okazaki fragment maturation: nucleases take centre stage. J.Mol.Cell.Biol. 2011Feb;3(1):23-30
86. Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu.Rev.Biochem. 2004;73:589-615
87. Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004Dec10;306(5703):1951-1953
88. Eller MS, Liao X, Liu S, Hanna K, Backvall H, Opresko PL. et al. A role for WRN in telomere-based DNA damage responses. Proc.Natl.Acad.Sci.U.S.A. 2006Oct10;103(41):15073-15078
89. Opresko PL, Sowd G, Wang H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PLoS One. 2009;4(3):e4825
90. Kusumoto-Matsuo R, Opresko PL, Ramsden D, Tahara H, Bohr VA. Cooperation of DNA-PKcs and WRN helicase in the maintenance of telomeric D-loops. Aging (Albany NY). 2010May;2(5):274-284
91. Laud PR, Multani AS, Bailey SM, Wu L, Ma J, Kingsley C. et al. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005Nov1;19(21):2560-2570
92. Noel JF, Wellinger RJ. The Smc5/6 complex and the difficulties cutting the ties of twin sisters. Aging (Albany NY). 2011Mar;3(3):186-188
93. Potts PR, Yu H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat.Struct.Mol.Biol. 2007Jul;14(7):581-590
94. Chavez A, George V, Agrawal V, Johnson FB. Sumoylation and the structural maintenance of chromosomes (Smc) 5/6 complex slow senescence through recombination intermediate resolution. J.Biol.Chem. 2010Apr16;285(16):11922-11930
95. Grewal SI, Jia S. Heterochromatin revisited. Nat.Rev.Genet. 2007Jan;8(1):35-46
96. Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011Jul1;25(13):1345-1358
97. Norris A, Boeke JD. Silent information regulator 3: the Goldilocks of the silencing complex. Genes Dev. 2010Jan15;24(2):115-122
98. Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M. et al. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat.Cell Biol. 2006Apr;8(4):416-424
99. Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK. et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008Nov28;135(5):907-918
100. Vaitiekunaite R, Butkiewicz D, Krzesniak M, Przybylek M, Gryc A, Snietura M. et al. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech.Ageing Dev. 2007;128(11-12):650-661
101. Kahyo T, Mostoslavsky R, Goto M, Setou M. Sirtuin-mediated deacetylation pathway stabilizes Werner syndrome protein. FEBS Lett. 2008Jul23;582(17):2479-2483
102. Sakao Y, Kato A, Tsuji T, Yasuda H, Togawa A, Fujigaki Y. et al. Cisplatin induces Sirt1 in association with histone deacetylation and increased Werner syndrome protein in the kidney. Clin.Exp.Nephrol. 2011Jun;15(3):363-372
103. Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol.Cell. 2007Jul6;27(1):149-162
104. Yuan Z, Seto E. A functional link between SIRT1 deacetylase and NBS1 in DNA damage response. Cell.Cycle. 2007Dec1;6(23):2869-2871
105. Lim DS, Kim ST, Xu B, Maser RS, Lin J, Petrini JH. et al. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000Apr6;404(6778):613-617
106. Palacios JA, Herranz D, De Bonis ML, Velasco S, Serrano M, Blasco MA. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J.Cell Biol. 2010Dec27;191(7):1299-1313
107. Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V. et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010Oct29;143(3):367-378
108. Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S. et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010Mar5;140(5):678-691
109. Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD. et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010Mar;20(3):351-360
110. Lafferty-Whyte K, Cairney CJ, Will MB, Serakinci N, Daidone MG, Zaffaroni N. et al. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene. 2009Oct29;28(43):3765-3774
111. Xue Y, Li L, Zhang D, Wu K, Chen Y, Zeng J. et al. Twisted Epithelial-to-Mesenchymal Transition Promotes Progression of Surviving Bladder Cancer T24 Cells with hTERT-Dysfunction. PLoS One. 2011;6(11):e27748
112. Stiller D, Path FW, Kaiser HE. Some characteristics of the mesenchymal stem cell of soft tissue tumors. In Vivo. 1992;6(4):477-480
113. Atkinson SP, Hoare SF, Glasspool RM, Keith WN. Lack of telomerase gene expression in alternative lengthening of telomere cells is associated with chromatin remodeling of the hTR and hTERT gene promoters. Cancer Res. 2005Sep1;65(17):7585-7590
114. Lafferty-Whyte K, Cairney CJ, Will MB, Serakinci N, Daidone MG, Zaffaroni N. et al. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene. 2009Oct29;28(43):3765-3774
115. Hao H, Nancai Y, Lei F, Xiong W, Wen S, Guofu H. et al. siRNA directed against c-Myc inhibits proliferation and downregulates human telomerase reverse transcriptase in human colon cancer Colo 320 cells. J.Exp.Clin.Cancer Res. 2008Aug12;27:27
116. Chang M, Dittmar JC, Rothstein R. Long telomeres are preferentially extended during recombination-mediated telomere maintenance. Nat.Struct.Mol.Biol. 2011Apr;18(4):451-456
117. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C. et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011Jul22;333(6041):425
118. McDonald KL, McDonnell J, Muntoni A, Henson JD, Hegi ME, von Deimling A. et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J.Neuropathol.Exp.Neurol. 2010Jul;69(7):729-736
119. Costa A, Daidone MG, Daprai L, Villa R, Cantu S, Pilotti S. et al. Telomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progression. Cancer Res. 2006Sep1;66(17):8918-8924
120. Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002Jan21;21(4):598-610
121. Villa R, Daidone MG, Motta R, Venturini L, De Marco C, Vannelli A. et al. Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin.Cancer Res. 2008Jul1;14(13):4134-4140
122. Subhawong AP, Heaphy CM, Argani P, Konishi Y, Kouprina N, Nassar H. et al. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod.Pathol. 2009Nov;22(11):1423-1431
123. Venturini L, Erdas R, Costa A, Gronchi A, Pilotti S, Zaffaroni N. et al. ALT-associated promyelocytic leukaemia body (APB) detection as a reproducible tool to assess alternative lengthening of telomere stability in liposarcomas. J.Pathol. 2008Mar;214(4):410-414
124. Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E. et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am.J.Pathol. 2011Oct;179(4):1608-1615
125. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C. et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011Jul22;333(6041):425
126. Silvestre DC, Pineda JR, Hoffschir F, Studler JM, Mouthon MA, Pflumio F. et al. Alternative lengthening of telomeres in human glioma stem cells. Stem Cells. 2011Mar;29(3):440-451
127. Lundberg G, Sehic D, Lansberg JK, Ora I, Frigyesi A, Castel V. et al. Alternative lengthening of telomeres--an enhanced chromosomal instability in aggressive non-MYCN amplified and telomere elongated neuroblastomas. Genes Chromosomes Cancer. 2011Apr;50(4):250-262
128. Au AY, Hackl T, Yeager TR, Cohen SB, Pass HI, Harris CC. et al. Telomerase activity in pleural malignant mesotheliomas. Lung Cancer. 2011Sep;73(3):283-288
129. Lundberg G, Sehic D, Lansberg JK, Ora I, Frigyesi A, Castel V. et al. Alternative lengthening of telomeres--an enhanced chromosomal instability in aggressive non-MYCN amplified and telomere elongated neuroblastomas. Genes Chromosomes Cancer. 2011Apr;50(4):250-262
130. Venturini L, Motta R, Gronchi A, Daidone M, Zaffaroni N. Prognostic relevance of ALT-associated markers in liposarcoma: a comparative analysis. BMC Cancer. 2010Jun3;10:254
131. Henson JD, Reddel RR. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010Sep10;584(17):3800-3811
132. Slatter T, Gifford-Garner J, Wiles A, Tan X, Chen YJ, MacFarlane M. et al. Pilocytic astrocytomas have telomere-associated promyelocytic leukemia bodies without alternatively lengthened telomeres. Am.J.Pathol. 2010Dec;177(6):2694-2700
133. Matsuo T, Shimose S, Kubo T, Fujimori J, Yasunaga Y, Ochi M. Alternative lengthening of telomeres as a prognostic factor in malignant fibrous histiocytomas of bone. Anticancer Res. 2010Dec;30(12):4959-4962
134. Yamamoto Y, Yamamoto N, Tajima K, Ohno A, Washimi Y, Ishimura D. et al. Characterization of human multicentric osteosarcoma using newly established cells derived from multicentric osteosarcoma. J.Cancer Res.Clin.Oncol. 2011Mar;137(3):423-433
135. Lundberg G, Sehic D, Lansberg JK, Ora I, Frigyesi A, Castel V. et al. Alternative lengthening of telomeres-An enhanced chromosomal instability in aggressive non-MYCN amplified and telomere elongated neuroblastomas. Genes Chromosomes Cancer. 2011Apr;50(4):250-62
136. Omori Y, Nakayama F, Li D, Kanemitsu K, Semba S, Ito A. et al. Alternative lengthening of telomeres frequently occurs in mismatch repair system-deficient gastric carcinoma. Cancer.Sci. 2009Mar;100(3):413-418
137. Venturini L, Daidone MG, Motta R, Collini P, Spreafico F, Terenziani M. et al. Telomere maintenance in Wilms tumors: first evidence for the presence of alternative lengthening of telomeres mechanism. Genes Chromosomes Cancer. 2011Oct;50(10):823-829
138. Dome JS, Chung S, Bergemann T, Umbricht CB, Saji M, Carey LA. et al. High telomerase reverse transcriptase (hTERT) messenger RNA level correlates with tumor recurrence in patients with favorable histology Wilms' tumor. Cancer Res. 1999Sep1;59(17):4301-4307
139. Dome JS, Bockhold CA, Li SM, Baker SD, Green DM, Perlman EJ. et al. High telomerase RNA expression level is an adverse prognostic factor for favorable-histology Wilms' tumor. J.Clin.Oncol. 2005Dec20;23(36):9138-9145
140. Wittmann S, Wunder C, Zirn B, Furtwangler R, Wegert J, Graf N. et al. New prognostic markers revealed by evaluation of genes correlated with clinical parameters in Wilms tumors. Genes Chromosomes Cancer. 2008May;47(5):386-395
Author contact
![]() Corresponding author: Stephen T Durant, Associate Principal Scientist, AstraZeneca - 33F41, Mereside, DNA Damage Response, Bioscience, Oncology iMed, Alderley Park, Cheshire, SK10 4TG, England, UK. Email: stephen.durantcom or stephentdurantcom; Phone: +44 (0)1625 519562.
Corresponding author: Stephen T Durant, Associate Principal Scientist, AstraZeneca - 33F41, Mereside, DNA Damage Response, Bioscience, Oncology iMed, Alderley Park, Cheshire, SK10 4TG, England, UK. Email: stephen.durantcom or stephentdurantcom; Phone: +44 (0)1625 519562.