Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(2):98-104. doi:10.7150/jca.10496 This issue Cite
Research Paper
Fibronectin Matrix Formation is a Prerequisite for Colonization of Kidney Tumor Cells in Fibrin
Lynn M. Knowles1*, Lisa A. Gurski1*, Jodi K. Maranchie1,2, Jan Pilch1,2,3 ![]()
1. Department of Urology, University of Pittsburgh School of Medicine, Shadyside Medical Center, 5200 Centre Avenue, Pittsburgh, PA15232, USA.
2. Prostate and Urological Cancers Program, University of Pittsburgh Cancer Institute, 5150 Centre Avenue, Pittsburgh, PA 15232, USA.
3. Institute of Clinical Hemostaseology and Transfusion Medicine, Saarland University Medical Center, Ringstr.52, D-66421 Homburg/Saar, Germany.
*LMK and LAG contributed equally.
Received 2014-9-6; Accepted 2014-10-28; Published 2015-1-1
Abstract
Fibrin plays an important role in lung metastasis. Here we show that fibrin promotes colony formation in primary kidney tumor cells from patients with kidney metastasis. In addition, we found that inhibition of fibrin formation with the thrombin inhibitor hirudin in nude mice in vivo significantly reduced the metastatic outgrowth of kidney tumor cells. Colony formation was significantly more efficient in tumor cells embedded in fibrin compared to matrigel and this effect correlates with the capacity of tumor cells to assemble a fibronectin matrix and generate stress fibers. Interestingly, stress fiber formation in fibrin was a specific function of metastatic kidney tumor cells while non-metastatic cells remained round. Inhibition of stress fiber formation with the Rho kinase inhibitor Y-27632, in turn, reduced fibronectin matrix assembly and colony formation in fibrin suggesting that spreading is a critical mechanism for the outgrowth of metastatic kidney tumor cells. Overall, our results indicate that adhesive interactions with fibrin play an important role for the progression of renal cell carcinoma and that inhibiting these interactions could be a promising strategy for treatment and prevention of kidney cancer metastasis.
Keywords: Fibronectin, Metastasis, Fibrin.
Introduction
Cancer is largely incurable once tumor cells begin to metastasize and colonize in distant organs. Metastasis is initiated by tumor cells dislodging from the primary tumor and intravasating into the blood stream. This process is supported by a specific genetic program, epithelial-mesenchymal transition (EMT), which enables tumor cells to detach from the tumor tissue and invade the surrounding tumor stroma as a single cell (1). Then, after entering the blood stream, tumor cells are carried into the vasculature of distant organs, where they extravasate or proliferate within the vasculature (2, 3). This stage represents an important switch from tumor cell dissemination to metastatic colonization, which depends on cell-autonomous processes such as tumor initiation and mesenchymal-epithelial transition (4, 5). Another important determinant for metastatic colonization is the nature of the tissue environment, which needs to provide critical survival and proliferative cues in order for tumor cell colonization to succeed (6-8). Failure to provide these cues can either result in tumor cell death or dormancy.
The ability of tumor cells to generate colonies in distant organs depends in large parts on their capacity to proliferate. Starting out as a single cell, this process requires adhesive interactions of integrins with the extracellular matrix, which result in focal adhesion formation and subsequent activation of MAP kinases or PI3-kinase (4, 9). In line with this, adhesive pathways that modulate F-actin and stress fiber assembly are often up-regulated in metastatic tumor cells and their inhibition has been shown to be anti-metastatic (2, 7). The capacity to generate productive adhesive interactions is also determined by the properties of the surrounding extracellular matrix (6, 10). To this end, it has been demonstrated that fibrin is significantly more efficient in supporting colony formation than collagen I (6). This effect is highly relevant in vivo as embedding in a 3-dimensional (3D) matrix of fibrin specifically conveys tumor- and metastasis-initiating capabilities while treatment with clotting inhibitors or knocking down fibrin(ogen) severely impairs the capacity of tumor cells to metastasize (6, 11, 12).
We previously showed that blood clotting specifically supports lung metastasis but not metastasis to the liver (13). Conversely, we found that tumor cells from cancers that predominantly metastasize to the lung, such as renal cell carcinoma (RCC) and soft tissue sarcoma (STS), display a specific fibrin-invasive phenotype (10). Invadopodia formation in these tumor types depends on a constitutively active form of integrin αvβ3, which promotes fibronectin matrix formation in fibrin-embedded tumor cells. Fibronectin in turn has been shown to activate dormant tumor cells and to induce tumor cell metastasis (7). In line with this, it has been shown that high levels of fibronectin correlate positively with poor outcome in patients with RCC and other malignancies (14, 15). Based on the apparent role of clotting in tumor metastasis, we aimed to define the function of fibronectin matrix formation for kidney tumor cell colonization in fibrin.
Materials and Methods
Tumor Cells. RCC4, 786-0 (renal cell carcinoma, RCC) and HT1080 cells (soft tissue sarcoma, STS) were purchased from ATCC and cultured per manufacturer's specifications. Primary human tumor cells were isolated from kidney tumors of 2 patients with metastatic RCC (pT3bN0M1, pT3bN2Mx) and 2 patients with localized RCC (ea. pT1aNxMx) as previously described (10).
Cell Invasion, Colony Formation and Western Blot. Tumor cells embedded in various 3D matrices were analyzed for invadopodia and colony formation using phase contrast or confocal microscopy as previously described (10, 13, 16, 17). Western blot analysis was performed as previously described (10, 17). Y-27632 Rho kinase inhibitor (EMD Millipore) or recombinant human HGF (R&D Systems) were added to cultures where indicated.
Experimental Metastasis. Lungs from female, 6-8 weeks old athymic nude mice (Charles Rivers) were isolated 6 weeks after intravenous injection (tail vein) with 2.5x105 786-0 kidney cancer cells in the presence or absence of 500 IU hirudin (EMD Millipore). Lungs were subsequently fixed in Bouin's solution and analyzed for tumor multiplicity as well as tumor size using a stereo microscope (Zeiss Stemi 2000-C).
shRNA Mediated Gene Silencing. To achieve stable knockdown of fibronectin, we transduced HT1080 cells with lentiviral shRNA vectors compared to a scrambled control shRNA (University of Pittsburgh Cancer Institute Vector Core Facility) as previously described (10). Target knockdown was confirmed by western blot analysis.
Statistical Analysis. Data were analyzed using unpaired two-tailed Student's t test or one-way ANOVA followed by the posthoc Tukey's multiple comparisons test (GraphPad Prism 5). Treatment differences with a two-sided p value < 0.05 were considered significantly different. Error bars show mean ± SEM.
Results
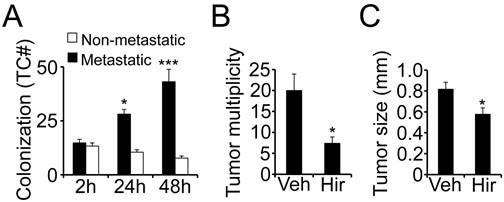
Metastatic kidney tumor cells colonize fibrin. Fibrin matrices have been shown to promote the tumorigenic potential in several tumor types (6). To determine the role of fibrin for tumor cell proliferation in kidney cancer, we embedded primary tumor cells from patients with clear cell RCC in fibrin and scored the clots for tumor colonization over time. Surrounded by 3D fibrin, tumor cells from metastatic RCC began to proliferate soon after embedding with a doubling in cell number after 24 hours and a tripling after 48 hours (Fig. 1A). Cells from non-metastatic tumors on the other hand did not grow at all in fibrin but rather slightly declined in numbers, demonstrating that the clotting cascade specifically promotes colony formation in an aggressive, metastatic subset of kidney tumors. To determine if clotting is also pro-metastatic in vivo, we injected 786-0 kidney tumor cells into the tail vein of athymic nude mice in the presence of the thrombin inhibitor hirudin. Notably, a single application of anticoagulant significantly reduced the overall appearance of metastatic kidney tumors in the lung over time (Fig. 1B). In addition, we found that lung tumors had a significantly smaller diameter after co-injection with hirudin indicating that inhibiting clot formation at the time of tumor cell seeding in the lung is associated with reduced tumor formation as well as growth (Fig. 1C). Moreover, the in vivo results are in line with our data that fibrin promotes colony formation of primary tumor cells from patients with metastatic RCC in vitro.
Metastatic kidney tumor cells colonize fibrin. (A), colony formation of fibrin-embedded primary RCC cells from patients with or without metastasis after 2, 24, and 48 hours. Diagram shows average number of tumor cells per optical field. * p < 0.05, *** p < 0.001, compared to 2 hours. * p < 0.05, 24 hours compared to 48 hours. (B-C), the number of tumor nodules on the surface of lungs (B; tumor multiplicity) and the average diameter of lung tumors (C; tumor size) was assessed on lungs isolated six weeks after i.v. injection of 5 x 105 786-0 RCC cells in the presence of 500 IU hirudin (Hir) or vehicle (Veh), * p < 0.05.
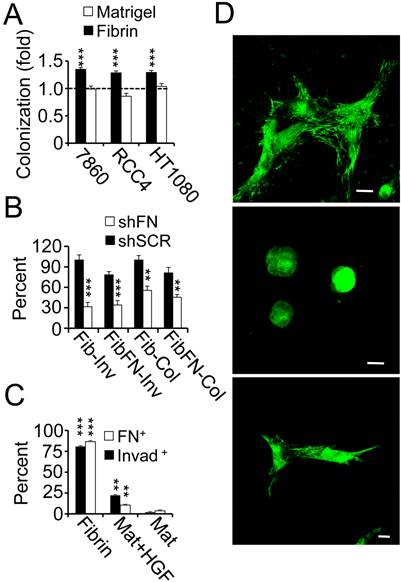
Fibronectin promotes colony formation of tumor cells in fibrin. To determine if the capacity of tumor cells to colonize in 3D culture depends on specific extracellular matrix properties, we embedded a panel of tumor cells side by side in fibrin versus matrigel. While 786-0, RCC4 and HT1080 tumor cells replicated the previously observed colony formation of clinical RCC cells in fibrin, cell numbers remained largely unchanged after embedding the same cell lines in matrigel (Fig. 2A). Paralleling this result, we made the observation that fibrin-embedded tumor cells generated an extensive fibronectin matrix, which was necessary to support tumor cell spreading as well as proliferation in fibrin (Fig. 2B-D; Supplementary Material: Fig. S1). Matrigel-embedded tumor cells, in contrast, displayed a round morphology and an apparent lack of fibronectin matrix deposition unless cells were treated with hepatocyte growth factor, a cytokine that has been shown to induce kidney tumor cell sprouting (Fig. 2C-D) (18). Interestingly, tumor cell growth in fibrin was impaired whether we embedded fibronectin shRNA-transformed tumor cells in fibrin or in fibrin-fibronectin suggesting that tumor cell sprouting depended on endogenous fibronectin (Fig. 2B). Together, these results suggest a close connection between fibronectin expression, tumor cell spreading and colony formation in fibrin compared to other extracellular matrices such as matrigel.
Fibrin promotes fibronectin matrix deposition. (A), 786-0, RCC4 and HT1080 cells embedded in fibrin and matrigel were analyzed for colony formation after 48 hours as fold change over 24 hours (dotted line). (B), fibrin (Fib)- and fibrin-fibronectin (FibFN)-embedded HT1080 cells transformed with fibronectin (shFN) or scrambled (shSCR) shRNA were analyzed for invadopodia (Inv; 24 hours) and colony formation (Col; 48 hours). Fib control was set to 100%. ***P < 0.001, **P < 0.01, shSCR versus shFN. (C), tumor cells embedded in fibrin, matrigel (Mat) or matrigel + 166.7 ng/ml HGF (Mat+HGF) were scored for invadopodia (invad+) or fibronectin matrix formation (FN+) as percent of total tumor cell count per optical field using phase contrast or confocal microscopy, respectively. ***P < 0.001, Fib vs. Mat+HGF and Mat, **P < 0.01, Mat+HGF vs. Mat. (D), confocal images of 786-0 cells 48 hours after embedding in fibrin (top), matrigel (middle), or matrigel + 166.7 ng/ml HGF (bottom) were stained for fibronectin (green). Nuclei are stained with draq5 (blue). Scale bar, 20 µm.
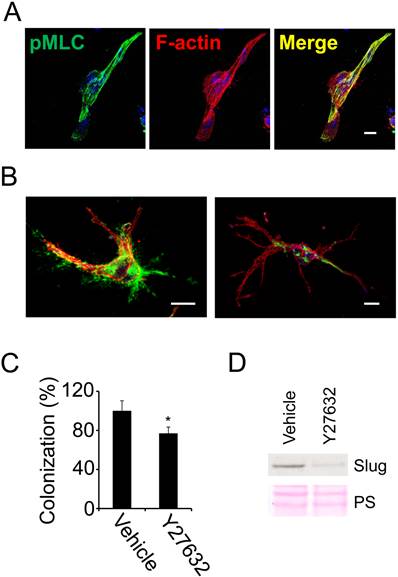
Tumor cell colonization and fibronectin assembly in 3D fibrin depends on actomyosin dynamics. We previously demonstrated that fibronectin is necessary for stress fiber formation in fibrin (10). To further evaluate the role of stress fiber formation in fibrin-embedded tumor cells, we stained 786-0 cells cultured for 48 hours in 3D fibrin with an antibody for phosphorylated myosin light chain (pMLC). Subsequent analysis with confocal microscopy documented that spread 786-0 cells expressed high levels of pMLC, which co-localized completely with F-actin in stress fibers (Fig. 3A). To determine the functional implications of stress fiber formation, we treated fibrin-embedded 786-0 cells with the Rho kinase inhibitor Y-27632, which inhibits stress fiber formation through dephosphorylation of MLC. As a consequence, fibronectin matrix assembly and colony formation were significantly impaired in Y-27632-treated cells (Fig. 3B-C). Moreover, the inability of cells to generate stress fibers correlated with a significant reduction in expression of Slug (snail2), a transcription factor involved in tumor initiation as well as epithelial-mesenchymal transition (EMT) (Fig 3D). Together, these results indicate that tumor cell spreading depends on Rho kinase activity, which in turn generates important signals for tumor colonization in 3D fibrin.
Tumor cell colonization of fibrin depends on stress fiber formation. (A), fibrin-embedded 786-0 cells were fixed after 48 hours and probed for phospho-myosin light chain (pMLC; green) as well as F-actin (red) using confocal microscopy. Nuclei are stained with draq5 (blue). (B), confocal microscopy images of 786-0 cells treated with Y-27632 (right) or vehicle (left) for 24 hours and then fixed and stained with anti-fibronectin (green) as well as phalloidin (red). Scale bar, 20 µm. (C), 786-0 cells treated with 25 µM Y-27632 or vehicle were analyzed for colony formation 48 hours after embedding in fibrin using phase contrast microscopy. *P < 0.05. (D), 786-0 cells were probed for Slug after treatment with Y-27632 for 24 hours.
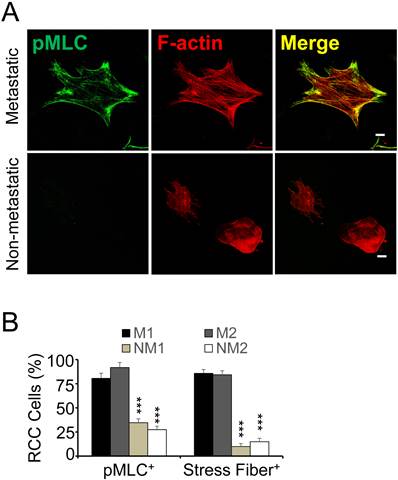
Stress fiber formation is a specific feature of metastatic RCC. Primary kidney cancer cells have been shown to generate an elaborate fibronectin matrix if they are derived from patients with metastasis, while non-metastatic RCC cells are largely devoid of a fibronectin matrix (10). To determine if fibronectin matrix assembly correlates with stress fiber formation in clinical cancer, we embedded primary tumor cells from RCC patients with or without metastasis in fibrin and stained with an antibody against phosphorylated myosin light chain (pMLC) as well as phalloidin to visualize F-actin. Subsequent confocal microscopy revealed co-localization of pMLC as well as F-actin in extensive stress fibers in metastatic RCC cells (Fig. 4A). In non-metastatic cells, pMLC staining was significantly weaker while F-actin was diffusely distributed throughout the cell (Fig. 4A-B). Together, these results show that stress fiber formation in fibrin is a specific function of metastatic RCC cells embedded in fibrin. Moreover the results indicate a link between cell spreading and colony formation in fibrin that correlates with kidney cancer metastasis.
Increased actomyosin dynamics in metastatic RCC. (A), primary RCC cells from patients with or without metastasis were fixed after 48 hours of embedding in fibrin and probed for phospho-myosin light chain (pMLC; green) and F-actin (red) using confocal microscopy. Scale bar, 20 µm. (B), microscopy fields were scored for stress fiber (SF)- and pMLC-positive RCC cells (M, metastastatic; NM, non-metastatic; 2 patients ea.) as percent of total after 48 hours embedding in fibrin. ***P < 0.001, M1 and 2 vs. NM1 and 2.
Discussion
Blood clotting and subsequent fibrin formation are important factors for tumor cell seeding to the lungs (11). The objective of this study was to determine the role of fibrin formation for kidney cancer metastasis in comparison to other 3-D matrices such as Matrigel™, an ECM mixture consisting of the basement membrane components collagen IV, laminin and fibulin. Here, we show that fibrin supports colonization of metastatic kidney tumor cells in an animal model in vivo as well as in 3D culture in vitro. Colony formation was more efficient in 3D fibrin than in matrigel and depended on the ability of metastatic cells to generate a fibronectin matrix as a prerequisite for subsequent spreading. Together our results show that fibrin promotes fibronectin matrix formation and kidney tumor cell spreading. Moreover, they suggest that these functions are important for the initiation of lung metastasis in renal cell carcinoma.
The role of the clotting system for tumor metastasis has long been established (12, 19). To this end, it has been demonstrated that tumor cell seeding is strongly reduced in transgenic mice deficient for platelet activators, fibrinogen or other coagulation factors (20, 21). More recently, it has been shown that blood clotting is specifically important for metastasis to the lung, which features blood vessels with a tight layer of endothelium and a continuous basement membrane (10, 22, 23). As a result of these distinct vascular features, tumor cell extravasation is much more elaborate in the lung vasculature than in the liver vasculature, which lacks a basement membrane underneath a fenestrated endothelium (24). In this situation, adhesive interactions with a provisional matrix of platelets and fibrin can provide critical cues for tumor cell survival and invasion in the lung vasculature (10). Consequently, fibrin formation plays a major role in promoting metastatic pathways in malignancies that predominantly metastasize to the lung such as renal cell carcinoma and soft tissue sarcoma (10).
The function of the clotting cascade is to protect tumor cells from the cytotoxic effects of circulating natural killer cells (19, 21). In addition, it has been shown that components of the clotting system support tumor cell extravasation by promoting epithelial to mesenchymal transition as well as the retraction of endothelial cells (3, 10, 25). Extravasation also requires tumor cells to penetrate the basement membrane of the pulmonary vasculature, which depends on a specific set of properties including the auto- or paracrine expression of HGF to promote c-met-dependent invasion and subsequent tumor cell sprouting (18). Alternatively, tumor cells can survive and proliferate in the lumen of pulmonary blood vessels (2). Such a mechanism for metastatic outgrowth is supported by our data showing that clotting promotes colony formation of primary metastatic kidney tumor cells in vitro and metastatic outgrowth in a model of experimental metastasis in vivo. This is also in line with a recent report demonstrating that fibrin promotes stem cell-like properties in tumor cells (6).
We and others have shown that colonization of fibrin with tumor cells depends on integrin αvβ3, which in its activated form participates in the generation of a fibronectin matrix (6, 10). This is significant as we previously demonstrated that spreading and fibronectin matrix formation in 3D fibrin is a prerequisite of metastatic RCC cells (10). Mechanistically, integrin αvβ3 and fibronectin promote the expression of the EMT master regulator Slug, which also has been shown to promote tumor initiation (10, 26). Here we show that Slug was reduced in kidney cancer cells when we inhibited stress fiber formation with the Rho kinase inhibitor Y-27632 suggesting that the tensile strength generated through cell matrix interaction is relevant for the up-regulation of a critical pro-metastatic transcription factor. This function correlates directly with fibronectin expression because in absence of fibronectin, fibrin or matrigel alone were unable to provide the tensile strength necessary to promote stress fiber formation in tumor cells. Interestingly, while both spreading and colony formation depended on the expression of tumor cell fibronectin, this function was not rescued by the addition of plasma fibronectin to fibrin. This difference in function could be owed to the specific expression of alternatively spliced modules in cellular fibronectin (27). Alternatively, it is conceivable that the formation of compact fibrin fibrils is impaired as free diffusion of plasma fibronectin is hindered following the FXIII-mediated cross-linking activity of fibronectin to fibrin (28).
Overall, our data suggest that fibronectin matrix formation is a prerequisite for tumor cell spreading and colonization in 3D. In agreement with this, it has been shown that 786-0 kidney cancer cells are unable to assemble fibronectin fibers when cultured on 2D plastic even though they generate an elaborate fibronectin matrix in 3D fibrin as well as matrigel (29). Lack of fibronectin matrix assembly in 2D has been attributed to the loss of functional von-Hippel-Lindau Protein (VHL), which supports fibronectin fibril formation through a distinct mechanism involving β1 integrin in combination with RhoA (29-31). However, while these VHL-related functions induce spreading and extracellular matrix formation in 2D, they are largely insufficient for mediating spreading of VHL-positive kidney tumor cells in 3D matrigel (18). VHL-negative 786-0 cells on the other hand spread in matrigel, albeit to a lesser extent than in fibrin, and this function correlates with fibronectin matrix assembly. Interestingly, kidney tumor cell spreading and fibronectin matrix formation in matrigel can be induced through stimulation of the receptor tyrosine kinase c-met, which has been shown to promote cell scattering through cross-talk with β1 integrin (32). A similar mechanism involving crosstalk between β3 integrin and other receptor tyrosine kinases activated in RCC such as PDGFR or EGFR could account for spreading and subsequent fibronectin assembly in 3D fibrin (33, 34).
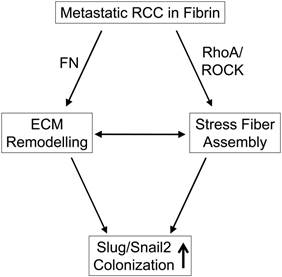
In addition to fibronectin matrix formation, we found that kidney tumor cells generated extensive stress fibers following embedding in fibrin. Both stress fiber and fibronectin matrix formation have been shown to be relevant for the metastatic outgrowth of dormant breast tumor cells (7). This process appears to be highly relevant because primary tumor cells from patients with kidney cancer metastasis where significantly more prone to stress fiber formation than cells from patients where the tumor was limited to the kidney. Moreover, inhibition of actomyosin contraction with the Rho kinase inhibitor Y-27632 inhibited fibronectin matrix assembly as well as colony formation in fibrin-embedded tumor cells. While the mechanism of fibronectin and stress fiber formation is relevant for tumor cell colonization, it does not appear to play a role in kidney tumor cell survival (17). Based on these results, we propose a mechanism where fibrin promotes adhesive interactions that result in fibronectin matrix and stress fiber assembly. This in turn appears to be a prerequisite for expression of the EMT master regulator and cancer stem cell factor Slug/Snail (Fig. 5) (26, 35). Therefore, inhibiting actomyosin dynamics in kidney cancer could be an attractive strategy to prevent and treat metastasis.
Diagram depicting the interaction of metastatic RCC with fibrin.
Supplementary Material
Fig. S1 Knockdown of fibronectin, western blotting for fibronectin (FN) to demonstrate knockdown in shFN HT1080 clone compared to shSCR control. Β-Actin shows protein loading.
Acknowledgements
This work was supported by National Institutes of Health grants CA134330 (JP), 5T32DK007774-14 (LAG) and P30CA047904 (UPCI CCSG). This project used the UPCI Cell and Tissue Imaging Facility, UPCI Animal Facility and the UPCI Vector Core Facility, which are supported by the UPCI Cancer Center Support Grant.
Competing Interests
None.
References
1. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679-95
2. Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med. 2000;6:100-2
3. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576-90
4. Shibue T, Brooks MW, Weinberg RA. An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell. 2013;24:481-98
5. Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271-8
6. Liu J, Tan Y, Zhang H, Zhang Y, Xu P, Chen J. et al. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat Mater. 2012;11:734-41
7. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ. et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68:6241-50
8. Jan Y, Matter M, Pai JT, Chen YL, Pilch J, Komatsu M. et al. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell. 2004;116:751-62
9. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906
10. Knowles LM, Gurski LA, Engel C, Gnarra JR, Maranchie JK, Pilch J. Integrin alphavbeta3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer Res. 2013;73:6175-84
11. Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL. et al. Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood. 2000;96:3302-9
12. Esumi N, Fan D, Fidler IJ. Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer Res. 1991;51:4549-56
13. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E. et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010;70:4327-34
14. Yao ES, Zhang H, Chen YY, Lee B, Chew K, Moore D. et al. Increased beta1 integrin is associated with decreased survival in invasive breast cancer. Cancer Res. 2007;67:659-64
15. Steffens S, Schrader AJ, Vetter G, Eggers H, Blasig H, Becker J. et al. Fibronectin 1 protein expression in clear cell renal cell carcinoma. Oncology letters. 2012;3:787-90
16. Knowles LM, Malik G, Pilch J. Plasma Fibronectin Promotes Tumor Cell Survival and Invasion through Regulation of Tie2. J Cancer. 2013;4:383-90
17. Gurski LA, Knowles LM, Basse PH, Maranchie JK, Watkins SC, Pilch J. Relocation of CLIC1 Promotes Tumor Cell Invasion and Colonization of Fibrin. Mol Cancer Res. 2014 [Epub ahead of print]
18. Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247-55
19. Gorelik E, Bere WW, Herberman RB. Role of NK cells in the antimetastatic effect of anticoagulant drugs. Int J Cancer. 1984;33:87-94
20. Palumbo JS, Barney KA, Blevins EA, Shaw MA, Mishra A, Flick MJ. et al. Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J Thromb Haemost. 2008;6:812-9
21. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW. et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178-85
22. Walker DC, MacKenzie A, Hosford S. The structure of the tricellular region of endothelial tight junctions of pulmonary capillaries analyzed by freeze-fracture. Microvasc Res. 1994;48:259-81
23. Coupland LA, Chong BH, Parish CR. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res. 2012;72:4662-71
24. Reichen J. The Role of the Sinusoidal Endothelium in Liver Function. News Physiol Sci. 1999;14:117-21
25. Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 2013;24:130-7
26. Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F. et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015-28
27. Guan JL, Trevithick JE, Hynes RO. Retroviral expression of alternatively spliced forms of rat fibronectin. J Cell Biol. 1990;110:833-47
28. Mosher DF. Cross-linking of plasma and cellular fibronectin by plasma transglutaminase. Ann N Y Acad Sci. 1978;312:38-42
29. Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN. et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959-68
30. Feijoo-Cuaresma M, Mendez F, Maqueda A, Esteban MA, Naranjo-Suarez S, Castellanos MC. et al. Inadequate activation of the GTPase RhoA contributes to the lack of fibronectin matrix assembly in von Hippel-Lindau protein-defective renal cancer cells. J Biol Chem. 2008;283:24982-90
31. Calzada MJ, Kuznetsova SA, Sipes JM, Rodrigues RG, Cashel JA, Annis DS. et al. Calcium indirectly regulates immunochemical reactivity and functional activities of the N-domain of thrombospondin-1. Matrix Biol. 2008;27:339-51
32. Mitra AK, Sawada K, Tiwari P, Mui K, Gwin K, Lengyel E. Ligand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;30:1566-76
33. Sargent ER, Gomella LG, Belldegrun A, Linehan WM, Kasid A. Epidermal growth factor receptor gene expression in normal human kidney and renal cell carcinoma. J Urol. 1989;142:1364-8
34. Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J. et al. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70:1053-62
35. Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ. et al. Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res. 2005;11:8070-8
Author contact
![]() Corresponding author: Institute of Clinical Hemostaseology and Transfusion Medicine, Saarland University Medical Center, Ringstr.52 / Geb. 1, D-66421 Homburg/Saar, Germany. Jan.Pilchde.
Corresponding author: Institute of Clinical Hemostaseology and Transfusion Medicine, Saarland University Medical Center, Ringstr.52 / Geb. 1, D-66421 Homburg/Saar, Germany. Jan.Pilchde.