Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(6):555-567. doi:10.7150/jca.11997 This issue Cite
Review
Hypothesis: Artifacts, Including Spurious Chimeric RNAs with a Short Homologous Sequence, Caused by Consecutive Reverse Transcriptions and Endogenous Random Primers
Zhiyu Peng1 ![]() , Chengfu Yuan2, Lucas Zellmer2, Siqi Liu3
, Chengfu Yuan2, Lucas Zellmer2, Siqi Liu3 ![]() , Ningzhi Xu4
, Ningzhi Xu4 ![]() , D. Joshua Liao2
, D. Joshua Liao2 ![]()
1. Beijing Genomics Institute at Shenzhen, Building No.11, Beishan Industrial Zone, Yantian District, Shenzhen 518083, P. R. China
2. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
3. CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, P. R. China
4. Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P. R. China
Received 2015-2-26; Accepted 2015-4-2; Published 2015-5-1
Abstract
Recent RNA-sequencing technology and associated bioinformatics have led to identification of tens of thousands of putative human chimeric RNAs, i.e. RNAs containing sequences from two different genes, most of which are derived from neighboring genes on the same chromosome. In this essay, we redefine “two neighboring genes” as those producing individual transcripts, and point out two known mechanisms for chimeric RNA formation, i.e. transcription from a fusion gene or trans-splicing of two RNAs. By our definition, most putative RNA chimeras derived from canonically-defined neighboring genes may either be technical artifacts or be cis-splicing products of 5'- or 3'-extended RNA of either partner that is redefined herein as an unannotated gene, whereas trans-splicing events are rare in human cells. Therefore, most authentic chimeric RNAs result from fusion genes, about 1,000 of which have been identified hitherto. We propose a hypothesis of “consecutive reverse transcriptions (RTs)”, i.e. another RT reaction following the previous one, for how most spurious chimeric RNAs, especially those containing a short homologous sequence, may be generated during RT, especially in RNA-sequencing wherein RNAs are fragmented. We also point out that RNA samples contain numerous RNA and DNA shreds that can serve as endogenous random primers for RT and ensuing polymerase chain reactions (PCR), creating artifacts in RT-PCR.
Keywords: artifacts, chimeric RNAs, endogenous random primers
Introduction
The swift spread of RNA technologies in recent years, mainly RNA sequencing (RNA-seq) [1-4] and associated bioinformatics [3,5], has led to identification of a huge number of “chimeric RNAs”, or RNA chimeras, which are those RNA molecules containing sequences from two different genes. For instance, a report from the ENCODE project in 2007 estimates that transcripts from 65%, i.e. about two-thirds, of the genes in the human genome, mainly those adjacent genes in the same chromosomal region, may be involved in formation of chimeric RNAs [6,7]. An analysis of expression sequence tags (ESTs) identifies over 30,000 putative human chimeric ESTs [8]. Another recently established database collects over 16,000 human RNA chimeras, along with chimeras of other species [9]. All these figures are astonishing, considering that the human genome encodes only slightly over 20,000 genes [10-14] and that chimeric RNAs have been thought for a long time to be rare in mammalian cells, although they are common in the unicellular and evolutionarily-lower multicellular organisms [15]. We suspect that the vast majority of the putative chimeras in human cells reported so far either are technical artifacts or should not be classified as chimeras. In this essay, we present our musings on these issues.
Chimeric RNAs derived from fusion genes
Chromosomal translocations are commonly seen in cancers and genetic diseases and often result in fusion genes [4]. These alterations have been a center of cancer genetics for over a century and are known to be hallmarks of cancer cells. However, in spite of numerous studies conducted, the underlying mechanisms are still largely unknown [16], although some mechanisms have been proposed, such as the “proximity mechanism” in which two genes that are far apart on the same chromosome may be near one another in the nucleus for recombination, as exemplified by RET-PTC fusion [17]. There have been about 800 different chromosomal rearrangements, including translocations [18-20], in association with about 1,000 fusion genes [20], documented in the literature. On the other hand, one reported microarray chip collects 548 chimeric RNAs that have been preliminarily verified [21,22], but not all of them are associated with a known fusion gene as the genomic basis. The best example of such fusion genes is the product of Philadelphia chromosome that results from a reciprocal translocation involving the long arms of chromosomes 9 and 22, t(9;22) (q34;q11) [23]. This translocation puts the c-ABL gene on chromosome 9 downstream of the breakpoint cluster region (BCR) on chromosome 22 [24]. There are three common BCRs, i.e. a major (M-bcr), a minor (m-bcr), and a micro (micro-bcr), thus forming at least three different BCR-ABL genes. One RNA transcript from a BCR-ABL fusion gene may be spliced alternatively to form different mature mRNAs. All these different versions of fusion and alternative splicing together produce many BCR-ABL mRNA variants and ensuing protein isoforms across the patients, with six most prevalent ones being b2a2, b3a2, b2a3, b3a3, e19a2, and e1a2 [25].
Cancer cells often have genomic DNA amplifications as well. If an amplified copy is translocated, it can also result in a fusion gene. Genomic DNA deletion is another common mechanism for fusion gene formation. One of the best examples is the deletion of 800 kilo-bases from chromosomal 4q12 that results in the fusion of FIP1L1 to PDGFRα. The FIP1L1-PDGFRα fusion gene encodes a new protein tyrosine kinase that plays an important role in the development of eosinophilia-associated myeloproliferative neoplasms but in the meantime is also a good target for treatment of this malignancy with tyrosine kinase inhibitor imatinib [26,27].
Fusion genes may occur occasionally in normal individuals as well, in part because evolution is a continuous process and evolutionarily occurring translocations can result in fusion genes. For instance, the tyrosine kinase fusion gene (TFG) can fuse with the G-protein-coupled receptor 128 (GPR128) [28]. The resulting TFG-GPR128 fusion gene, which produces a protein tyrosine kinase, occurs in 0.02% of healthy Europeans but has so far not yet been detected in Asians [28]. Another mechanism for fusion gene formation in healthy individuals has also developed evolutionally but does not involve genomic translocation, as exemplified by those POTE-actin fusion genes in the POTE family that emerged evolutionarily very recently and only in primates [29,30]. Another example is the PIPSL gene on chromosome 10 that is actually an intron-less copy of the intergenic splicing between the neighboring PIP5K1A and PSMD4 (also known as S5a) on chromosome 1 [31,32]. However, because this type of fusion does not involve genetic rearrangement, it can also be regarded as evolutionarily new genes, but not fusion ones.
Clearance of confusions on chimeric RNAs formed between two adjacent genes
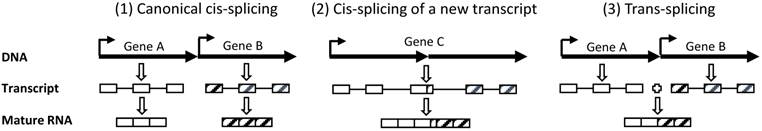
Although chimeric RNAs are defined as those RNAs containing sequences from two different genes, “two different genes” has actually not been well-defined when they locate in an adjacent manner in the same chromosomal locus in eukaryotic cells. Canonically, genes A and B that are neighbors and are encoded by the same strand of the DNA double helix in the same chromosomal region have their RNA transcripts individually cis-spliced to different mature mRNAs (scenario 1 in figure 1), which is the situation most familiar to biologists although in most cases 'cis-splicing” is simplified as “splicing” [33]. Circular RNAs, which may also be abundant in human cells [34]. are also regarded as cis-spliced product. However, when there is only one RNA transcript running from gene A to gene B (scenario 2 in figure 1), it may be regarded in three different ways: 1) it is an RNA variant of gene A whose transcription fails to terminate at the canonical site but, instead, reads through to gene B, resulting in a 3' extension, 2) it is an RNA variant of gene B whose transcription is initiated from an upstream site that belongs to gene A, resulting in a 5' extension, and 3) it is actually a transcript from an unannotated gene (gene C) that covers genes A and B and may or may not share the same exon-intron organization with gene A or gene B. We favor the third definition. In our opinion, if an RNA is formed by splicing these two genes' transcripts together (scenario 3 in figure 1), it is an authentic chimera; otherwise it is just an RNA of an unannotated gene (gene C).
Our definitions of “two neighboring genes” and trans-splicing. (1): Two canonically annotated genes (genes A and B) in the same genomic region are transcribed to two individual RNA molecules, followed by independent splicing to two individual mature RNAs. This is a canonical cis-splicing. (2): If this chromosomal region produces only one single RNA transcript, regardless of whether it has the same exon-intron organization as gene A or B, we prefer to consider it as an unannotated gene (gene C). Its splicing is also a canonical cis-splicing and the resultant mature mRNA should not be classified as a chimera. (3): If the two RNA transcripts from genes A and B, respectively, are spliced to one single RNA molecule, it is defined as trans-splicing and the resulting RNA is a chimera. (Boxes indicate exons while lines connecting boxes indicate introns. Dark arrows indicate the 5'-to-3' orientation of transcription).
Chimeric RNA formed at the RNA level without a DNA basis
Chimeric RNAs do not need to have a DNA basis, as they can be formed at the RNA level. One of the hypothetical mechanisms is the so-called “transcriptional slippage” [8], which theorizes that transcription machinery can slip from one gene to another during transcription elongation, even when this other gene is on another chromosome, resulting in a chimeric transcript. The slippage is supposed to occur at a region where the two genes are homologous in the DNA sequence, which is usually short and thus referred to as “short homologous sequence (SHS)” [8]. Indeed, we found an SHS in about 67% of the putative human chimeric ESTs in the NCBI database and Li et al found it in about half of the chimeric ESTs deposited in different databases [8,35]. However, while there may be some experimental data showing the possible existence of intramolecular slippage, i.e. slipping to a downstream gene on the same chromosome during transcription elongation [36], so far there has not been any experimental evidence for the existence of intermolecular slippage, i.e. slipping from one chromosome to another, during transcription. Therefore, why so many putative chimeras contain a SHS remains unknown, although there are discussions and hypotheses about it [8,36,37].
While the well-studied cis-splicing removes introns from a pre-mRNA molecule so that exons join together to form a mature mRNA, splicing can also occur to join two pre-mRNA molecules together, which is coined “trans-splicing” [38]. Trans-splicing events are common in unicellular organisms and some evolutionarily-lower multicellular organisms wherein it occurs between a leader sequence and a target RNA [15]. In addition, some chloroplasts and mitochondria of lower eukaryotes and plants manifest another type of trans-splicing to remove so-called discontinuous group II introns [33,39]. However, for decades, trans-splicing has been considered rare, and thus has not been well defined, in mammalian cells. With regard to the chimeric RNA formation, we propose that cis-splicing is a biochemical reaction that utilizes only one RNA molecule as the substrate and produces one RNA molecule as the product, whereas trans-splicing is a biochemical reaction that utilizes two RNA molecules as the substrates but produces only one RNA molecule as the product (Scenario 3 in figure 1). The two substrate or precursor RNA molecules can be two copies of the same one; in this case trans-splicing results in an RNA containing duplicated exons seen in some human genes' mature mRNAs [40,41], such as the 77-kD estrogen receptor alpha (ERα)[42,43] and the ERα variant with exon 1A repeat [44]. The two RNAs can also be transcribed from the two opposite strands of the DNA double helix of the same gene, resulting in an RNA that may be considered a chimera because the antisense transcript, coding for a protein or not, is actually from another gene (encoded by the opposite DNA strand), although its two partners are partially identical, but oppositely oriented, to each other, as exemplified by a human KLK4 mRNA variant[45] and some chimeric RNA variants of mdg4 in drosophila [46,47]. However, in most cases of trans-splicing, the two substrate RNAs are transcribed from different genes located on the same chromosome or different chromosomes, resulting in a canonically defined chimeric RNA [6,48]. It is worth mentioning that in some strains of Giardia, a minimalistic protozoan which is a common cause of diarrhea worldwide, each of the two RNA substrates may contain a poly A tail, indicating that the trans-splicing occurs after polyadenylation of the RNA substrates [49-51], although whether it also occurs in mammalian cells remains unknown. All these complex trans-splicing products raise a serious question as to whether 'gene” needs to be redefined [52-54]. For example, the mouse Msh4 gene produces several chimeric mRNAs formed between a transcript from chromosome 3 and a transcript from chromosome 16, 2 or 10, respectively [55]. Some of its chimeric variants are bicistronic while one of the variants contains antisense sequence [55]. How to define this Msh4 gene that involves four different chromosomes and produces bicistronic, chimeric, and antisense-containing mRNAs is a challenge to today's biology.
Most putative chimeras identified by the ENCODE and other researchers are derived from two adjacent genes in the same chromosomal locus [5,56,57]. If the RNAs are cis-splicing products of single RNA transcript, illustrated as scenario 2 in figure 1, they should not be classified as chimeras by our definition, although they are genuine RNAs truly expressed in cells. Only those that are trans-splicing products of two different RNA molecules, as the scenario 3 in figure 1, are authentic chimeras. Unfortunately, none of the RNA-seq reports provides information of whether the chimeras are derived from one or two precursor RNA molecules; thus it is unclear how many of them are genuine by our definition. However, such a huge number of reported putative chimeric RNAs, which much outnumbers the about 1,000 known fusion genes [20], suggest that most of them are formed without a DNA basis. Therefore, the vast majority of the putative chimeras either are trans-splicing products or are unauthentic. Those unauthentic ones may in turn be either cis-splicing products or technical artifacts. Since many researchers consider the chimeras they identified as “read-though” products of transcription [58-63], we assume that a significant portion of them are cis-splicing products and thus should not be classified as chimeric RNAs, but, unlike technical artifacts to be described later, they are RNA transcripts truly-expressed in cells.
It needs to be mentioned that the vast majority of reported chimeras have not been verified with a vigorous method, have not been cloned for their full-length sequences, and have not been known for their open reading frames [64], and thus still remain putative and basically meaningless to us so far. This drawback is due mainly to the lack of reliable and efficient approaches of cloning and verification, since current RNA-seq technologies are reliant on RT and PCR [65], provide only short sequences, and have poor strand-specificity [66], and thus are only suitable for screening, but not for verification, of long RNA.
From fusion RNA to fusion gene: the cart before the horse in carcinogenesis?
While gene fusion derived from various forms of chromosomal rearrangement rarely occurs in normal cells, there are inklings, as summarized by Kowarz et al [19], that link trans-splicing occurring in normal cells to fusion gene formation in cancer cells. For instance, the fusion genes formed between immunoglobulin heavy chain gene (IGH) and the BCL2 or c-MYC gene are known to be common in cancers [67,68]. Surprisingly, the IGH-BCL2 fusion has also been observed in normal spleen [69] or normal individuals at surprisingly high frequencies, varying between 16-55% among different populations, as reviewed by Brassesco [67]. IGH-MYC chimeric RNA has been detected in mouse B lymphocytes[70] and in Peyer's patch follicles as well [71]. Other fusion genes such as the aforementioned BCR-ABL fusion derived from the Philadelphia chromosome have also been detected in lymphocytes from normal individuals [72], and the TEL-AML1 (also known as ETV6-RUNX1) or AML1-ETO fusions can occur during normal fetal development.[73-77] Moreover, Ig-BCL6 translocations have been found in human germinal center B lymphocytes in human lymphoid tissues as well [78]. On the other hand, some chimeric RNAs in prostate cancer do not have corresponding genetic rearrangements detected [79], such as the SLC45A3-ELK4 RNA that is present in normal prostate and does not primarily arise from a chromosomal rearrangement in prostate cancer [80]. A PML-RARα chimeric transcript has also been detected in promyelocytic leukemia without the corresponding fusion gene detected in the genomic DNA [81-83]. The abovementioned RET-PTC fusion gene is a marker for thyroid cancer but can often be detected in inflammatory and benign thyroid diseases as well [84,85]. During the developmental stage, stromal cells in normal uterine endometrium show a trans-splicing event between the JAZF1 pre-mRNA from chromosomal 7p15 and the JJAZ1 pre-mRNA from 17q11 [86-88]. The resulting JAZF1-JJAZ1 chimeric mRNA encodes a fusion protein with anti-apoptotic function. Neoplastic stromal cells of the endometrium also highly express this chimeric mRNA and protein but, besides the trans-splicing mechanism, the chimeric RNA can also be transcribed from a fusion gene derived from a chromosomal translocation [86]. Collectively, these data seem to suggest an intriguing possibility that a trans-splicing event may be a precursor of chromosomal rearrangement occurring more often during carcinogenesis [6,19,86], including its early stages before the malignant transformation, and some of the resulting chimeric RNAs may eventually lead to formation of fusion genes in chromosomes [19]. This actually means that “a gene is derived from an RNA”, at least during carcinogenesis, which opposes the “gene gives rise to RNA” dogma and thus seems to be “the cart before the horse”, as pointed out by Rowley [86].
For decades, fusion genes have been thought to occur mainly in lymphomas, leukemias and myelomas but rarely in solid tumors [89]. However, recent advances in cancer genomics and ribonomics suggest that prostate and lung cancers, and probably breast cancer as well [58,90-94], also express many chimeric RNAs, although some of them do not seem to be associated with a corresponding fusion gene [19,80,95]. Albeit there lacks a survey of the frequencies of fusion RNAs in different malignancies, at least prostate cancer is among those that are reported at the highest frequency to have fusion genes or fusion RNAs among all types of malignancy, including epithelial cancers, lymphomas, leukemias and various sarcomas, as extensively reviewed by many investigators [16,96-99].
Although the above “the cart before the horse” hypothesis sounds intriguing, convincing supportive evidence is still lacking and many concerns still need to be addressed. For example, most supporting data just suggest a correlative, but not a causative, relation between the fusion gene and the fusion RNA, especially in the cases wherein the fusion occurs at the breakpoint cluster regions. Convincing evidence is also needed to prove that the cells that have the fusion genes or the chimeric RNAs are really normal. Moreover, a technical detail deserves mentioning that in recent years fusion genes are not usually studied using traditional hybridization-based techniques such as FISH (fluorescent in-situ hybridization) and southern blots but, instead, are mainly studied using RT-PCR and RNA-seq technologies that detect fusion RNAs, but not fusion genes per se. RT and PCR may create many artifacts, as to be discussed later, making it possible that some so detected are artifacts [75,94,100,101]. Also, in many cases it is actually unclear whether the detected chimeric RNAs, even if they are authentic, are associated with a corresponding fusion gene or are just formed at the RNA level. PCR amplification of genomic DNA has great technical difficulties and limitations, and thus may easily fail, in detecting fusion genes on the chromosomal DNA. Since absence of evidence is not evidence of absence, in some cases fusion genes may actually exist although they are not detected.
“Consecutive RTs” scenario for spurious RNA chimeras
Although tens of thousands of putative chimeric RNAs have been reported, only very few of them can be verified, especially those that are formed at the RNA level sans a fusion gene as a genomic basis [65,102,103]. A large and comprehensive study analyzed 7424 human chimeric RNAs selected from basically all major databases but could only confirm 175 (2.36%) of them [64], many of which have a DNA basis. Another study aiming to identify interchromosomal trans-splicing products with a new methodology identified only 16 human chimeras [56]. Similarly, we also tried to verify those chimeras reported in the literature or deposited in the EST database of the NCBI using nested PCR of RT products from many cancer cell lines of the breast, prostate, pancreas and other organ origins, but failed to confirm the vast majority of them. For instance, we were not able to verify the true existence of several ERα-containing RNA chimeras [104,105], the CCND1-Trop2 RNA chimera [106], and the fatty-acid synthase (FAS)-ERα chimera [107], in any of the cell lines we studied. Those that we could verify are all associated with known fusion genes as the genomic basis, such as the BCAS4-BCAS3 chimeric mRNAs [108].
Virtually all ESTs were obtained via RT-PCR. Similarly, most data derived from RNA-seq also involve RT and PCR, because direct RNA-seq technology has a low efficiency and cannot sequence deeply [109-111]. In a routine sample-preparation procedure for RNA-seq, RNAs need to be fragmented, usually by metal ion hydrolysis, to a length of several-hundred nucleotides, followed by conversion to the first strand of cDNA in RT using random hexamers that contain an adaptor sequence at the 5'-end [112-114]. In some cases, RT is performed using so-called gene-specific primer with or without a 5'-adaptor sequence, which is used for sequencing one or some specific genes' transcripts or for determining strand-specificity, because both Watson and Crick strands of the DNA double helix may be expressed to sense and antisense transcripts, respectively. Sometimes RT is performed first with poly-dT primers and the cDNA is fragmented. The second cDNA strand is then synthesized and PCR ensues to amplify the cDNA library [112], which may or may not be followed by ligation to a vector or specific sequencing primers (depending on whether the prior primers contain an adaptor or not). During these RT and PCR procedures, artificial chimeric cDNAs may be formed [66,115-122], in part because template switching may occur to skip the region in a secondary structure during RT [102,115,116,123,124] and mis-priming can occur in PCR, as having been well discussed in the literature [116,117,125-128].
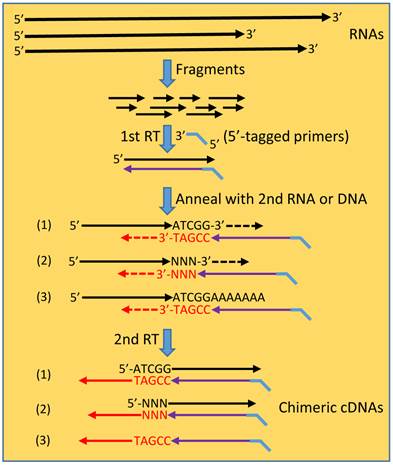
The continuous frustrations from endless failure in verifying those reported chimeras that are formed at the RNA level without a fusion as a genomic basis lead us to a new hypothetical scenario for possible formation of artificial chimeras during RT, which to our knowledge has not been described before: After an RT primed by the intended (usually 5'-tagged) primer is finished and the RNA template has degraded or has been digested by the RNA-H activity of the reverse transcriptase, the 3'-end of the resulting cDNA may anneal to a new RNA fragment via a SHS, as illustrated in figure 2. The SHS of this new RNA will serve as the primer to initiate a second RT reaction that elongates the cDNA, thus creating a chimera. This is possible since a retrovirus uses cellular tRNA to prime mRNA for reverse transcriptases to synthesize the first DNA strand [129,130] and reverse transcriptases are known to be able to utilize endogenous small RNAs to efficiently prime cDNA synthesis in vitro [131-133]. Since pentamers are often used in PCR, we assume that the random annealing only requires a SHS as short as five nucleotides, resulting in a chimeric cDNA in which the two partners share this SHS. This scenario of “consecutive RTs”, i.e. another RT reaction following the previous one, enlightens us in that many SHS-containing chimeric RNAs obtained by RT-based approaches may simply be technical artifacts [108]. In this scenario the artificial chimera occurs during a second RT reaction primed by a new primer, which differs completely from the well-described 'template switching” mechanism for artifact formation that occurs within a single RT reaction and involves only a single primer [115,116,125]. Unfortunately, until now we still have not yet figured out a strategy to prove that the second RT indeed occurs and to prevent its occurrence (thus preventing formation of spurious chimera), since both the first and the second RTs are all finished in probably just seconds in the same tube. Moreover, although we propose “consecutive RTs” as a potential reason for technical spuriousness, theoretically it may still truly occur in retrovirus-infected human cells, because these cells may express the viral reverse transcriptase. This scenario, in which authentic chimeras may occur in vivo, also needs to be tested.
The hypothetic scenario of “consecutive RTs” for formation of spurious chimeric RNAs. In a typical procedure for RNA-seq sample preparation, RNAs are fragmented to many smaller fragments. RT reaction, usually primed by 5'-tagged random primers, engenders the 1st cDNA strand. After the RNA template has degraded or has been digested by the RNase-H activity of the reverse transcriptase, the cDNA may anneal to another RNA fragment at the 3' end by five or more nucleotides, say ATCGG/TAGCC (scenario 1) that is referred to as “short homologous sequence” (SHS). This annealing initiates a second RT reaction, producing a chimeric cDNA in which the two partners are joined at the SHS. In many cases, reverse transcriptase may append, at the cDNA end in a non-template manner, one or several nucleotides, referred to as “NNN” (scenario 2). In this situation, the resulting chimera has the two partners joining with a shorter SHS or even without a SHS when five or more bases are appended that alone constitute the primer. DNA residuals in the RNA samples may also be molten to single-stranded oligos, which then anneal to the cDNAs via a SHS, resulting in DNA-cDNA hybrids in the second RT reaction. Annealing to the second RNA or DNA can occur at any place of the molecule before the poly-A or poly-dT tail (scenario3).
RT is usually conducted using reverse transcriptase from MMLV (Moloney murine leukemia virus) that often appends one or several nucleotides, usually CCC or GGG but also other base or bases [134,135], at the cDNA end in a non-template manner. Actually, some other DNA polymerases also have similar properties. For example, sometimes Taq and Tth DNA polymerases can append, in a non-template manner as well, poly-dA or poly-dT [136-140], although more often only a single dA nucleotide is added [141], to the DNA end. These properties have been utilized as a strategy to synthesize the second strand of cDNA [142,143], to detect RNA or specific DNA strand, [139] or to clone cDNAs [141]. Although these appended nucleotides do not belong to the original RNA template, they may constitute a primer or part of the primer to anneal to a second RNA fragment and initiate the second RT reaction, creating a chimera in which the SHS is shorter or is absent (when five or more nucleotides are appended that alone constitute the primer). It deserves mentioning that the cDNA can actually anneal to a SHS at any position of a second RNA before the poly-A tail.
RNA samples usually contain DNA residuals, especially mitochondrial DNAs that are small and in a circular structure, because one cell has hundreds or even thousands of mitochondria. DNase treatment of the RNA sample can decrease the amount of, but usually cannot completely remove, DNA residuals [35,144]. Some of these DNA fragments may be molten to single-stranded oligos and serve as templates. The cDNA may also anneal to these single-stranded genomic or mitochondrial DNA oligos, resulting in DNA-cDNA chimeras in the second RT reaction, because reverse transcriptase from MMLV has DNA-dependent DNA polymerase activity, i.e. can use DNA as the template [145-147]. Moreover, RT using MMLV reverse transcriptase is error-prone due to the lack of proofreading mechanism [148,149]. If mutations occur at the cDNA end, the resulting chimera may have mismatches in the SHS, and there are many chimeric ESTs that contain such mismatches [8].
It is worth mentioning that the “consecutive RTs” scenario described above can also occur in routine RT, and many chimeric ESTs may be technical artifacts so derived. Moreover, if two RNAs may be artificially fused in this way, so can three or more RNAs as well. Supporting this inference, we recently identified some trimeric or tetrameric ESTs, i.e. cDNAs containing sequences from three or four genes, including mitochondrial genes [35], some of which may be such artifacts. However, the number of RNA fragments in routine RNA samples is smaller, and the resulting cDNA fragments are fewer; therefore there are fewer anneals to occur, relative to the RT for RNA-seq sample preparation that involves RNA fragmentation. Moreover, RNA and DNA have fragile sites at which breakage occurs much more easily. Thus chimeras can be formed much more often at these sites, which is reflected by higher reads in RNA-seq. In other words, spurious chimeras may also be highly recurrent or repeatable.
Using RNA-seq, many mRNA chimeras have also been identified in bacteria [94,101,150]. Because it is a consensus that the bacterial genome is intron-less and thus its transcripts should not undergo splicing, these RNA chimeras “must be artifacts”. Conversely, these RNA chimeras may be explained as a novel finding suggesting that bacterial RNAs also undergo sort of previously unknown splicing. This explanation duality will continue baffling us until new methodology is established to prove either the “spuriousness” or the “bacterial RNA splicing”. If most bacterial chimeras are artifacts, so would be most of those identified in human cells by using the same technology.
Possible artifacts caused by endogenous random primers
It has been reported for a long time but has not yet been alerted to most RNA researchers that cDNA can be produced in RT reactions without addition of primers [151-154]. Recently we found that products of RT reactions without adding random hexamers or poly-dT primers could still be good template resources, almost as good as the RT products with random hexamers, for PCR amplification of mRNA of many genes [108]. The mechanism for generation of these cDNAs in no-primer RT reaction is still unclear, although several possibilities have been discussed [155]. In our cogitation, the most likely possibility is that the cDNAs are primed by endogenous random primers (ERPs) in the RNA samples, some of which prime the abovementioned second RT reaction that produces an artificial chimera. Indeed, RNA samples contain a huge number of long and short RNA fragments, such as short noncoding RNAs, excised introns and other processed mRNAs, as well as degraded RNAs. These RNA shreds, albeit many of them have been modified at the 5'-end [156], can efficiently prime a primary, i.e. the first, RT reaction in the same principle as the above-described second RT (Fig. 3). Moreover, RNA samples contain a lot of short DNA fragments as well, especially those derived from hundreds or even thousands of copies of mitochondrial DNA as aforementioned, even after the RNA samples have been treated with DNase, since very short DNA shreds (as short as five nucleotides to be a pentamer) are hard to remove. Some of these DNA shreds, especially those very short ones, may be molten to single-stranded oligos to serve as ERPs. Each DNA piece makes two oligo primers.
Spuriousness caused by endogenous random primers (ERPs): When an antisense is expressed and partly overlaps (blue area) with the sense transcript at either the 5'-end (top panel) or the 3'-end (bottom panel), ERPs (short red arrows) will prime both RNA transcripts in an RT. If the two oppositely-oriented RNAs overlap at the 3'-end, each RNA can serve as an endogenous primer to convert the opposite RNA strand to cDNA during RT, resulting in an artificial “full-length” cDNA of either gene, similar to the result from an RT using poly-dT primer. When the two RNAs overlap at the 5'-end, the same “full-length” cDNA will still be made if PCR ensues. RT with a gene-specific primer (green or purple arrow), usually 5'-tagged, can still specifically convert the targeted RNA transcript to cDNA. However, the oppositely-oriented transcript, along with numerous other RNA transcripts expressed in the cells, will also be converted to cDNA simultaneously, due to the ERP-primed RT reaction.
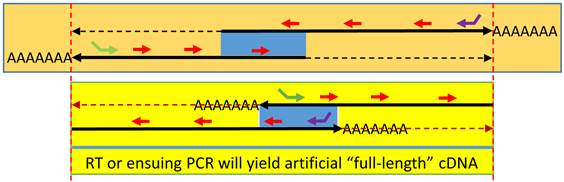
ERPs should not affect the RT products primed by intended poly-dT primers and 5'-tagged random primers usually used for RNA-seq. RT products primed by gene-specific or strand-specific primers that usually contain a 5'-tag should not be intervened either. However, these cDNAs of interest are mingled with a huge number of cDNAs, virtually a whole cDNA library, primed by ERPs, which will intervene with the ensuing PCR amplification of the targeted cDNAs (figure 3). Therefore, there is no such thing called “strand-specific primer” or “gene-specific primer” if PCR is later involved in cloning or detecting a transcript from a specific strand of the DNA double helix, as explained earlier [108]. To our knowledge, only the direct RNA-seq without the involvement of PCR amplification can be gene- or strand-specific, although this newly emerging technique has a poor efficiency because the template is not amplified [109-111]. Gene-specific primers have been widely used in cloning and expression studies for decades, but ERP-caused artifacts have seldom been addressed. Since routine RT primed by gene-specific primers is, likely, neither gene- nor strand-specific, whether those published data that also involve PCR need to be reevaluated or reinterpreted becomes an uncomfortable but unavoidable question that requires a serious consideration, in our humble opinion.
Unvanquished obstacles for cloning the 3'-end of antisense-accompanied transcripts
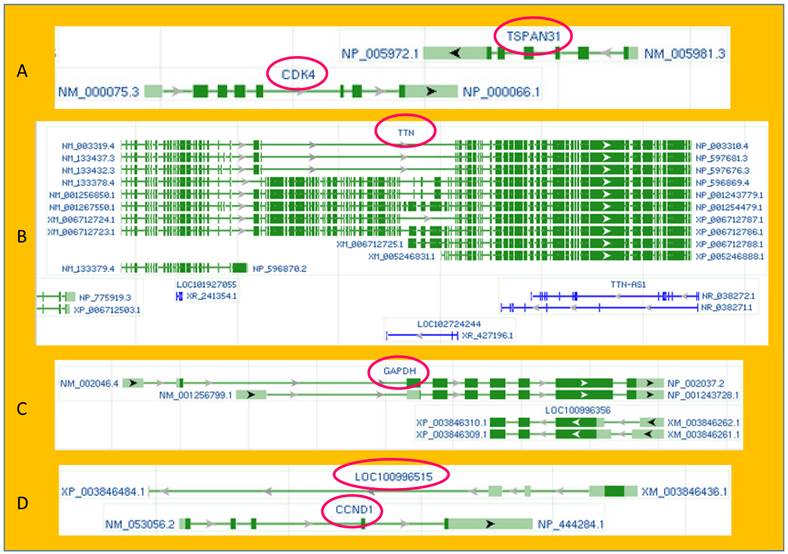
There is now a consensus that virtually the entire non-repeat part of the human genome is transcribed [6,7], to a total of over 161,000 transcripts [157], although the actual number should be much larger if minor coding and noncoding RNAs are also counted, since the TTN (Titin) gene alone may be expressed to over one million mRNA variants [158,159]. The Unigene database of the NCBI contains over 123,000 human antisense entries [160], and another study estimates that about 63% of RNA transcripts are accompanied by antisense counterparts [161]. These figures indicate that, for most genomic loci, both the Watson strand and the Crick strand of the DNA double helix are transcribed [162,163]. Although the intron-exon organization usually differs between the sense and antisense transcripts, in many occasions the two oppositely oriented RNAs have their 5'-end or 3'-end overlapped. For instance, the cyclin dependent kinase-4 (CDK4) and TSPAN31 genes, which are encoded by the opposite DNA strands in the same genomic locus, have their last 571 nucleotides of the RNAs overlapped (figure 4) [108]. The THRA and NR1D1 genes also have their transcripts overlapped, as shown in the NCBI database. If the overlap occurs at the 3' end, each RNA will serve as the primer to convert the opposite RNA strand to cDNA in RT, engendering an artificial chimeric cDNA that may be mistaken as the full-length cDNA of either gene, as depicted in figure 3. This still remains, today, an unvanquished obstacle for using RT-PCR to clone the 3'-end of overlapped transcripts that do not have a poly-A tail for being primed by poly-dT primers and thus require using random hexamers in the RT. For instance, it may not be easy to clone the 5' and 3' ends of the ncRNACCND1, a CCND1 related RNA [164], and to determine whether it is transcribed from the same strand as the CCND1 or from the opposite DNA strand.
Illustrations of exon-intron organization and sense-antisense relationship of some genes copied from the current or a previous version of the GenBank database of the NCBI. A: TSPAN31 and CDK4, two different oncogenes, are encoded by the two opposite strands of the DNA double helix. Their mRNAs overlap at the 3' end. B: The DNA strand opposite to the TTN (Titin) coding one not only produces two TTN antisenses (NR_038271.1 and NR_038272.1) but also harbors two unannotated genes (LOC102724244 and LOC101927055), each encoding an RNA as well. C: The GAPDH gene has two mRNA variants (NM-002046.4) and NM_001256799.1). In a previous version of the NCBI, the opposite DNA strand was shown to encode two protein-coding mRNAs (XM_003846262.1 and XM_003846261.1), and some of their exons were identical to the two GAPDH mRNAs except for their opposite orientation. D: Also in a previous version of the NCBI, the DNA strand opposite to the CCND1-coding one was shown to harbor an unannotated gene (LOC100996515) that coded for an mRNA (XM_003846436.1). The antisense mRNAs of the GAPDH and the CCND1 have been deleted from the latest NCBI database. (By the NCBI's annotation, “NM”, “XM” and “NR” indicate mRNA, predicted mRNA, and noncoding RNA, respectively.)
If an antisense transcript overlaps with the mRNA in the middle region, but not at the 5'- or 3'-end, either transcript may still serve as an endogenous primer to convert the overlapped part of the opposite strand to cDNA. Besides, like the mRNA, the antisense transcript can also be primed by ERPs described above. As a result, there will be some sections of double-stranded cDNA sequence, which may intervene with the later cloning or expression studies. If routine or quantitative RT-PCR is used to determine the expression level of an RNA that is accompanied by an overlapped antisense transcript, no matter whether the overlap occurs at the 5'- or 3'-end or at the middle region, PCR with primers at the overlapped region starts with two templates and thus will falsely double the expression level, as explicated before [108]. Therefore, the locations of the primers matter, and primers at different regions of the RNA should be used. For instance, the TTN gene consists of 364 exons that together constitute an over 100-kb long wild type mRNA and may produce over one million alternatively cis-spliced mRNA variants [158,159,165,166]. According to the NCBI database, the opposite DNA strand not only produces two antisense RNAs (NR_038271.1 and NR_038272.1) but also harbors two unannotated genes (LOC102724244 and LOC101927055), each producing an RNA (figure 4). These four antisense transcripts will likely be primed by ERPs and by parts of the TTN mRNAs and converted to cDNAs in RT, making it more difficult to use PCR to analyze the already too many (over one million) TTN mRNA variants [158,159]. Since over 63% of RNA transcripts may be accompanied by antisense counterparts [161], we need to be well aware of this potential pitfall.
The NCBI updates its database virtually on a daily basis and very often deletes some prior antisense transcripts, likely because antisenses are so often spurious and so difficult to verify. For instance, a previous version of the NCBI database showed that the GAPDH gene had several antisense transcripts that not only overlap with the mRNAs at the 3'-end but also have some exons identical, but oppositely oriented, to the corresponding exons in the GAPDH mRNAs (figure 4). Similarly, the CCND1 mRNA was also shown to be accompanied by an antisense transcript dubbed as LOC100996515 (figure 4). These GAPDH and CCND1 antisense transcripts are likely artifacts as they are deleted from the latest NCBI website, which not only indicates that mistakes can easily occur to antisense detection but also emphasizes the importance of checking with the NCBI database once more before initiating a project or submitting a manuscript. Given the human CDK4-and-TSPAN31 relationship as an example: the first version of their mRNAs in the NCBI did not show any overlap, the second version showed a 24-nucleotide overlap, and the third (current) version shows a 517-nucleotide overlap.
Conclusion
The swift spread of RNA-seq technology and associated bioinformatics has led to identification of tens of thousands of putative chimeric RNAs in human, mainly cancerous, cells. Since most of the reported chimeras are derived from two neighboring genes in the same chromosomal region, we redefine “two neighboring genes” as those producing two individual transcripts whereas those together producing only one transcript are redefined as an unannotated gene. There are only two known mechanisms for chimeric RNA formation, i.e. trans-splicing of two RNA molecules or transcription from a fusion gene that is formed due to chromosomal translocation, deletion or amplification occurring mainly in cancers and genetic diseases. Because there have only been about 1,000 known fusion genes documented in the literature [20], we surmise that the vast majority of the reported chimeras are either trans-splicing products or technical artifacts. Most of those reported RNA chimeras that are derived from neighboring genes are described to result from “read-through” transcription [58-63]. Therefore, we further infer that trans-splicing events are rare in human cells as thought previously, meaning that most of the reported chimeras either are technical artifacts, i.e. non-existing, or are cis-splicing products of unannotated genes that should not be classified as chimeric RNAs by our definition. We further propose a “consecutive RTs” hypothesis for how most spurious chimeric RNAs, especially those containing a SHS that is actually seen in most putative chimeras, may be generated during RT reactions, especially in RNA-seq wherein RNAs are fragmented. Our definition and stratification of authentic and unauthentic chimeras as well as their relationship to chromosomal DNA are summarized in table 1, along with our conjecture, which awaits experimental verification, on the portion occupied by each subgroup in all (both authentic and unauthentic) the reported chimeras. We hope our definition and classification help in clearance of some confusions in chimeric RNA research. We also want to point out that RNA samples contain a huge amount of RNA shreds and probably also short single-stranded DNA oligos that can serve as ERPs for RT and ensuing PCR to create various technical artifacts. Therefore, there basically is no such thing called “gene-specific primer” or “strand-specific primer” in RT-PCR based RNA cloning or sequencing.
Definition and classification of chimeric RNAs in human cells
| Transcript | Genomic DNA | Number of precursor transcript | Chimera | Frequency |
|---|---|---|---|---|
| Artifact | None | No true precursor, fake by RT or PCR | Artifact | Majority |
| Authentic | Intrachromosomal | True adjacent genes (two transcripts) | Authentic | Rare |
| Unannotated gene (one transcript) | Unauthentic | Frequent | ||
| Fusion gene | One transcript | Authentic | Frequent | |
| Interchromosomal | Two transcripts | Authentic | Rare |
Note: "Frequency" indicates the estimated portion occupied by the subgroup with all putative chimeras, all intrachromosomal transcripts, or all authentic chimeras as the whole. The vast majority of all putative chimeras are artifacts whereas the vast majority of authentic chimeras are derived from fusion genes. In most cases, one fusion gene is on a single chromosome that has an alteration (translocation, deletion or amplification), and thus its transcript still belongs to the "intrachromosomal" group.
Acknowledgements
We would like to thank Dr. Fred Bogott at Austin Medical Center, Austin of Minnesota, for his excellent English edition of this manuscript. This work was supported by a grant from the Department of Defense of United States (DOD Award W81XWH-11-1-0119) to D.J. Liao. The funding agency had no role in study design, data collection and analysis as well as in decision to publish or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X. et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97-101
2. Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S. et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci U S A. 2009;106:12353-12358
3. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012 doi: 10.1093/bioinformatics/bts635
4. Panagopoulos I, Thorsen J, Gorunova L, Micci F, Heim S. Sequential combination of karyotyping and RNA-sequencing in the search for cancer-specific fusion genes. Int J Biochem Cell Biol. 2014:462-465
5. Akiva P, Toporik A, Edelheit S, Peretz Y, Diber A, Shemesh R. et al. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30-36
6. Gingeras TR. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206-211
7. Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799-816
8. Li X, Zhao L, Jiang H, Wang W. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J Mol Evol. 2009;68:56-65
9. Frenkel-Morgenstern M, Gorohovski A, Lacroix V, Rogers M, Ibanez K, Boullosa C. et al. ChiTaRS: a database of human, mouse and fruit fly chimeric transcripts and RNA-sequencing data. Nucleic Acids Res. 2013;41:D142-D151
10. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745-755
11. Belizario JE. The humankind genome: from genetic diversity to the origin of human diseases. Genome. 2013;56:705-716
12. Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL. et al. The reality of pervasive transcription. PLoS Biol. 2011;9:e1000625. doi: 10.1371/journal.pbio.1000625
13. Pennisi E. Genomics. ENCODE project writes eulogy for junk DNA. Science. 2012;337:1159 1161. doi: 10.1126/science.337.6099.1159
14. Skipper M, Dhand R, Campbell P. Presenting ENCODE. Nature. 2012;489:45. doi: 10.1038/489045a
15. Lasda EL, Blumenthal T. Trans-splicing. Wiley Interdiscip Rev RNA. 2011;2:417-434
16. Rabbitts TH. Commonality but diversity in cancer gene fusions. Cell. 2009;137:391-395
17. Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138-141
18. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233-245
19. Kowarz E, Merkens J, Karas M, Dingermann T, Marschalek R. Premature transcript termination, trans-splicing and DNA repair: a vicious path to cancer. Am J Blood Res. 2011;1:1-12
20. Mertens F, Tayebwa J. Evolving techniques for gene fusion detection in soft tissue tumours. Histopathology. 2014;64:151-162
21. Lovf M, Thomassen GO, Bakken AC, Celestino R, Fioretos T, Lind GE. et al. Fusion gene microarray reveals cancer type-specificity among fusion genes. Genes Chromosomes Cancer. 2011;50:348-357
22. Lovf M, Thomassen GO, Mertens F, Cerveira N, Teixeira MR, Lothe RA. et al. Assessment of fusion gene status in sarcomas using a custom made fusion gene microarray. PLoS One. 2013;8:e70649
23. Bennour A, Ouahchi I, Moez M, Elloumi M, Khelif A, Saad A. et al. Comprehensive analysis of BCR/ABL variants in chronic myeloid leukemia patients using multiplex RT-PCR. Clin Lab. 2012;58:433-439
24. Ma W, Kantarjian H, Yeh CH, Zhang ZJ, Cortes J, Albitar M. BCR-ABL truncation due to premature translation termination as a mechanism of resistance to kinase inhibitors. Acta Haematol. 2009;121:27-31
25. Neumann F, Herold C, Hildebrandt B, Kobbe G, Aivado M, Rong A. et al. Quantitative real-time reverse-transcription polymerase chain reaction for diagnosis of BCR-ABL positive leukemias and molecular monitoring following allogeneic stem cell transplantation. Eur J Haematol. 2003;70:1-10
26. Walz C, Score J, Mix J, Cilloni D, Roche-Lestienne C, Yeh RF. et al. The molecular anatomy of the FIP1L1-PDGFRA fusion gene. Leukemia. 2009;23:271-278
27. Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J. et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201-1214
28. Chase A, Ernst T, Fiebig A, Collins A, Grand F, Erben P. et al. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica. 2010;95:20-26
29. Liu XF, Bera TK, Liu LJ, Pastan I. A primate-specific POTE-actin fusion protein plays a role in apoptosis. Apoptosis. 2009;14:1237-1244
30. Lee Y, Ise T, Ha D, Saint FA, Hahn Y, Liu XF. et al. Evolution and expression of chimeric POTE-actin genes in the human genome. Proc Natl Acad Sci U S A. 2006;103:17885-17890
31. Babushok DV, Ohshima K, Ostertag EM, Chen X, Wang Y, Mandal PK. et al. A novel testis ubiquitin-binding protein gene arose by exon shuffling in hominoids. Genome Res. 2007;17:1129-1138
32. Ohshima K, Igarashi K. Inference for the initial stage of domain shuffling: tracing the evolutionary fate of the PIPSL retrogene in hominoids. Mol Biol Evol. 2010;27:2522-2533
33. Jacobs J, Glanz S, Bunse-Grassmann A, Kruse O, Kuck U. RNA trans-splicing: identification of components of a putative chloroplast spliceosome. Eur J Cell Biol. 2010;89:932-939
34. Hentze MW, Preiss T. Circular RNAs: splicing's enigma variations. EMBO J. 2013;32:923-925
35. Yang W, Wu JM, Bi AD, Ou-Yang YC, Shen HH, Chirn GW. et al. Possible Formation of Mitochondrial-RNA Containing Chimeric or Trimeric RNA Implies a Post-Transcriptional and Post-Splicing Mechanism for RNA Fusion. PLoS One. 2013;8:e77016. doi: 10.1371/journal.pone.0077016
36. Ritz K, van Schaik BD, Jakobs ME, Aronica E, Tijssen MA, van Kampen AH. et al. Looking ultra deep: short identical sequences and transcriptional slippage. Genomics. 2011;98:90-95
37. Al-Balool HH, Weber D, Liu Y, Wade M, Guleria K, Nam PL. et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011;21:1788-1799
38. Horiuchi T, Aigaki T. Alternative trans-splicing: a novel mode of pre-mRNA processing. Biol Cell. 2006;98:135-140
39. Glanz S, Kuck U. Trans-splicing of organelle introns--a detour to continuous RNAs. Bioessays. 2009;31:921-934
40. Rigatti R, Jia JH, Samani NJ, Eperon IC. Exon repetition: a major pathway for processing mRNA of some genes is allele-specific. Nucleic Acids Res. 2004;32:441-446
41. Dixon RJ, Eperon IC, Samani NJ. Complementary intron sequence motifs associated with human exon repetition: a role for intragenic, inter-transcript interactions in gene expression. Bioinformatics. 2007;23:150-155
42. Pink JJ, Wu SQ, Wolf DM, Bilimoria MM, Jordan VC. A novel 80 kDa human estrogen receptor containing a duplication of exons 6 and 7. Nucleic Acids Res. 1996;24:962-969
43. Pink JJ, Fritsch M, Bilimoria MM, Assikis VJ, Jordan VC. Cloning and characterization of a 77-kDa oestrogen receptor isolated from a human breast cancer cell line. Br J Cancer. 1997;75:17-27
44. Flouriot G, Brand H, Seraphin B, Gannon F. Natural trans-spliced mRNAs are generated from the human estrogen receptor-alpha (hER alpha) gene. J Biol Chem. 2002;277:26244-26251
45. Lai J, Lehman ML, Dinger ME, Hendy SC, Mercer TR, Seim I. et al. A variant of the KLK4 gene is expressed as a cis sense-antisense chimeric transcript in prostate cancer cells. RNA. 2010;16:1156-1166
46. Gabler M, Volkmar M, Weinlich S, Herbst A, Dobberthien P, Sklarss S. et al. Trans-splicing of the mod(mdg4) complex locus is conserved between the distantly related species Drosophila melanogaster and D. virilis. Genetics. 2005;169:723-736
47. Labrador M, Mongelard F, Plata-Rengifo P, Baxter EM, Corces VG, Gerasimova TI. Protein encoding by both DNA strands. Nature. 2001;409:1000
48. Zaphiropoulos PG. Trans-splicing in Higher Eukaryotes: Implications for Cancer Development? Front Genet. 2011;2:92. doi: 10.3389/fgene.2011.00092
49. Nageshan RK, Roy N, Ranade S, Tatu U. Trans-spliced heat shock protein 90 modulates encystation in Giardia lamblia. PLoS Negl Trop Dis. 2014;8:e2829
50. Nageshan RK, Roy N, Hehl AB, Tatu U. Post-transcriptional repair of a split heat shock protein 90 gene by mRNA trans-splicing. J Biol Chem. 2011;286:7116-7122
51. Kamikawa R, Inagaki Y, Tokoro M, Roger AJ, Hashimoto T. Split introns in the genome of Giardia intestinalis are excised by spliceosome-mediated trans-splicing. Curr Biol. 2011;21:311-315
52. Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO. et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17:669-681
53. Finta C, Warner SC, Zaphiropoulos PG. Intergenic mRNAs. Minor gene products or tools of diversity? Histol Histopathol. 2002;17:677-682
54. Portin P. The elusive concept of the gene. Hereditas. 2009;146:112-117
55. Hirano M, Noda T. Genomic organization of the mouse Msh4 gene producing bicistronic, chimeric and antisense mRNA. Gene. 2004;342:165-177
56. Herai RH, Yamagishi ME. Detection of human interchromosomal trans-splicing in sequence databanks. Brief Bioinform. 2010;11:198-209
57. Parra G, Reymond A, Dabbouseh N, Dermitzakis ET, Castelo R, Thomson TM. et al. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37-44
58. Varley KE, Gertz J, Roberts BS, Davis NS, Bowling KM, Kirby MK. et al. Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res Treat. 2014;146:287-297
59. Nacu S, Yuan W, Kan Z, Bhatt D, Rivers CS, Stinson J. et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med Genomics. 2011;4:11-doi 10.1186/1755-8794-4-11
60. Shiga Y, Sagawa K, Takai R, Sakaguchi H, Yamagata H, Hayashi S. Transcriptional readthrough of Hox genes Ubx and Antp and their divergent post-transcriptional control during crustacean evolution. Evol Dev. 2006;8:407-414
61. Kim HP, Cho GA, Han SW, Shin JY, Jeong EG, Song SH. et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene. 2013;33:5434-5441
62. Bruno AE, Miecznikowski JC, Qin M, Wang J, Liu S. FUSIM: a software tool for simulating fusion transcripts. BMC Bioinformatics. 2013;14:13. doi: 10.1186/1471-2105-14-13
63. Kumar-Sinha C, Kalyana-Sundaram S, Chinnaiyan AM. SLC45A3-ELK4 chimera in prostate cancer: spotlight on cis-splicing. Cancer Discov. 2012;2:582-585
64. Frenkel-Morgenstern M, Lacroix V, Ezkurdia I, Levin Y, Gabashvili A, Prilusky J. et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231-1242
65. Djebali S, Lagarde J, Kapranov P, Lacroix V, Borel C, Mudge JM. et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 2012;7:e28213. doi: 10.1371/journal.pone.0028213
66. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87-98
67. Brassesco MS. Leukemia/lymphoma-associated gene fusions in normal individuals. Genet Mol Res. 2008;7:782-790
68. Janz S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair (Amst). 2006;5:1213-1224
69. Janz S, Potter M, Rabkin CS. Lymphoma- and leukemia-associated chromosomal translocations in healthy individuals. Genes Chromosomes Cancer. 2003;36:211-223
70. Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, Bolland DJ. et al. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 2007;5:e192
71. Muller JR, Mushinski EB, Williams JA, Hausner PF. Immunoglobulin/Myc recombinations in murine Peyer's patch follicles. Genes Chromosomes Cancer. 1997;20:1-8
72. Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood. 1995;86:3118-3122
73. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C. et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99:8242-8247
74. Greaves M, Colman SM, Kearney L, Ford AM. Fusion genes in cord blood. Blood. 2011;117:369-370
75. Lausten-Thomsen U, Madsen HO, Vestergaard TR, Hjalgrim H, Nersting J, Schmiegelow K. Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood. 2011;117:186-189
76. Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3:639-649
77. Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K. et al. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. 2011;117:368-369
78. Yang X, Lee K, Said J, Gong X, Zhang K. Association of Ig/BCL6 translocations with germinal center B lymphocytes in human lymphoid tissues: implications for malignant transformation. Blood. 2006;108:2006-2012
79. Kannan K, Wang L, Wang J, Ittmann MM, Li W, Yen L. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc Natl Acad Sci U S A. 2011;108:9172-9177
80. Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, de la TA. et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734-2738
81. Soriani S, Cesana C, Farioli R, Scarpati B, Mancini V, Nosari A. PML/RAR-alpha fusion transcript and polyploidy in acute promyelocytic leukemia without t(15;17). Leuk Res. 2010;34:e261-e263
82. Grimwade D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M. et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood. 2000;96:1297-1308
83. Matsouka P, Sambani C, Giannakoulas N, Symeonidis A, Zoumbos N. Polyploidy in acute promyelocytic leukemia without the 15:17 translocation. Haematologica. 2001;86:1312-1313
84. Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol. 2002;13:3-16
85. Marotta V, Guerra A, Sapio MR, Vitale M. RET/PTC rearrangement in benign and malignant thyroid diseases: a clinical standpoint. Eur J Endocrinol. 2011;165:499-507
86. Rowley JD, Blumenthal T. Medicine. The cart before the horse. Science. 2008;321:1302-1304
87. Li H, Wang J, Mor G, Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321:1357-1361
88. Li H, Wang J, Ma X, Sklar J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle. 2009;8:218-222
89. Wang L. Identification of cancer gene fusions based on advanced analysis of the human genome or transcriptome. Front Med. 2013;7:280-289
90. Natrajan R, Wilkerson PM, Marchio C, Piscuoglio S, Ng CK, Wai P. et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol. 2014;232:553-565
91. Edgren H, Murumagi A, Kangaspeska S, Nicorici D, Hongisto V, Kleivi K. et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011;12:R6. doi: 10.1186/gb-2011-12-1-r6
92. Giacomini CP, Sun S, Varma S, Shain AH, Giacomini MM, Balagtas J. et al. Breakpoint analysis of transcriptional and genomic profiles uncovers novel gene fusions spanning multiple human cancer types. PLoS Genet. 2013;9:e1003464
93. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405-409
94. Doose G, Alexis M, Kirsch R, Findeiss S, Langenberger D, Machne R. et al. Mapping the RNA-Seq trash bin: unusual transcripts in prokaryotic transcriptome sequencing data. RNA Biol. 2013;10:1204-1210
95. Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR. et al. Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res. 2008;68:73-80
96. Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6:429-439
97. Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA. et al. ETS gene fusions in prostate cancer: from discovery to daily clinical practice. Eur Urol. 2009;56:275-286
98. Brenner JC, Chinnaiyan AM. Translocations in epithelial cancers. Biochim Biophys Acta. 2009;1796:201-215
99. Prensner JR, Chinnaiyan AM. Oncogenic gene fusions in epithelial carcinomas. Curr Opin Genet Dev. 2009;19:82-91
100. Kusk MS, Lausten-Thomsen U, Andersen MK, Olsen M, Hjalgrim H, Schmiegelow K. False positivity of ETV6/RUNX1 detected by FISH in healthy newborns and adults. Pediatr Blood Cancer. 2014;61:1704-1706
101. Llorens-Rico V, Serrano L, Lluch-Senar M. Assessing the hodgepodge of non-mapped reads in bacterial transcriptomes: real or artifactual RNA chimeras? BMC Genomics. 2014;15:633
102. McManus CJ, Duff MO, Eipper-Mains J, Graveley BR. Global analysis of trans-splicing in Drosophila. Proc Natl Acad Sci U S A. 2010;107:12975-12979
103. Wu CS, Yu CY, Chuang CY, Hsiao M, Kao CF, Kuo HC. et al. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014;24:25-36
104. Dotzlaw H, Alkhalaf M, Murphy LC. Characterization of estrogen receptor variant mRNAs from human breast cancers. Mol Endocrinol. 1992;6:773-785
105. Murphy LC, Dotzlaw H, Hamerton J, Schwarz J. Investigation of the origin of variant, truncated estrogen receptor-like mRNAs identified in some human breast cancer biopsy samples. Breast Cancer Res Treat. 1993;26:149-161
106. Guerra E, Trerotola M, Dell' AR, Bonasera V, Palombo B, El-Sewedy T. et al. A bicistronic CYCLIN D1-TROP2 mRNA chimera demonstrates a novel oncogenic mechanism in human cancer. Cancer Res. 2008;68:8113-8121
107. Ye Q, Chung LW, Li S, Zhau HE. Identification of a novel FAS/ER-alpha fusion transcript expressed in human cancer cells. Biochim Biophys Acta. 2000;1493:373-377
108. Yuan C, Liu Y, Yang M, Liao DJ. New methods as alternative or corrective measures for the pitfalls and artifacts of reverse transcription and polymerase chain reactions (RT-PCR) in cloning chimeric or antisense-accompanied RNA. RNA Biol. 2013;10:958-967
109. Ozsolak F, Milos PM. Single-molecule direct RNA sequencing without cDNA synthesis. Wiley Interdiscip Rev RNA. 2011;2:565-570
110. Ozsolak F, Milos PM. Transcriptome profiling using single-molecule direct RNA sequencing. Methods Mol Biol. 2011;733:51-61
111. Yu WH, Hovik H, Olsen I, Chen T. Strand-specific transcriptome profiling with directly labeled RNA on genomic tiling microarrays. BMC Mol Biol. 2011;12:3. doi: 10.1186/1471-2199-12-3
112. Langevin SA, Bent ZW, Solberg OD, Curtis DJ, Lane PD, Williams KP. et al. Peregrine: A rapid and unbiased method to produce strand-specific RNA-Seq libraries from small quantities of starting material. RNA Biol. 2013;10:502-515
113. Sendler E, Johnson GD, Krawetz SA. Local and global factors affecting RNA sequencing analysis. Anal Biochem. 2011;419:317-322
114. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57-63
115. Houseley J, Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271
116. Cocquet J, Chong A, Zhang G, Veitia RA. Reverse transcriptase template switching and false alternative transcripts. Genomics. 2006;88:127-131
117. Mader RM, Schmidt WM, Sedivy R, Rizovski B, Braun J, Kalipciyan M. et al. Reverse transcriptase template switching during reverse transcriptase-polymerase chain reaction: artificial generation of deletions in ribonucleotide reductase mRNA. J Lab Clin Med. 2001;137:422-428
118. Brakenhoff RH, Schoenmakers JG, Lubsen NH. Chimeric cDNA clones: a novel PCR artifact. Nucleic Acids Res. 1991;19:1949
119. Paabo S, Irwin DM, Wilson AC. DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem. 1990;265:4718-4721
120. Qiu X, Wu L, Huang H, McDonel PE, Palumbo AV, Tiedje JM. et al. Evaluation of PCR-Generated Chimeras, Mutations, and Heteroduplexes with 16S rRNA Gene-Based Cloning. Appl Environ Microbiol. 2001;67:880-887
121. Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R. et al. A large genome center's improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005-1010
122. Shammas FV, Heikkila R, Osland A. Fluorescence-based method for measuring and determining the mechanisms of recombination in quantitative PCR. Clin Chim Acta. 2001;304:19-28
123. Zaphiropoulos PG. Template switching generated during reverse transcription? FEBS Lett. 2002;527:326
124. Ro S, Kang SH, Farrelly AM, Ordog T, Partain R, Fleming N. et al. Template switching within exons 3 and 4 of KV11.1 (HERG) gives rise to a 5' truncated cDNA. Biochem Biophys Res Commun. 2006;345:1342-1349
125. Roy SW, Irimia M. When good transcripts go bad: artifactual RT-PCR 'splicing' and genome analysis. Bioessays. 2008;30:601-605
126. Roy SW, Irimia M. Intron mis-splicing: no alternative? Genome Biol. 2008;9:208. doi: 10.1186/gb-2008-9-2-208
127. Gao R, Zhao AH, Du Y, Ho WT, Fu X, Zhao ZJ. PCR artifacts can explain the reported biallelic JAK2 mutations. Blood Cancer J. 2012;2:e56-doi 10.1038/bcj.2012.2
128. Zheng W, Chung LM, Zhao H. Bias detection and correction in RNA-Sequencing data. BMC Bioinformatics. 2011;12:290. doi: 10.1186/1471-2105-12-290
129. Mak J, Kleiman L. Primer tRNAs for reverse transcription. J Virol. 1997;71:8087-8095
130. Marquet R, Isel C, Ehresmann C, Ehresmann B. tRNAs as primer of reverse transcriptases. Biochimie. 1995;77:113-124
131. Colett MS, Larson R, Gold C, Strick D, Anderson DK, Purchio AF. Molecular cloning and nucleotide sequence of the pestivirus bovine viral diarrhea virus. Virology. 1988;165:191-199
132. Gubler U. Second-strand cDNA synthesis: mRNA fragments as primers. Methods Enzymol. 1987;152:330-335
133. Herzig E, Voronin N, Hizi A. The removal of RNA primers from DNA synthesized by the reverse transcriptase of the retrotransposon Tf1 is stimulated by Tf1 integrase. J Virol. 2012;86:6222-6230
134. Che S, Ginsberg SD. Amplification of RNA transcripts using terminal continuation. Lab Invest. 2004;84:131-137
135. Ginsberg SD, Che S. RNA amplification in brain tissues. Neurochem Res. 2002;27:981-992
136. Hanaki K, Nakatake H, Yamamoto K, Odawara T, Yoshikura H. DNase I activity retained after heat inactivation in standard buffer. Biotechniques. 2000;29:38-40 42
137. Hanaki K, Nishihara T, Odawara T, Nakajima N, Yamamoto K, Yoshikura H. RNAse A treatment of Taq and Tth DNA polymerases eliminates primer/template-independent poly(dA-dT) synthesis. Biotechniques. 2001;31:734 736, 738
138. Hanaki K, Odawara T, Nakajima N, Shimizu YK, Nozaki C, Mizuno K. et al. Two different reactions involved in the primer/template-independent polymerization of dATP and dTTP by Taq DNA polymerase. Biochem Biophys Res Commun. 1998;244:210-219
139. Nakajima N, Hanaki K, Shimizu YK, Ohnishi S, Gunji T, Nakajima A. et al. Hybridization-AT-tailing (HybrAT) method for sensitive and strand-specific detection of DNA and RNA. Biochem Biophys Res Commun. 1998;248:613-620
140. Hanaki K, Odawara T, Muramatsu T, Kuchino Y, Masuda M, Yamamoto K. et al. Primer/template-independent synthesis of poly d(A-T) by Taq polymerase. Biochem Biophys Res Commun. 1997;238:113-118
141. Zhou MY, Gomez-Sanchez CE. Universal TA cloning. Curr Issues Mol Biol. 2000;2:1-7
142. Alldred MJ, Che S, Ginsberg SD. Terminal continuation (TC) RNA amplification without second strand synthesis. J Neurosci Methods. 2009;177:381-385
143. Wellenreuther R, Schupp I, Poustka A, Wiemann S. SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genomics. 2004;5:36. doi:10.1186/1471-2164-5-36
144. Sun Y, Li Y, Luo D, Liao DJ. Pseudogenes as Weaknesses of ACTB (Actb) and GAPDH (Gapdh) Used as Reference Genes in Reverse Transcription and Polymerase Chain Reactions. PLoS One. 2012;7:e41659. doi:10.1371/journal.pone.0041659
145. Gubler U. Second-strand cDNA synthesis: classical method. Methods Enzymol. 1987;152:325-329
146. Gubler U. Second-strand cDNA synthesis: mRNA fragments as primers. Methods Enzymol. 1987;152:330-335
147. Spiegelman S, Burny A, Das MR, Keydar J, Schlom J, Travnicek M. et al. DNA-directed DNA polymerase activity in oncogenic RNA viruses. Nature. 1970;227:1029-1031
148. Roberts JD, Preston BD, Johnston LA, Soni A, Loeb LA, Kunkel TA. Fidelity of two retroviral reverse transcriptases during DNA-dependent DNA synthesis in vitro. Mol Cell Biol. 1989;9:469-476
149. Varadaraj K, Skinner DM. Denaturants or cosolvents improve the specificity of PCR amplification of a G + C-rich DNA using genetically engineered DNA polymerases. Gene. 1994;140:1-5
150. Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G. et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494-504
151. Haddad F, Qin AX, Bodell PW, Zhang LY, Guo H, Giger JM. et al. Regulation of antisense RNA expression during cardiac MHC gene switching in response to pressure overload. Am J Physiol Heart Circ Physiol. 2006;290:H2351-H2361
152. Stahlberg A, Hakansson J, Xian X, Semb H, Kubista M. Properties of the reverse transcription reaction in mRNA quantification. Clin Chem. 2004;50:509-515
153. Adrover MF, Munoz MJ, Baez MV, Thomas J, Kornblihtt AR, Epstein AL. et al. Characterization of specific cDNA background synthesis introduced by reverse transcription in RT-PCR assays. Biochimie. 2010;92:1839-1846
154. Frech B, Peterhans E. RT-PCR: 'background priming' during reverse transcription. Nucleic Acids Res. 1994;22:4342-4343
155. Haddad F, Qin AX, Giger JM, Guo H, Baldwin KM. Potential pitfalls in the accuracy of analysis of natural sense-antisense RNA pairs by reverse transcription-PCR. BMC Biotechnol. 2007;7:21. doi:10.1186/1472-6750-7-21
156. Fejes-Toth K. et al. Post-transcriptional processing generates a diversity of 5'-modified long and short RNAs. Nature. 2009;457:1028-1032
157. Kowalczyk MS, Higgs DR, Gingeras TR. Molecular biology: RNA discrimination. Nature. 2012;482:310-311
158. Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity--alternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675. doi: 10.1155/2010/753675
159. Kruger M, Linke WA. The giant protein titin: a regulatory node that integrates myocyte signaling pathways. J Biol Chem. 2011;286:9905-9912
160. Li K, Ramchandran R. Natural antisense transcript: a concomitant engagement with protein-coding transcript. Oncotarget. 2010;1:447-452
161. Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M. et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564-1566
162. Grinchuk OV, Jenjaroenpun P, Orlov YL, Zhou J, Kuznetsov VA. Integrative analysis of the human cis-antisense gene pairs, miRNAs and their transcription regulation patterns. Nucleic Acids Res. 2010;38:534-547
163. Beiter T, Reich E, Williams RW, Simon P. Antisense transcription: a critical look in both directions. Cell Mol Life Sci. 2009;66:94-112
164. Wang X, Arai S, Song X, Reichart D, Du K, Pascual G. et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126-130
165. Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M. et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065-1072
166. Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De SJ. et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492-500
Author contact
![]() Corresponding authors: Zhiyu Peng, Beijing Genomics Institute at Shenzhen, Building No.11, Beishan Industrial Zone, Yantian District, Shenzhen 518083, P.R. China. Email: pengzhiyucn; Siqi Liu, CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, P.R. China. Email: siqiliuorg.cn; Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Academy of Medical Science, Beijing 100021, P.R. China. Email: xningzhibta.net.cn; D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Email: djliaoumn.edu
Corresponding authors: Zhiyu Peng, Beijing Genomics Institute at Shenzhen, Building No.11, Beishan Industrial Zone, Yantian District, Shenzhen 518083, P.R. China. Email: pengzhiyucn; Siqi Liu, CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, P.R. China. Email: siqiliuorg.cn; Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Academy of Medical Science, Beijing 100021, P.R. China. Email: xningzhibta.net.cn; D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Email: djliaoumn.edu