Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(6):583-592. doi:10.7150/jca.11567 This issue Cite
Research Paper
In Vitro Administration of Gold Nanoparticles Functionalized with MUC-1 Protein Fragment Generates Anticancer Vaccine Response via Macrophage Activation and Polarization Mechanism
Teodora Mocan2, 3, Cristian Matea1,2, Flaviu Tabaran2, 4, Cornel Iancu1, 2, Remus Orasan3, Lucian Mocan1, 2 ![]()
1. 3rd Surgery Clinic, “Iuliu Hatieganu” University of Medicine and Pharmacy, 19-21 Croitorilor St., 400162 Cluj-Napoca, Romania.
2. Department of Nanomedicine, Institute of Gastroenterology and Hepatology, 19-21 Croitorilor St., 400162 Cluj-Napoca, Romania.
3. Department of Physiology, “Iuliu Hatieganu” University of Medicine and Pharmacy, no. 1 Clinicilor Street., 400006 Cluj-Napoca, Romania.
4. Department of Pathology, Faculty of Veterinary Medicine, University of Agricultural Sciences and Veterinary Medicine, Cluj-Napoca, Romania; Calea Mănăștur 3-5, Cluj-Napoca 400372.
Received 2015-1-11; Accepted 2015-4-13; Published 2015-5-15
Abstract
Therapeutic cancer vaccines (or active immunotherapy) aim to guide the patient's personal immune system to eradicate cancer cells. An exciting approach to cancer vaccines has been offered by nanoscale drug delivery systems containing tumor associated antigens (TAAs). Their capacity to stimulate the immune system has been suggested during late years. However, the role of the macrophages as key-elements in antigen-presentation process following TAAs-containing nanosystems is not completely understood. We aimed to evaluate the effect of gold nanoparticles functionalized with mucin-1 peptide (MUC-1) on murine peritoneal macrophages.
Gold nanoparticles, obtained using a modified Turkevich method, were functionalized with MUC-1 protein using Clealand's reagent. The obtained GNP-MUC-1 solution was used to treat at various concentrations monolayers of peritoneum-derived macrophages that were further analyzed using confocal and hyperspectral microscopy, ELISA assays and spectroscopic techniques. The GNP-MUC-1 nano-construct had proven to function as a powerful macrophage activator with consequent release of cytokines such as: TNF-ɑ, IL-6, IL-10 and IL-12 on peritoneal macrophages we have isolated from mice. Our results demonstrate optimization of antigen-presenting process and predominant M1 polarization following exposure GNP-MUC-1. To our best knowledge this is the first study to evaluate the anticancer effects of a newly designed nano-biocompound on the complex antigen- processing apparatus of peritoneal macrophages.
Keywords: cancer vaccine, peptides, monoclonal antibodies, macrophages
Introduction
The development of nanoscale drug delivery systems represents an exciting and new approach to cancer treatment.1-3 One remarkable property of these nano-systems is represented by the activation of the immune system, which could form an attractive basis for cancer vaccine development.4 Although such drug delivery systems hold a tremendous potential for the future prevention of cancer, the research for a true anticancer vaccine remains elusive. Nanotechnology has already brought to light promising results in the field of anticancer vaccines. For instance, inert nanobeads, recombinant virus-like particles (VLPs), and immunostimulating complexes, are being used in cancer vaccine research due to their efficacy at eliciting both cellular and humoral immune responses. 5
There are several encouraging data that some classes of nanostructures may be used in initiating and maintaining immune responses. When bound to tumor antigens, they elicit a specific antitumor response in animal models.6 Furthermore, data shown that peptide-functionalized nanomaterials can act as proficient immunomodulators and consequently generate specific antibody responses.7 Moreover, ex vivo clonal expansion of T cells with antibody-linked nanoparticles results in T-cell activation and might lead to the development of novel immunotherapies.8
It has been shown that significant secretion of cytokines may help to rebuild the host`s immunity against cancer and consequently to generate obvious antitumor immunity.9 Despite their proven role in proliferation of T lymphocytes, especially the proliferation of CD8+ lymphocytes which constitutes the main part of antitumor effector cells, there are currently no studies that explore the concept of cancer prophylaxis mediated by specific antigens bound to gold nanoparticles.
Preliminary research data suggest that gold nanostructures may function as antigen presenting molecules.10 There is also increasing evidence sustaining the activator role of tumor associated antigens (TAAs) in antitumor immunity.7 We have, therefore, reasoned that binding of a low immunogenic peptide antigen onto the gold nanoparticle could lead to an essential mount in immune response intensity. We reasoned that MUC-1 (CD227), a membrane-tethered mucin glycoprotein that is normally expressed on the apical surfaces of normal glandular epithelia, could become an adequate conjugator for GNPs, capable to produce the expected effect. Recent description of MUC-1 as a target for cytotoxic T lymphocytes (CTLs) has raised interest in using this protein as a possible target for immunotherapy, suggesting a good potential for its application of nanomediated cancer vaccine. 11
We therefore hypothesize that a novel vaccine, containing MUC-1-functionalized GNPs could lead to potent antitumor effect by inducing macrophage activation, thus opening new avenues in the prophylaxis of cancer. The key role of macrophages as specialized antigen presenting cells has previously been suggested.7 The newly designed GNP-MUC-1 nanostructure was tested for efficiency as macrophage activator with consequent release of cytokines such as: TNF-ɑ, IL-6, IL-10 and IL-12 on peritoneral macrophages we have isolated from CD1 mice. To our best knowledge this is the first study to test the cytokine production effects of a newly designed nano-biocompound on the complex antigen -processing apparatus of peritoneal macrophages.
Materials and methods
Materials and Reagents
HAuCl4 (≥99.9%), tri-sodium citrate (≥99%), DL-dithiotreitol (DTT) (≥99%) were purchased from Sigma-Aldrich™ (Darmstadt, Germany). MUC-1 protein fragment (14kDa) having the following aminoacid sequence - RPGSVVVQLTLAFREGTIN VHDVETQFNQYKTEAASRYNLTISDVS, was purchased from Abcam (Austin, USA). All reagents were used as received, without further purification.
Synthesis of gold nanoparticles (GNPs)
Citrate capped gold nanoparticles (GNPs) were synthesized according to a modified Turkevich method. Briefly, 36mg HAuCl4 were dissolved in 75mL distilled H2O and heated to 100°C, to this a 4mg/mL sodium citrate solution (5 mL) was rapidly injected under vigorous stirring. The solution was kept under reflux for 2 hours and vigorous stirring, during this stage the solution's color turned from pale yellow to dark-red.
Mucin-1 (MUC-1) functionalization of Gold Nanoparticles
In order to functionalize the obtained GNPs with the MUC-1 protein Clealand's reagent was used. Thus, 100μL of MUC-1 protein was mixed with 300μL DTT 100mM (pH=8.5) and was added to 10mL GNP solution and kept under vigorous agitation for 1h. The obtained GNP-MUC-1 solution was subjected to a centrifugation step at 16000RPM/30min and the resulting pellet re-dispersed in distilled H2O. The MUC-1 functionalized GNP solution presented good stability over several weeks at room temperature.
Attenuated total reflectance fourier transform infrared spectroscopy (ATR-FT-IR) characterization of newly sinthetized structure
Universal attenuated total reflectance fourier transform infrared spectroscopy (ATR-FT-IR) measurements were conducted on a Perkin-Elmer Spectrum Two® instrument with an UATR single reflection diamond. Spectra processing and baseline corrections were done using the Spectrum 10™ software.
Atomic force microscopy (AFM) characterization of nanobioconstruct
Atomic force microscopy (AFM) measurements were performed on Workshop TT-AFM® (AFMWorkshop, CA, USA) in vibrating mode using ACTA-SS cantilevers (AppNano, CA, USA). The recorded data was further processed with the Gwyddion® 2.36 software.
Isolation of mouse peritoneal cavity cells
Collection of mouse peritoneal cavity cells was performed as previously described.12 In short, mice (n=5) were euthanized and dissection with exposure of inner skin lining of the peritoneal cavity was completed. Following, intraperitoneal injection of solution (ice-cold PBS+3%fetal calf serum) was performed, followed by gentle massage of the peritoneum for further dislodgement of attached cells into solution. Next, collection of the fluid into 15mL Falcon tubes was completed. The remaining fluid from the cavity was collected using a Pasteur pipette. Cell pelleting by centrifugation (1500rpm, 8 min) was performed. According to a previously validated protocol13, re-suspension of cells in cell culture media (Dulbecco's Modified Eagle Medium/F12 (Invitrogen), supplemented with 10 mM L-glutamine, 100 IU/ml penicillin, and 100 µg/ml streptomycin, 10% fetal bovine serum) was completed afterward. Cell concentration was adjusted to 2X106 total peritoneal cells/mL by automated counting (RevSci Counter RS-RC-100), and kept on ice.
Isolation of Murine Peritoneum-Derived Macrophages
To obtain monolayers of peritoneum-derived macrophages, cell-containing fluid was added to 4 chamber microscopic slides (LabTeck Chamber Slides, Nunc) for fixation/staining fixation procedures, according to standard protocol 13. For ELISA/UV-VIS intracellular detection assays, cells were seeded into 6-well uncoated cell culture plates. Cells were also seeded onto a 96 well plate for proliferation/viability testing. Cell seeding was followed by standard incubation (3 hrs. 37ᴼC, 5% CO2).
Macrophages were immediately used for testing. All animal experiments were approved by the Institutional Ethics Committee.
Cell treatment
Cell medium removal was performed at 24 hrs. after isolation and seeding. Non-adherent cells were removed by washing (three times, pre-warmed PBS). For microscopic assays, 300μL of the appropriate solution were added to each cell culture chamber. Also, 1.2 mL of each exposure solution was added to each well on the uncoated 6 well plate. 50 μl of the appropriate solution was added to each well on the 96 well plates for further proliferation assays. The four different solutions were: cell culture medium (control group); 50μg/mL GNP-MUC-1; 25μg/mL GNP-MUC-1; 12.5μg/mL GNP-MUC-1, respectively. Following incubation (6 hrs. 37° C, 5% CO2), cell culture chamber slides and 6 well chambers were further included into different processing protocols, appropriate to the specific analysis to be performed.
Golgi Apparatus (GA), Endoplasmic Reticulum(ER) and Nucleus(N) Detection Assay
Multiple organelle staining was performed using a Orgenelle-ID RGB III Assay kit (Enzo Life Sciences) which allows selective ER, GA and N staining, according to manufacturer's instructions. In short, cells were washed with 100μL assay solution and further incubated with multiple staining solution (Organelle-ID RGB Reagent III) (100 μL/chamber, 30 minutes, 4°C, dark room). Next, a second washing step was performed (3 x ice-cold medium) followed by fresh ice-cold medium incubation (37°C, 30 minutes). Following, cells were washed with 100μL 1X assay solution. After removal of buffer excess, fixation (formaldehyde 10%, 5 min) and washing (deionized water) were performed. Next, the medium chamber was detached from the slide, coverslip was placed and mounted (Water-based Mountant, Magnacol Ltd.). The slide was further analyzed using microscopy techniques. To detect the distinct fluorescent emissions signals, the following standard filter sets were used: FITC (Golgi apparatus), DAPI (nucleus) and Texas Red (endoplasmic reticulum), respectively.
Microscopic evaluation
Microscopic analyses were conducted using the Olympus FSX100 Bio Imaging Navigator all-in-one High Performance microscope system. Multi-excitation filter (red, blue, green) fluorescence detection modes were used.
Interleukin 6 concentration assessment
Determination of IL 6 serum level was performed using a commercially available solid-phase sandwich Enzyme Linked Immunosorbent Assay (ELISA) (Life Technologies, no. catalog. KMC0061). According to the manufacturer protocol antigen was bound to the pre-coated antibody by addition of 50μL of sample mixed with 50 μL diluent into each well of the 96 -well plate followed by incubation (RT, 2 hrs.) and washing(4x, washing buffer). Next, detector antibody was bound by addition of 100 μL Ms IL-6 Biotin Conjugate solution onto each well, followed by a second incubation (30 min, RT) washing step (4x, washing buffer). Following, 100 μL of streptavidin-horse radish peroxidase (HRP) was added to each well except for the well corresponding to chromogenic blanks. Again, incubation (30 min, RT) and washing (4X, washing buffer) steps were pursued. Next, chromogenic addition was performed by adding 100μL of Stabilized Tetramethylbenzidine (TMB) Chromogen to each well followed by incubation (30 min, dark). Last, reaction was stopped by 100μL/well addition of Stop Solution. Absorbance reading was performed (Perkin Elmer Viktor 3 multilabel multitask plate reader, 450nm) Background absorbance was subtracted from all samples. Final concentration was calculated using the curve-fitting feature of Microsoft Office2010 Excel application.
Interleukin 10 concentration assessment
IL-10 concentration was assessed using a commercially available solid phase sandwich ELISA kit (Life Technologies, No. Catalog. KMC0101). The manufacturer protocol was followed for all procedures. In short, assay procedures started with antigen binding by 100μL sample addition in each well. Incubation (1 hr, 37ᴼC) and washing (4X, washing buffer) followed. Next, by adding 100μL of IL-10 Biotin Conjugate solution, incubation (1 hr, RT), and washing (4X, washing buffer), the biotinated antibody was added to each well. Next 100μL streptavidin-HRP addition was added to the wells followed by incubation (30 min, RT) and washing(4X, washing buffer).100 μL of Stabilized Tetramethylbenzidine (TMB) Chromogen was furher added to each well, incubated(30 min, RT, dark) and washed (4X, washing buffer). Reaction was stopped using 100μL of Stop Solution Absorbance reading was performed (450nm, Perkin Elmer Viktor 3 multilabel multitask plate reader), with background absorbance subtraction from all samples. Curve fitting (Microsoft Office 2010 Excel) with equation generation were used to calculate Il-10 concentration.
Interleukin 12 concentration assessment
Determination of serum IL-12 concentration was performed using a standardized kit (Life Technologies, No Catalog KMC0121). In short, the manufacturer steps were followed for a sandwich solid phase ELISA assay. The antigen binding step was achieved by addition of 50 μL of sample diluted with 50μL diluent. Next, the biotinylated detection antibody was bound by pipetting 50μL of Ms IL-12 Biotin Conjugate Solution /well. Incubation (2hrs, RT) and washing (4X, washing buffer) followed. Tetramethylbenzidine (TMB) Chromogen addition was completed,(100μL/well Stabilized Chromogen Solution) followed by incubation (30 min, dark). The reaction interruption was performed (100μL Stop Solution/well). Absorbance reading (450nm, Perkin Elmer Viktor 3 multilabel multitask plate reader) with background absorbance subtraction was performed next. The concentration calculus was performed using curve-fitting and equation generation by means of Microsoft Office 2010 Excel application.
Alpha TNF concentration assessment
Alpha-TNF concentration assessment was performed using a commercially available solid phase sandwich ELISA kit (Life Technologies, No. Catalog. KMC3011). In short, a first step of antigen binding was performed by addition of 50μL of sample together with 50μL of standard diluent. Then, we added the detection antibody (50μl/well of biotinylated Ms TNF-α Biotin Conjugate). The step was followed by incubation (90 min, RT) and washing (4x, washing buffer). Next, 100μL of streptavidin -HRP addition, with incubation (30 min, RT) and washing (4x, washing buffer) was performed. The addition of Tetramethylbenzidine (TMB) Chromogen (100μL/well) was next, followed by incubation (30 min, dark). The reaction was stopped using 100μL/well stop solution. Absorbance reading was performed at 450nm, using a Perkin Elmer Viktor 3 multilabel multitask plate reader. Concentration calculation was performed based on Microsoft Office 2010 Excel curve-fitting and equation determination
Cellular internalization detection
Hyperspectral and optical images were acquired utilizing a Olympus BX-43 microscope optical microscope equipped with the patented CytoViva150 Ultrahigh Resolution Imaging (URI) System (Auburn, AL). Image capture was performed using a 1600 × 1200 pixel/8 bit DAGE-MTI XL16 digital camera (DAGE-MTI, Michigan City). Image processing was performed using the Exponent 7 Software. The high signal-to-noise ratio allowed the direct intracellular visualization of GNP-MUC-1 nanoparticles.
Intracellular presence of GNP-MUC-1 was also examined using UV-VIS spectroscopy. Cells were previously cleared from exposure solution and washed (PBS, 3X). Next, addition of dH2O was performed (1 mL/chamber), followed by mechanical cell detachment from the plate. Cell lysate was further obtained by means of probe-sonication (Virtis VirSonic 100 Ultrasonic Cell Disruptor; 3x3sec; 10W, on ice). Next, the obtained solution was examined using the standard UV-VIS spectroscopy protocol, as described below.
Image analysis
Quantification of fluorescence staining levels were performed in ImageJ (NIH, ver. 1.43 u) by applying a linear stretch of the pixel intensity histogram corresponding to each slice in the z stack to adjust the number of low and high intensity pixels thus increasing the contrast. To remove the spaces between each slice of the z-stack and to create a single image from all slices the image was further converted into a z projection. A 7×7 top-hat filter was further applied, followed by a median filter and a thresholding step. The thresholded image was then converted to a binary image, which results in white structures (Annexin V/green fluorescence) on a black background that was further analyzed in ImageJ.
Statistical analysis
Data were expressed as mean (std error) and were analyzed after previous normality testing (Kolmogorov-Smirnov test) Differences in continuous data between independent groups were tested using independent sample Student test. A threshold of p<0.05 was selected.
Results
Characterization of GNP-MUC-1
Gold nanoparticles and MUC-1 functionalized GNPs were characterized by means of UV-Vis, ATR-FT-IR, DLS and AFM techniques. Figure S1 (Supplementary Material) presents the UV-Vis spectra for GNP and GNP-MUC-1 solutions. The GNP sample presents a λmax=521 nm while the λmax for the MUC-1 functionalized GNPs is recorded at 536 nm. This bathochromic shift of the surface plasmon resonance peak for the GNP-MUC-1 sample is attributed to the protein coupling on the gold nanoparticle surface.
Dynamic light scattering measurements were performed on both GNP and GNP-MUC-1 samples and their respective size distribution curves are presented in Figure S2 (Supplementary Material). Analyzes indicate that, the MUC-1 protein formed a monolayer around the nanogold cores during the functionalization step. The increase in size suggested by the UV-Vis spectroscopy measurements, through the 15nm λmax red shift of gold nanoparticles, is confirmed by the DLS technique. Citrate stabilized GNPs were recorded to have a 15.7 nm mean diameter while GNP-MUC-1 had a mean diameter of 44nm.
ATR-FT-IR spectroscopy was employed to investigate the binding of the MUC-1 protein on the surface of the gold nanoparticles, spectra for GNP and GNP-MUC-1 samples are presented in Figure 1-II. The citrate capped GNPs present two absorption bands at 1586 and 1395 cm-1, attributed to the antisymmetric and symmetric stretching of COO- of citrate ions. In the case of GNP-MUC-1 sample bands at 1628 cm-1 and 1542 cm-1 are attributed to amide I (COO- stretching vibration) and amide II (N-H bending vibrations), respectively, from the MUC-1 protein fragment. Thus, in the case of GNP-MUC-1 sample, the disappearance of citrate attributed bands, correlated with the appearance of amide I and amide II bands confirms the successful functionalization of GNPs with the MUC-1 protein.
I. AFM images GNP-MUC-1: A. 2D image of MUC-1 functionalized GNPs; B. Cross section graph of a single gold nanoparticle functionalized with MUC-1; C. 3D image of MUC-1 functionalized GNPs; II. ATR-FT-IR spectra of GNP and GNP-MUC-1 samples (GNP sample absorption bands at 1586 cm-1 and 1395 cm-1 (red line); GNP-MUC-1 sample absorption bands at 1628 cm-1 and 1542 cm-1 (blue line).
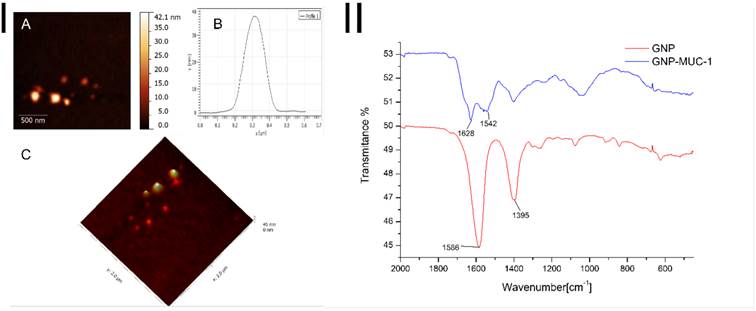
GNP-MUC-1 sample was subjected to AFM measurements in order to investigate the shape and size of the obtained nanostructures. To this end, Figure 1-IA and Figure 1-IC depict a 2D and 3D representation of the synthesized nanoparticles, which present themselves as spherical with a diameter of ~37nm.
Evaluation of organelle morphology (Golgi apparatus, Endoplasmic reticulum)
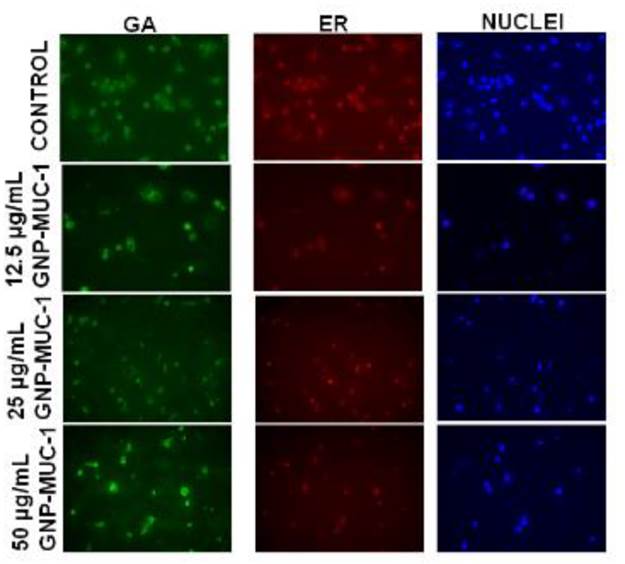
To further localize the activated structures and pathways within the cells as a result of treatment, we have used a specially designed, GA and ER- selective staining assay. Organelle morphology microscopic assay revealed loss of membrane continuity as a result of antigen trafficking, with reducing intensity of green (GA) and red fluorescence (ER) in GNP-MUC-1 exposed samples as compared to control. The decreasing trend in fluorescence staining for both types of organelles was stronger with increasing concentration of GNP-MUC-1 exposure. The appearance of non-cellular fragments, especially in high concentration- exposed samples, has also been detected. (Figure 2) Image J analysis revealed a significant decrease in fluorescence for both ER and GA compared to control in a dose dependent manner (p<0.005 in all slides).
Peritoneal macrophages following different exposures. Organelle staining (GA- green; ER-red) and nuclei staining (DAPI). (30X)
Evaluation of macrophages secretory products
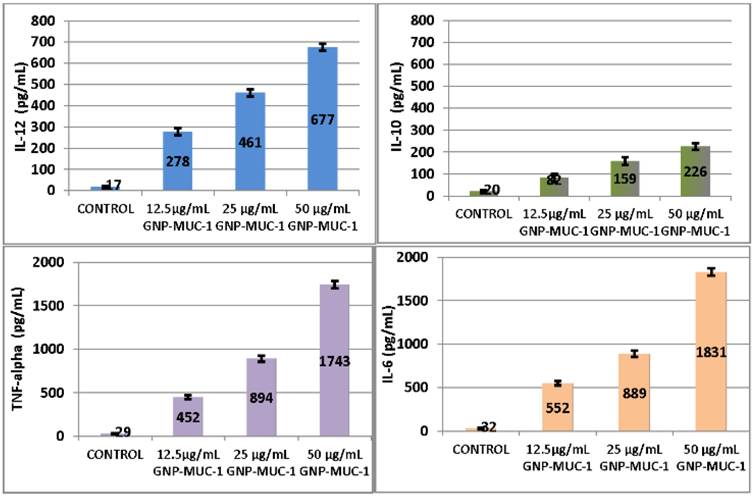
Significant serum level increase in IL-10, IL-12, TNF-alpha and IL-6 was obtained for 50 μg/mL GNP-MUC-1 (p<0.05 for all). (Figure 3)
Peritoneal macrophages following different exposures. ELISA results for I-10, IL-12, Tnf-alpha amd IL-6 serum levels. Bars represent mean. Error bars represent standard error of mean.
Evaluation of ubiquitin-proteasome pathway
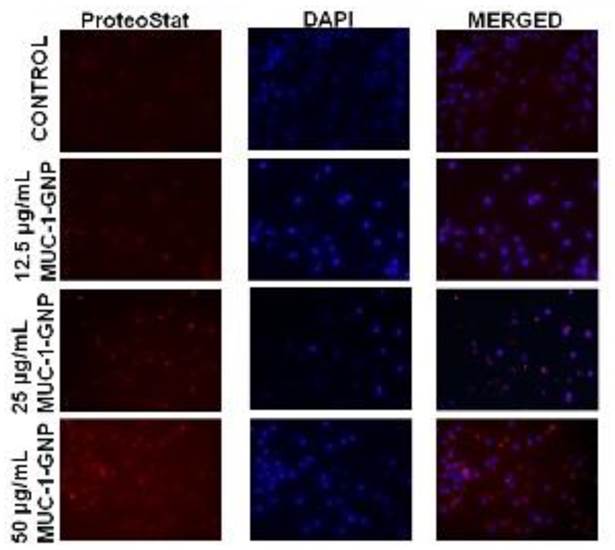
To further depict the full antigen trafficking pathway, we have used a homogenous assay for monitoring peptide and protein aggregation during manufacturing. GNP-MUC-1 exposed groups recorded enhanced fluorescence intensity as compared to control group. (Figure 4) As suggested by Image J analysis, cells presented significantly (p<0005) increased staining with increasing concentration compared with control, suggesting that GNP-MUC-1 exposure activates proteasome machinery.
Peritoneal macrophages following different exposures. Aggresome formation (Proteostat red staining) and nuclei staining (DAPI). (25X)
Viability/ Proliferation Assay
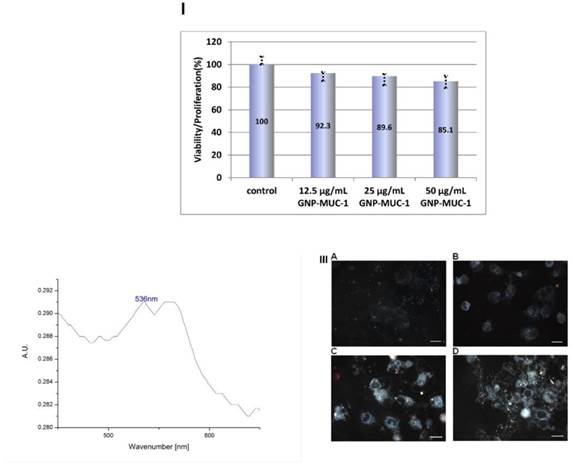
To test the cytotoxic effect of the newly designed nano-biocompund, the MTT assay was performed for all samples. The test allows the detection and quantification of viable cells by means of their ability to form insoluble formazan using oxidative processes. Limited reduction in formazan-containing, viable cells, were observed following exposure to nano-construct (Figure 5.I). Maximal reduction in viability of cell culture was obtained for 50μg/mL (100% viability in control, 85.1% viability for 50μg/mL), but the difference did not reach statistical significance. (p>0.05)
I. Viability/proliferation assay results. Cells were exposed to GNP-MUC-1 in respective concentration or cell culture media alone (control), for 6 hours, 37ᴼC, 5%CO2. Bars represent mean. Error bars represent standard error of mean. II. UV-Vis spectra of cell lysate following GNP-MUC-1 exposure (6 hrs. 50μg/mL GNP-MUC-1) III. Dark-field image at 40X magnification of peritoneal macrophages culture incubated with GNP-MUC-1 for 6 hrs. Cells have been subject to washing, fixing and immobilization before images capture with CytoViva system. Four panels: A) control group; B) cells exposed to 12.5μg/mL; C) cells exposed to 25μg/mL; D) cells exposed to 50 μg/mL. .Scale bars represent 10μM.
Nanoparticles internalization studies
In order to evaluate the presence and extent of internalization, dark-field images with hyperspectral acquisition was used. GNP-MUC-1 exposed cells demonstrated nanomaterial internalization in a dose-dependent manner, as one can observe in Figure 5.III. The particles tend to agglomerate and form intracellular aggregates, especially when exposed to elevated concentrations (25, 50 μg/mL). By contrast, the control sample presented negligible particulate light reflection. To further confirm the presence of intracellular GNP-MUC-1, UV-VIS spectroscopy was used. After 50μg/mL GNP-MUC-1 solution exposure and media removal, cell lysate was prepared.
The UV-Vis spectral analysis of the cell lysate is presented in the Figure 5.II. An absorption peak can be observed at λ=536nm which is consistent with surface plasmon resonance peak (SPR) of GNP-MUC-1. This corroborated with the Cytoviva images of the cells treated with GNP-MUC-1 confirms the successful internalization of the bio-nanocomposite.
Discussions
The active immunotherapy against neoplastic diseases using tumor associated antigens (TAAs) has made considerable progress lately, holding promise for ameliorating the outcome in cancer patients.14
Amongst professional antigen presenting cells (APCs), the macrophages, have recently revealed their distinct role and potential for active immunotherapy development. New data argue that a part of macrophages are able to activate CD8+ T cells in response to dead cell-associated antigens in lymph nodes and act by relocating antigen to dendritic cells (dc) localized in the spleen.15,16 Other authors have demonstrated that, likewise DCs, macrophages are able to present citrullinated peptides by means of autophagy.17
There have been comprehensive investigations of the use of nanoparticles as pharmaceutical drug carriers enhancing therapeutic agent potency with many of these carriers being successfully developed until now. The main cargo of nanocarriers in tumor immunotherapy are usually peptides or DNA encoding tumor-associated antigens (TAAs). TAAs are abnormal proteins aberrantly expressed by tumor cells, but which don not normally appear in normal tissues. Nowadays, most tumors are assumed to express TAAs, this being proved by multiple animal tumor reviews. The immune system can be stimulated to detect these TAAs as non- self, thus affecting specific anti-tumor responses. For immune therapy, there is a high need for novel TAA carriers that might both actively or passively target professional APCs, such as macrophages, thus generating a powerful tumor-specific cytotoxic CD8+ T lymphocyte (CTL) response.
We developed a novel nano-biosystem using GNPs functionalized with MUC-1 as immune activators to be used as anticancer vaccine. The reasoning behind this vaccine design was to combine the effect of gold nanoparticles as well as that of MUC-1 (two elements previously used as immune-active structures), therefore intending to outperform the efficiency of other previously used immune boosters. The newly designed structure was tested for efficiency as macrophage activator. To our best knowledge this is the first study to test the effect of a newly designed nano- biocompound on the complex antigen -processing apparatus of peritoneal macrophages.
The initial growth and viability testing demonstrates no significant toxic effects of GNP-MUC-1. Our results demonstrate the ability of GNP-MUC-1 administration to activate the peritoneal macrophages.
Golgi apparatus (GA) involvement in antigen processing was established by corresponding green fluorescence, related to nanomaterial exposure. Interestingly, the endoplasmic reticulum(ER) also presented strong red fluorescence capture with significantly reduced staining for elevated concentrations. Moreover, our results suggest the implication of proteasome machinery in antigen processing. Images collected using a red fluorescence proteasome staining suggests the involvement of ubiquitin-proteasome pathway in degradation of antigens. The regular exogenous (endocytic) antigen processing pathway usually follows the acquiring of major histocompatibility complex class II (MHC-II) within the Golgi apparatus region (trans-Golgi network) followed by membrane presentation. The process is followed by recognition by CD 4+ helper T lymphocytes. However, the pathway does not involve proteasome antigen degradation or intense involvement of rough ER. The respective compartments represent a distinctive part of the endogenous antigen processing pathway, characterized by major histocompatibility complex class I (MHC-I) and membrane presentation for further recognition by CD8+ T lymphocytes. Therefore we can reason that, in part, this might be the case of a cross-presentation of exogenous antigens. Similar to a process described by other researchers in dendritic cells, we believe that part of our nanomaterial escapes lysosomal degradation and is further translocated into the cytosol, following classical endogenous antigen degradation and presentation pathway.18 This escape might be facilitated by the recently demonstrated presence of ER- resident proteins in phagosomes structures.19 Also, evidences exist that in the case of murine macrophages, the phagosomes acquire the entire MHC-I loading complex as well as other important molecules for peptide processing and translocation.20 By generation of small sized peptides into proteasomes, they become suitable for TAP-mediated import into the endoplasmic reticulum and access the MHC-I pathway.21 Therefore, we can conclude that the designed nanomaterial can induce antigen processing and presentation by using a dual- trafficking route: exogenous and cross-presentation pathway.
To further depict the full spectrum of macrophages polarization following GNP-MUC-1 exposure, the concentration of IL-6, IL-10 and alpha-TNF was calculated. Although both IL-10 and Il-12 presented increasing levels with concentrations, the amplified IL-12 response as compared to IL-10, as well as increased alpha-TNF levels suggests a high M1/M2 ratio. The predominant M1 polarization of macrophages as a result of GNP-MUC-1 exposure represents an important finding, since it has already been demonstrated that M1 macrophages are important in efficient antigen presentation and tumor vaccine generation.22, 23 The macrophages manipulation and reprogramming for enhanced anti-tumor effect has been recently become an important research goal.24 We hereby present a novel nano- biosystem, namely gold nanoparticles functionalized with MUC-1 protein fragment that is able to induce macrophage activation, increased antigen presentation and predominant M1 polarization. Data presented may serve as a solid base for manipulation of peritoneal macrophages through administration of GNP-MUC-1 for anticancer immunoprophylaxy.
Conclusions
Our results may open new avenues for nanoparticle-based anti-cancer vaccine development, with special importance in adenocarcinomas such as colorectal, pancreatic or breast tumors.
Supplementary Material
Fig.S1 - Fig.S2.
Acknowledgements
This paper was published under the frame of European Social Fund, Human Resources Development Operational Programme 2007-2013, project no. POSDRU/159/1.5/S/138776. Also, we wish to acknowledge grant numbers PN-II-PT-PCCA-2011-3.1-1586 and PN II-ID-PCE-2012-4-0243.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mocan L, Ilie I, Tabaran FA. et al. Surface plasmon resonance-induced photoactivation of gold nanoparticles as mitochondria-targeted therapeutic agents for pancreatic cancer. Expert opinion on therapeutic targets. 2013:1-11
2. Iancu C, Ilie IR, Georgescu CE. et al. Applications of Nanomaterials in Cell Stem Therapies and the Onset of Nanomedicine. Particul Sci Technol. 2009;27(6):562-574
3. Nanda R, Chennamaneni P, Gibson J. et al. Abstract P2-16-21: A randomized phase I trial of nanoparticle albumin bound paclitaxel (nab-paclitaxel, Abraxane®) with or without mifepristone for advanced breast cancer. Cancer Res. 2013;73(24 Supplement):P2-16 -21-P2-16-21
4. Fuller S, Stewart S, Lebowitz M, Malhotra K, Ghanbari H. Inhibition of tumor growth in vivo by a nanoparticle-based therapeutic cancer vaccine targeting HAAH. Journal for immunotherapy of cancer. 2013;1(Suppl 1):P269
5. Hobo W, Novobrantseva TI, Fredrix H. et al. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunology, Immunotherapy. 2013;62(2):285-297
6. Liu S, Wei W, Yue H. et al. Nanoparticles-based multi-adjuvant whole cell tumor vaccine for cancer immunotherapy. Biomaterials. 2013;34(33):8291-8300
7. Fang RH, Hu CJ, Luk BT. et al. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano letters. 2014;14(4):2181-2188
8. Sehgal K, Ragheb R, Fahmy TM, Dhodapkar MV, Dhodapkar KM. Nanoparticle-mediated combinatorial targeting of multiple human dendritic cell (DC) subsets leads to enhanced T cell activation via IL-15-dependent DC crosstalk. J Immunol. 2014;193(5):2297-2305
9. Jeanbart L, Ballester M, de Titta A. et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol Res. 2014;2(5):436-447
10. Ahn S, Lee I, Kang S. et al. Gold Nanoparticles Displaying Tumor-Associated Self-Antigens as a Potential Vaccine for Cancer Immunotherapy. Advanced healthcare materials. 2014
11. Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nature Reviews Clinical Oncology. 2013
12. Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp. 2010(35):pii1488
13. Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Current protocols in immunology. 2008:14.1.1-14.1.14
14. Koido S, Enomoto Y, Apostolopoulos V, Gong J. Tumor regression by CD4 T-cells primed with dendritic/tumor fusion cell vaccines. Anticancer Res. 2014;34(8):3917-3924
15. Martinez-Pomares L, Gordon S. CD169 macrophages at the crossroads of antigen presentation. Trends Immunol. 2012;33(2):66-70
16. Asano K, Nabeyama A, Miyake Y. et al. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34(1):85-95
17. Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med. 2011;208(13):2625-2632
18. Rodriguez A, Regnault A, Kleijmeer M, Ricciardi-Castagnoli P, Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol. 1999;1(6):362-368
19. Garin J, Diez R, Kieffer S. et al. The phagosome proteome: insight into phagosome functions. J Cell Biol. 2001;152(1):165-180
20. Houde M, Bertholet S, Gagnon E. et al. Phagosomes are competent organelles for antigen cross-presentation. Nature. 2003;425(6956):402-406
21. Brode S, Macary PA. Cross-presentation: dendritic cells and macrophages bite off more than they can chew!. Immunology. 2004;112(3):345-351
22. Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174(2):636-645
23. Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. The Lancet. 2008;371(9614):771-783
24. Lyamina S, Kruglov S, Vedenikin TY. et al. Alternative Reprogramming of M1/M2 Phenotype of Mouse Peritoneal Macrophages In Vitro with Interferon-γ and Interleukin-4. Bull Exp Biol Med. 2012;152(4):548-551
Author contact
![]() Corresponding author: Lucian Mocan, email: lucian.mocanro
Corresponding author: Lucian Mocan, email: lucian.mocanro