Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(5):523-531. doi:10.7150/jca.13821 This issue Cite
Review
Immunochemical Assays and Nucleic-Acid Detection Techniques for Clinical Diagnosis of Prostate Cancer
Prosper Kanyong ![]() , Sean Rawlinson, James Davis
, Sean Rawlinson, James Davis
School of Engineering, Ulster University, Jordanstown, Northern Ireland, BT37 0QB.
Received 2015-9-11; Accepted 2015-12-8; Published 2016-2-10
Abstract
Prostate cancer (PCa) is a significant cause of morbidity and mortality and the most common cancer in men in Europe, North America, and some parts of Africa. The established methods for detecting PCa are normally based on tests using Prostate Specific Antigen (PSA) in blood, Prostate cancer antigen 3 (PCA3) in urine and tissue Alpha-methylacyl-CoA racemase (AMACR) as tumour markers in patient samples. Prior to the introduction of PSA in clinics, prostatic acid phosphatase (PAP) was the most widely used biomarker. An early diagnosis of PCa through the detection of these biomarkers requires the availability of simple, reliable, cost-effective and robust techniques. Immunoassays and nucleic acid detection techniques have experienced unprecedented growth in recent years and seem to be the most promising analytical tools. This growth has been driven in part by the surge in demand for near-patient-testing systems in clinical diagnosis. This article reviews immunochemical assays, and nucleic-acid detection techniques that have been used to clinically diagnose PCa.
Keywords: Prostate Cancer, Biopsy, Immunoassays, Clinical Diagnosis, PAP, PSA.
Introduction
Prostate cancer is the second most common cancer in men worldwide [1] and in 2012, there were approximately 758,000 new prostate cancer cases in developed countries - more than double that found in developing countries; this is a particularly worrying statistic considering that developed countries contain only 17% of the world's population [2]. The disease incidence varies more than 25-fold worldwide, but tends to be highest in Northern and Western Europe, Northern America, New Zealand and Australia. The greater incidence within these regions can be attributed, in part, to the greater awareness of the disease through the increasing use of prostate specific antigen (PSA) as a screening tool [1]. As a consequence, it is little surprise to find that when comparing incidence and mortality rates, the numbers of deaths from PCa are notably greater in less developed regions. It is widely recognised that mortality rates are higher in predominantly black populations such as sub-Saharan Africa and the Caribbean while remaining very low in Asia - especially in Southern and Central Asia [4-6]. At age 85, it has been estimated that the cumulative risk of developing PCa ranges widely from 0·5% to 20% worldwide [3, 7]. Irrespective of demographics, it universally accepted that the most critical point to achieve the best outcome in cancer treatment is to diagnose the disease at an early stage [8] and therefore the aims of the present review are to provide an overview of the diagnostic options currently being employed and to highlight and critically appraise new and emerging prognostic systems.
Prostate cancer is a particularly problematic case as it is not until the cancer is in the advanced stages that symptoms can manifest and there can be reluctance on the part of the patient to seek medical advice. Clinical assessment can also be fraught with a considerable ambiguity and many of the symptoms are not directly specific to prostate cancer and can often occur due to other non-cancerous conditions such as infection and inflammation or enlargement of the prostate. As such, there are considerable hurdle to early diagnosis and it is often the case that the cancer is not detected until further symptoms occur such as pain in the ribs, spine or hips arise indicating that the cancer has spread from the prostate to the bones [9]. There is a pressing need for objective strategies that enable early diagnosis not simply of the presence of cancerous prostate but rather tests that can offer prognostic assessments of the severity of the condition.
Despite this considerable burden, the exact aetiology and pathogenesis of PCa remains largely unexplained. Epidemiological studies suggest that environmental and lifestyle risk factors are determinants of disease risk [12]. Despite the fact that age, race and family history have long been recognised as risk factors for PCa, there has been no conclusive evidence for direct links to its development [12].
Diagnosis of prostate cancer
The clinical approach to the diagnosis of prostate cancer (PCa) has changed significantly over time. Prior to the introduction of PSA testing in the 1980s (Figure 1), PCa was diagnosed by assessing prostatic acid phosphatase (PAP) activity, digital rectal examination (DRE) or as an incidental histological finding in prostatic chips from transurethral sections of the prostate - usually performed for benign prostatic hyperplasia [4]. In the present era, most men in the UK who are diagnosed with PCa are asymptomatic. It is clinically detected by abnormalities in serum PSA level and/or DRE. The elevation of serum PSA and/or abnormal DRE subsequently prompts further investigation - typically transrectal ultrasound-guided prostate needle biopsy (TRUS), usually with procurement of a Gleason core [13]. Histopathological examination of needle core sections (biopsy) is presently the definitive procedure for the establishment of the diagnosis of PCa [4].
Schematic representation of PAP timeline (adapted from [27]).
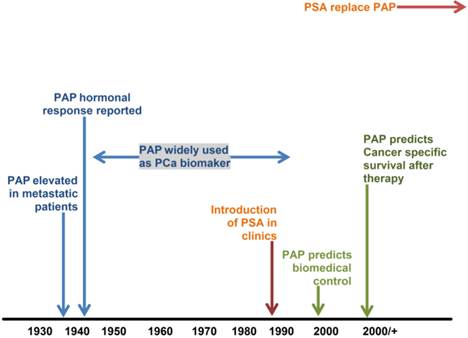
Currently, PSA, prostate health index (PHI), prostate cancer antigen 3 (PCA3) and alpha-methylacyl-CoA racemase (AMACR) are increasingly being utilised in the workup of difficult biopsies. This article provides a review of the immunochemical assays for PAP and PSA, and nucleic-acid detection techniques for PCA3 and AMACR that have been used to clinically diagnose PCa.
Detection of prostate cancer biomarkers in clinical samples
Prostatic Acid Phosphatase
Acid phosphatases are a group of tissue isoenzymes that can hydrolyse organic monophosphate esters and are found in red blood cells, kidney, lung, liver, platelets and osteoclasts. In healthy patients, the concentration of serum prostatic acid phosphatase (PAP) typically lies is in the range 8 - 16 μg/l [15] but can be over a hundredfold more abundant in the prostate than in other tissues [14]. Evaluation of PAP activity particularly in human serum became clinically relevant after it was discovered that its activity is not only high as a consequence of PCa [16, 17], but also increases with disease progression [18]. Consequently, tremendous effort was made in developing colorimetric enzymatic assays using various substrates such as p-nitrophenylphosphate and α-naphthylphosphate, inhibitors such as L-tartrate, or isoenzymes for PAP but these were only partly successful because the tests could not differentiate between prostatic and other tissue sources [19, 20]. Early methodological developments were devoted to distinguishing PAP from acid phosphatases originating in other tissues. To improve the assay specificity, Roy and colleagues in 1971 utilised sodium thymolphthalein monophosphate as a PAP selective substrate resulting in what is now considered to be the most prostate-specific of the enzymatic reactions [19]. The activity of PAP in the sera of 85 healthy individuals was found to be in the range of 0.56-110 U/l and when the sera of 87 patients (27 women with breast cancer, 23 patients with liver/biliary tract disease, 7 patients with kidney disease and 36 patients with cancer of non-prostatic, non-hepatic, non-mammary and non-renal origin) were analysed using their assay, there were no incidences of false positive results [19]. This clearly demonstrated that thymolphthalein monophosphate was the substrate least affected by non-prostatic acid phosphatases. However, the sensitivity of their method was only superior to other enzymatic assays when used to analyse serum where the activity of PAP was noticeably high [19]. Since these enzymatic assays were measuring PAP activity, any available inactive PAP could not be determined and a universal 'normal' serum PAP activity in healthy individuals could not be established because the substrates used were of varying specificity.
Given the clear deficiencies in the enzyme assays, the developments of immunoassays were pursued as a more specific measure of PAP [21]. In 1977, Foti and his colleagues developed one of the first PAP specific radioimmunoassays (RIA) which demonstrated only 5.5% (5/90) false positives and which was in marked contrast to the 26.6% (24:90) arising from the enzymatic assays [14]. Various antisera to PAP were successfully raised and used to develop other RIAs [14, 22-24]. A double-antibody RIA was used to measure PAP in serum and the incidence of false positive results further reduced to 3%, the normal values were found to be 2.69 ±1.8 μg/l and levels higher than 6.2 μg/l were found for prostatic cancer [25]. The RIAs offered extreme sensitivity, high specificity, quantifiable results and even the capability of determining 'inactivated' PAP. The RIA approach had numerous limitations: radioactive labels, complex instrumentation and a requirement for a high degree of user expertise. The tests were also time-consuming (48-hr duration) and clearly unsuitable as a screening technique. [14]. In contrast to RIA, another immunoassay for PAP known as counter-immunoelectrophoresis (CIEP) [15, 26] was favoured for its simplicity, higher sensitivity (as little as 1.0 ng of PAP could be detected in serum) and the short period (2 hrs) required to complete a test.
The measurement of serum 'prostatic' acid phosphatase (PAP) has been used in the diagnosis of PCa for over 45 years until it was replaced by PSA (Figure 1). However, recent work suggesting it plays a major role in the prognosis of intermediate and high-risk PCa has led to renewed interest in this biomarker [27]. This has resulted in the development of a barcode lateral immunochromatographic strip which could be used for rapid testing in clinical laboratories [28]. Combining PAP and PSA tests could further improve the diagnostic accuracy of PCa [27].
Prostate Specific Antigen (PSA)
PSA and its clinical relevance in prostate cancer
PSA is a member of the human kallikrein family, of which some of them are prostate specific [28,29]. It is known to play a vital role in the liquefaction of semen and exists in high concentrations in seminal fluid [30]. Consequently, it is an important biomolecule for normal human reproduction and exists in different molecular forms; namely, complexed PSA and free PSA within human sera [31]. PSA is a serine protease found in and secreted by the epithelial cells of the prostate (Figure 2). Normal secretory epithelial tissues which are surrounded by basal cells and a membrane releases proPSA into the lumen where it is cleaved by human kallikrein 2 (hK2) to generate active PSA [29]. Part of the active PSA can diffuse into the bloodstream where it is bound by protease inhibitors e.g. alpha1-antichymotrypsin (ACT) (bound PSA) while the rest can undergo proteolysis in the lumen to produce inactive PSA (this can enter the bloodstream and circulate as free PSA, fPSA). The remaining active PSA can also enter the bloodstream as fPSA. In PCa, the basal cells, basement membrane, and the normal architecture of the lumen is lost; thus, there is a decrease in the bioprocessing of proPSA to active PSA, and active PSA to inactive PSA. There is also an increase in the levels of bound PSA and proPSA in the bloodstream. In PCa there is a significant loss of the basal membrane.
Schematic representation of the biosynthesis of PSA in normal and cancerous epithelial tissues of the prostate (adapted from [29]).
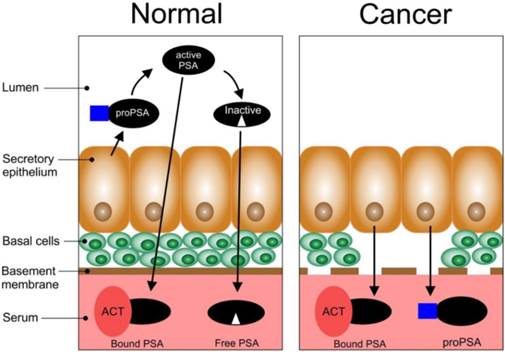
Studies in the early 1990s confirmed that serum PSA levels could be used to identify patients with prostate cancer [16, 32, 33]. The serum PSA is the most commonly used tumour biomarker for PCa and has a relevant role, despite possessing some considerable limitations in the detection, staging and post therapy monitoring of PCa [34, 35]. Currently, most men diagnosed with PCa have clinically impalpable carcinoma with elevated levels of serum PSA in the range of 2.5 -10.0 ng/ml. PSA is also detectable in the urine but this assay is regarded as having limited diagnostic value [4]. Unfortunately, elevated levels of serum PSA are not specific for histological diagnosis of PCa and it has been estimated that some 75% of men with elevated serum PSA are found not to have the disease but, rather another abnormality, benign prostatic hyperplasia (BPH) [4] or prostatic infarction and inflammation [36].
There is however a positive predictive value (PPV) of elevated serum PSA for the detection of PCa but this largely depends on the definition of an optimal upper limit of 'normal' PSA levels [34]. The PPV for histological diagnosis of PCa for serum PSA >4 ng/ml is ~31-51% but the rate of detection is 10% at 0.6-1.0 ng/ml; 17% at 1.1-2.0 ng/ml; 24% at 2.1-3.0 ng/ml and 27% at 3.1-4.0 ng/ml [37]. Additionally, about 13-25% of PCa detected at 4 ng/ml have high histological Gleason grades (a score ≥7). These data creates the challenge of setting lower limit cut-off points for 'normal' serum PSA levels. At high serum PSA levels (>10 ng/ml), the probability of disease detection is ~60% even though most men present with levels lower than 10 ng/ml. In an attempt to improve the PPV of serum PSA, variations in PSA assays have been extensively developed. These variations include proPSA, PSA doubling time (PSADT), age-specific PSA, PSA velocity (PSAV, rate of change of PSA levels over time), PSA density (PSAD, serum PSA ÷ ulstrasound volume of the prostate gland), free PSA (fPSA), percent free PSA (%fPSA), complexed PSA, and benign PSA (BPSA) [38, 39]. More recently, another isoform of proPSA, pro2PSA, was identified as the most PCa-specific biomarker [40].
PSAV >0.75 ng/ml per year is associated with the presence of PCa while a rise of >2 ng/ml per year is related with a more aggressive form of the cancer [41]. The percentage of fPSA is often used to counsel men whose total PSA ranges from 4 ng/ml to 10 ng/ml about their risk of PCa [34]. Total PSA (tPSA), which includes all types of PSA (whether free or bound to other proteins) circulating in the bloodstream, has also been used to screen for PCa [42, 43]. Sadly, there is no PSA value below which a man can be assured that he has no risk of PCa but professional organisations, such as the American Cancer Society, recommends that PSA testing be offered to men over 50 years. It also suggests that PSA testing could begin at a younger age (45 years) for high risk men, especially those with positive family history [44].
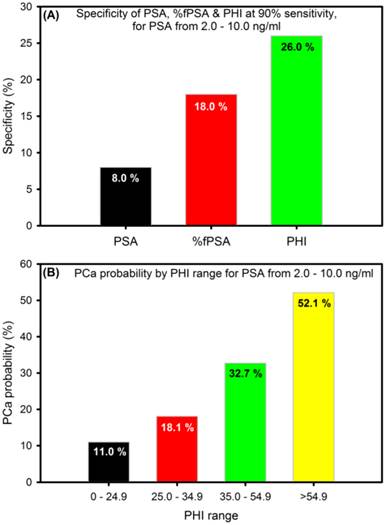
To enhance the specificity for early detection, a new mathematical formula (the Prostate Health Index, PHI), that combines tPSA, fPSA and pro2PSA into a single score that can be used to aid in clinical-decision making was developed in 2011. PHI has been approved in Europe, US and Australia [45] and is calculated using the following formula: (pro2PSA / fPSA) × √tPSA) [46]. PHI makes sense in that men with a higher tPSA and pro2PSA, and lower fPSA are more likely to have clinically significant PCa. It is a simple, non-invasive blood test that is shown to be a more accurate way to assess PCa risk than any other known PSA-based biomarker alone; thus, reducing unnecessary biopsies by ~26% especially for men with 2-10 ng/ml PSA values (Figure 3A). Higher PHI values are associated with increased probability of PCa (Figure 3B) and the inclusion of pro2PSA means that PHI preferentially detects more aggressive form of the disease [46].
The specificity and probability of prostate health index (PHI) (adapted from [46]).
Immunoassays for the detection of PSA
There are a range of commercially available assays currently used for PSA testing, the most popular being manufactured by Abbott Diagnostics, Bayer Diagnostics and Beckman Coulter. Although such systems are highly sensitive (possessing detection limits in the range of 0.005-0.05 ng/ml), they are very much a centralised option and Therapeutic Turnaround Time (TTAT) becomes a significant issue.
The TTAT is defined as the time between the physicians' stated need for the test and the therapeutic action taken on the test result [47-50] and in the case of PSA can take up to several weeks before the results are received by the physician. Early diagnosis of prostate cancer is crucial for the successful treatment of the disease [51]; therefore the long waiting time is not an ideal situation.
The development of point of care testing (POCT) for PSA has the potential to revolutionise the patients experience as results could be obtained within a matter of minutes, meaning fewer clinical visits will be needed and proffering the possibility of a better clinical outcome. A number of immunostrip (immunochromatographic membrane or lateral flow) tests have been developed in recent years to provide POCT for PSA. Immunostrip tests are popular because of their low cost, rapid result and ease of use. There are a number of commercially available PSA immunostrip tests: BioSign® PSA test by Princeton BioMeditech Corp and the SERATEC® PSA Semiquant test by Seratec Diagnostics, however they offer semi-quantitative diagnostics producing only a positive (PSA> 4ng/ml) or negative result. In order to replace centralised laboratory testing regimes, new POCTs need to be both rapid and quantitative. Two such devices commercially available are the PSAwatch™ and the FastPack® IP System. These immunoassay analysers are significantly less expensive than conventional autoanalyser systems and do not require any specialist knowledge or training to operate. Each device can produce a quantitative result with a detection range of 0.5 - 25 µg/L for the PSAwatch™ and 0.04 - 50 µg/L for the FastPack® IP System whereas most conventional autoanalyser systems have an upper detection limit in the region of 25 - 100 µg/L [48, 52]. Results can be produced from the immunoassay in approximately 15 minutes enabling the physicians to perform the test and make an informed decision on any further treatment within a single visit. Although these devices have started to pave the way for point of care (POC) PCa testing, an evidence review on POC PSA assays by the Centre for Evidence-Based Purchasing for the UK National Health Service concluded that none of the POC PSA assays tested (including the PSAwatch and FastPack® IP System) satisfied the criteria for acceptable performance and the POC PSA assays were deemed to have performed poorer that those in centralised laboratories [52].
Current status of PSA testing
Prostate cancer screening based on the use of the PSA test remains highly controversial since its introduction [53, 54]. The test was not initially intended to be a screening tool but as a way to measure treatments responses in men with the disease [54]. Far in advance of evidenced-based practice, the widespread utilisation of PSA screening globally, especially in Europe and USA, was in the first instance driven by the logical assumption that the earlier one detects a malignancy, the more likely treatment is to be curative and that attendant harms could be concurrently minimised. However, there is now a significant growing body of observational evidence that shows that there is a substantial burden of associated over-diagnosis and overtreatment of PCa from PSA screening tests [53-56]; this evidence suggests that screening for PCa using the PSA test confers a modest mortality benefits but at the cost of over-diagnosis and overtreatment. There have been varied responses to this by clinicians, researchers and medical societies and many of these have updated their recommendation statements. For example, the American Cancer Society now emphasises the need for informed decision-making as a prerequisite to PSA screening for PCa and that the test should not be automatic [44] while the American Urological Association (AUA) recommends against PSA screening in men under 40 or over 70 years. However, the AUA recommends PSA screening for men aged 55 to 69 years and recognises that the decision to undertake PSA screening should be individualised [57]. It should be mentioned that the scientist who initially discovered the PSA biomarker, Dr Richard J. Ablin, recently wrote that he has come to believe that the automatic and widespread use of PSA screening for men aged 50 years was “a public health disaster” [58]. In view of the above, clinicians would need to discuss the continued uncertainty of the overall value of the PSA test, particularly regarding the well documented potential harms resulting from over-diagnosis and overtreatment.
Detection of prostate cancer antigen 3
Prostate cancer antigen 3 (PCA3) was identified in 1999 as a gene that expresses a non-protein coding RNA [59]. It is only expressed in human prostate tissue, and is highly overexpressed in PCa. PCA3 expression is not correlated with prostatic volume and/or other prostatic disease such as prostatitis. Because of its restricted expression profile, the PCA3 mRNA is a very useful tumour marker [60-62]. Consequently, quantification of the gene expression is a promising tool that assists in PCa diagnosis. In contrast to surrogate tumour makers, including serum PSA, the PCA3 assay directly detects cancerous cells released into the urine following DRE. The assay (Figure 4) involves the purification of PCA3 and PSA mRNAs in urine samples through capture onto magnetic particles-coated with target-specific oligonucleotides and amplification of the captured targets using transcription-mediated amplification (TMA). The gene amplification products are then detected using a chemiluminescent DNA probe via a hybridisation protection assay [62]. The final output of the test (PCA3 score) is usually a ratio ([PCA3 mRNA/PSA mRNA] x 1000); this measures the degree to which PCA3 is overexpressed in the urine.
The PCA3 test assay and score.
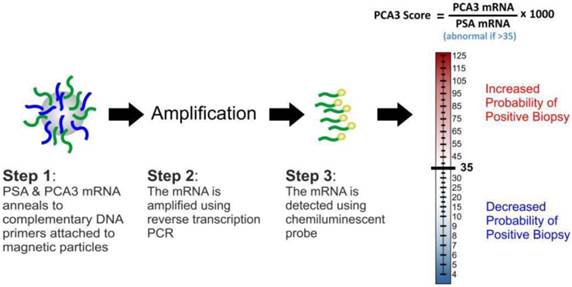
The diagnostic value of the PCA3 score in patients demonstrated a sensitivity of 46-82%, specificity of 56-89%, PPV of 59-97% and negative predictive value (NPV) of 87-98% [63-66]. Such variability can be explained by the fact that different inclusion criteria, cut-off values and protocols were used in these studies. However, the PCA3 score at cut-off of 35 tend to provide an optimal balance between sensitivity and specificity.
Prior to this, a time-resolved fluorescence (TRF) reverse transcription-polymerase chain reaction (RT-PCR) method was used to quantify PCA3 and PSA mRNAs in urine sediments [67]. The analytical performance of the PCA3 assays have been extensively characterised [65-70] and the test is emerging as the first fully translated PCa diagnostic assay [71].
Analysis at the mRNA level has shown to be difficult because of the limited sensitivity of most of the more traditional time-consuming techniques such as hybridisation protection assays (RPAs) [72] and RT-PCR [72, 73]. Thus, trained personnel as well as specialised laboratories are required before the PCA3 test can be performed. The assay usually takes about 7 days to be completed; thus, rendering it unsuitable for point-of-care applications [73]. Theoretically, RT-PCR can amplify a single nucleic acid sequence a million-fold but optimisation of primer sets tend to prolong the assay time and different genes in equal amounts in a starting mRNA mixture may not be equally amplified because of selective and non-linear target amplification [74]. These limitations affect the quality of the amplification data; thus, distorting the information on the gene expression. Consequently, it is imperative that rapid progress is made towards the development of sensitive, reliable and rapid gene detection assays for near-patient clinical diagnostics.
Detection of Alpha-methylacyl-CoA racemase (AMACR)
Alpha-methylacyl-CoA racemase (AMACR) is a peroxisomal and mitochondrial enzyme that plays a vital role in the biosynthesis of bile acid [75] and has been identified as a gene that is significantly expressed in PCa relative to benign prostate epithelium [76-79]. Enormous effort has been made towards early detection of PCa through mass screening and this has resulted in the discovery of an increasing number of small foci of cancers on needle biopsy specimens [80-82]. The diagnosis of these small foci of PCa in biopsy specimens is a major diagnostic challenge because these small lesions are either under or over-diagnosed; causing unfortunate consequences for patients. It has been shown that using AMACR as a positive biomarker in conjunction with other PCa tumour makers can help confirm PCa diagnosis [83-85]. Using AMACR alone as a positive biomarker could be misleading because expressions of AMACR have been seen in benign glands, high grade prostatic intraepithelial neoplasia (PIN) [78, 79, 86] and adenomatous hyperplasia [87].
Different kinds of techniques have been used to measure the expression of AMACR including Western blotting, immunohistochemistry, cDNA expression arrays and RT-PCR [89-92]. The main drawback of immunohistochemcial staining techniques for small lesions of cancer is that atypical glands could disappear on further sectioning. More recently, a fluorescent-based assay was used to measure the biomarker in tissue samples [93]. Therefore, the development of simple and accurate nucleic-acid assays is needed to help identify small foci of cancers to better support clinical decisions.
Multipanel Biomarker Arrays
The use of a single biomarker is clearly of little use and as mentioned in the previous sections there is a continuing demand for diagnostic systems that offer a much more refined clinical picture and a greater opportunity for personalised medicine [94, 95 96]. While multiparametric testing can be achieved through sequential autoanalyser assays, there is, at present, very little available as a POCT within the clinic. There is however an increasing effort to harness the use of microfluidic processing as the first steps in the development of systems that can assay a range of biomarkers.
There have been several recently commercialised systems enabling automated or semi-automated analysis for multi-protein determination and typically employ fluorescence (Luminex corp.; Rules Based Medicine Inc.) electrochemiluminescence (ECL) (Roche Diagnostics; Meso Scale Discovery), and surface plasmon resonance (Horiba Inc.; BIO-RAD) measurement technologies. These systems offer panels of up to 10 proteins per sample with detections in the region of 1-100 pg/ml (Rusling et al. 2010; Quansys Biosciences) [97]. These systems are still prohibitively expensive for decentralised POCT use involving both high capital outlay and high cost per sample. Moreover, the nature of their addition still requires a degree of expertise that is unlikely to be found within a general clinic setting.
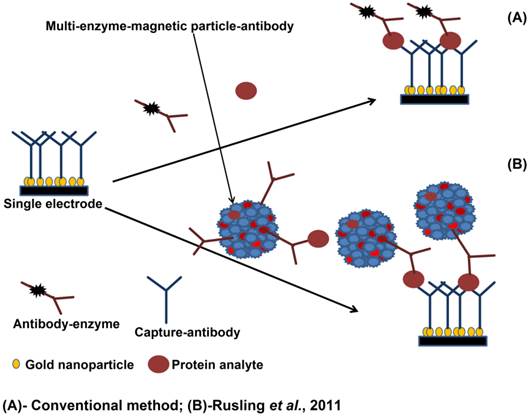
Rusling and coworkers however have pioneered the use of small foot print microarray technologies that can capture multiple proteins and have advanced the microfluidic processing to reduce the sample cost and the technical overhead associated with the autoanalyser and multiprotein chips, The system uses a combination of magnetic particles and enzyme labels (detailed in Figure 5) to provide a highly sensitive detection option[98-100] for the detection of PSA, prostate specific membrane antigen (PSMA), platelet factor-4 (PF-4), and interlukin-6 (IL-6).
Multi-enzyme magnetic particle-labelled antibody detection strategies for the detection of serum biomarkers: (A) immunosensor after incubating with protein analyte in a flowing conventional single-enzyme labelled antibody; (B) immunosensor after offline capture of protein analyte with heavily labelled antibody and enzyme. The enzyme-antibody-protein analyte is trapped by capture antibodies (adapted from [98]).
The system has been adapted for use with a number of detection technologies, electroanalytical[99] and electrochemiluminescent (ECL)[100] measurements and while these approaches are still very much at the lab bench prototype stage - they nevertheless serve as potent indicators of future diagnostic systems that could eventually make transition to the clinic and offer a more viable POCT option.
Conclusion
There is little debate over the need for routine measurement of PCa tumour markers and the availability of simple, rapid and low-cost diagnostics. Presently, clinical diagnosis of the disease involves a combination of blood (PSA), urine (PCA3) and tissue (AMACR) based tests as well as histopathological examination of needle sections. Immunochemical assays and nucleic acid detection techniques are the ideal approaches for measuring these biomarkers in biological fluids. But most of the traditional nucleic acid detection techniques at the mRNA level are relatively low in sensitivity, time-consuming, labourious and demands high technical skills. Therefore, it is imperative that rapid progress is made towards the development of simple, accurate and low-cost platforms for near-patient testing of these tumour biomarkers.
Acknowledgements
The authors would like to thank the Department of Employment and Learning Ireland (Grant No.: USI035) and the National Institutes of Health (Grant No.: 5R01ES003154-30) for the funding.
Conflict of interest
The authors declare no conflict of interest.
References
1. Ferlay J, Shin HR, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893-917
2. Population Reference Bureau. World Population Data Sheet in 2014.
3. GLOBOCAN. Prostate cancer, estimated incidence, mortality and prevalence worldwide in 2012. International Agency for Research on Cancer (World Health Organisation).
4. Humprey PA, Androiole G L. Prostate cancer diagnosis. Mo Med. 2010;107:107-12
5. Parkin DM, Whelan SL, Ferlay J, Teppo L, Thomas DB. Cancer incidence in five continents. International Agency for Research on Cancer (World Health Organisation). 1997
6. Marugame T, Katanoda K. International comparisons of cumulative risk of breast and prostate cancer, from cancer incidence in five continents Vol. VIII. Jpn J Clin Oncol. 2006;36:399-400
7. Parkin DM, Bray FI, Devesa SS. Cancer burden in the year 2000: the global picture. Eur J Cancer. 2001;37:4-66
8. Altintas Z, Tothill I. Biomarkers and biosensors for the early diagnosis of lung cancer. Sensors Actuators B Chem. 2013;188:988-98
9. American Cancer Society. Cancer Facts & Figures in 2010.
10. Groenberg H. Prostate cancer epidemiology. Lancet. 2003;361:859-64
11. Cancer Research UK. Prostate Cancer Statistics in 2015.
12. Eeles R, Goh C, Castro E, Bancroft E, Guy M, Al Olama AA, Easton D, Kote-Jarai Z. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol. 2014;11:18-31
13. Cancer Research UK. Factors in deciding treatment for prostate cancer. 2015.
14. Foti AG, Herschman H, Cooper JF. Comparison of human prostatic acid phosphatase by measurement of enzymatic activity and by radioimmunoassay. Clin Chem. 1997;23:95-9
15. McDonald I, Rose NR, Pontes EJ, Choe BK. Human prostatic acid phosphatases: III. Counterimmunoelectrophoresis for rapid identification. Arch Androl. 1978;1:325-29
16. Gutman AB, Gutman EB. An “acid” phosphatase occurring in the serum of patients with metastasizing carcinoma of the prostate gland. J Clin Invest. 1938;17:473-78
17. Gutman EB, Sproul EE, Gutman AB. Significance of increased phosphatase activity of bone at the site of osteoplastic metastases secondary to carcinoma of the prostate gland. Am J Cancer. 1936;28:485-95
18. Murphy GP, Reynoso G, Kenny GM, Gaeta JF. Comparison of total and prostatic fraction serum acid phosphatase levels in patients with differentiated and undifferentiated prostatic carcinoma. Cancer. 1969;23:1309-14
19. Roy AV, Brower ME, Hayden JE. Sodium thymolphathlein monophosphate: a new acid phosphatase substrate with greater specificity for the prostatic enzyme in serum. Clin Chem. 1971;17:1093-102
20. Yam LT. Clinical significance of the human acid phosphatase: A review. Am J Med. 1974;56:604-16
21. Milisauskas V, Rose NR. Immunochemical quantification of prostatic phosphatase. Clin Chem. 1972;18:1529-31
22. Griffiths J. Prostate-specific acid phosphatase: re-evaluation of radioimmunoassay. Clin Chem. 1980;26:433-36
23. Quinones G, Rohner T, Drago J, Demers L. Will prostatic acid phosphatase determinations by radioimmunoassay increase the diagnosis of early prostatic cancer? J Urol. 1981;125:361-64
24. Bruce AW, Mahan DE, Belville WD. The role of the radioimmunoassay for prostatic acid phosphatase in prostatic carcinoma. Urol Clin North Am. 1980;7:645-52
25. Choe BK, Pontes EJ, Morrison MK, Rose NR. Human prostatic acid phosphatase. II. A double-antibody radioimmunoassay. Arch Androl. 1978;1:227-33
26. Romas NA, Hsu KC, Tomashefsky P, Tannenbaum M. Counterimmunoelectrophoresis for detection of human prostatic acid phosphatase. Urology. 1978;12:79-83
27. Taira A, Merrick G, Wallner K, Dattoli M. Reviving the acid phosphatase test for prostate cancer. Oncology. 2007;21:1003-10
28. Fang C, Chen Z, Li L, Xia J. Barcode lateral flow immunochromatographic strip for prostate acid phosphatase determination. J Pharm Biomed Anal. 2011;56:1035-40
29. Balk SP, Ko YJ, Bubley G J. Biology of prostate-specific antigen. J Clin Oncol. 2003;15:383-91
30. Lilja H, Oldbring J, Rannevik G, Laurell CB. Seminal-secreted proteins and their reactions during gelation and liquefaction of human semen. J Clin Invest. 1987;80:281-85
31. Lilja H. Significance of different molecular forms of serum PSA: the free, noncomplexed form of PSA versus that complexed to alpha-1-antichymotrypsin. Urol Clin North Am. 1993;20:681-86
32. Catalona WJ, Smith DS, Ratliff TL, Dodds KM, Coplen DE, Yuan JJJ, Petros JA, Andriole GL. Measurement of prostate-specific antigen in serum as a screening test for prostate cancer. N Engl J Med. 1991;324:1156-61
33. Brawer MK, Chetner MP, Beatie J, Buchner DM, Vessella RL, Lange PH. Screening for prostatic carcinoma with prostate specific antigen. J Urol. 1992;147:841-5
34. Gretzer MB, Partin AW. Prostate cancer tumour markers. In: (ed.) Wein AJ, Kavoussi LR, Novick AC, Partin AW, Peters CA. Campbell-Walsh Urology, vol 27. Philadelphia: Saunders Elsevier. 2007:2896-2911
35. Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer. 2008;8:268-78
36. Ornstein DK, Smith DS, Humphrey PA, Catalona WJ. The effect of prostate volume, age, total prostate specific antigen level and acute inflammation on the percent free serum prostate specific antigen levels in men without clinically detectable prostate cancer. J Urol. 1998;159:1234-37
37. Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL. et al. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0 ng per millilitre. N Engl J Med. 2004;350:2239-46
38. D'Amico AV, Chen MH, Roehl KA, Catalona WJ. Preoperative PSA velocity and the risk of death from prostate cancer after radical prostatectomy. N Engl J Med. 2004;351:125-35
39. Catalona WJ, Partin AW, Slawin KM, Brawer MK, Flanigan RC, Patel A. et al. A multi-centre clinical trial evaluation of free PSA in the differentiation of prostate cancer from benign disease. J Am Med Assoc. 1998;279:1542-7
40. Mikolajczyk SD, Millar LS, Wang TJ, Rittenshouse HG, Marks LS, Song W. et al. A precursor form of prostate-specific antigen is more highly elevated in prostate cancer compared with benign transition zone prostate tissue. Cancer Res. 2000;60:756-9
41. Oesterling JE, Jacobsen SJ, Chute CG, Guess HA, Girman CJ, Panser LA. et al. Serum prostate- specific antigen in a community-based population of healthy men: establishment of age-specific reference ranges. J Am Med Assoc. 1993;270:860-4
42. Andriole GL, Crawford ED, Grubb RL, Buys SS, Chia D, Church TR. et al. Mortality results from a randomised prostate-cancer screening trial. N Engl J Med. 2009;360:1310-9
43. Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V. et al. For the ERSPC Investigators, Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320-8
44. American Cancer Society Guidelines. Finding cancer early. 2015.
45. Loeb S, Sokoll L, Broyles D, Bangma CH, van Schaik RH, Klee GG. et al. Prospective multicenter evaluation of the Beckman Coulter Prostate Health Index using WHO calibration. J Urol. 2013;189:1702-6
46. Catalona WJ, Partin AW, Sanda MG, Wei JT, Klee GG, Bangma CH. et al. A multi-centre study of [-2]Pro-Prostate-specific antigen (PSA) in combination with PSA and free PSA for prostate cancer detection in the 2.0 to 10.0 ng/ml PSA range. J Urol. 2011;185:1650-5
47. Hicks JM, Haeckel R, Price CP, Lewandrowski K, Wu AHB. Recommendations and opinions for the use of point-of-care testing for hospitals and primary care: summary of a 1999 symposium. Clin Chim Acta. 2001;303:1-17
48. Healy DA, Hayes CJ, Leonard P, McKenna L, O'Kennedy R. Biosensor developments: application to prostate-specific antigen detection. Trends Biotechnol. 2007;25:125-31
49. Seamonds B. Medical, economic, and regulatory factors affecting point-of-care testing. A report of the conference on factors affecting point-of-care testing, Philadelphia, PA 6-7 May 1994. Clin Chim Acta. 1996;249:1-19
50. Fermann GJ, Suyama J. Point of care testing in the emergency department. J Emerg Med. 2002;22:393-404
51. Crawford ED, DeAntoni EP, Etzioni R, Schaefer VC, Olson RM, Ross CA. Serum prostate-specific antigen and digital rectal examination for early detection of prostate cancer in a national community-based program. The Prostate Cancer Education Council. Urology. 1996;47:863-9
52. National Health Service UK. Evidence review of point-of-care total PSA assays. 2010.
53. Hayes JH, Barry MJ. Screening for prostate cancer with the prostate specific antigen test: a review of current evidence. JAMA. 2014;311:1143-9
54. Crosswell JM, Kramer BS, Crawford ED. Screening for prostate cancer with PSA testing: current status and future directions. Oncology (Williston Park). 2011;25(6):452-60
55. Kilpelainen TP, Tammela TL, Maatanen L. et al. false-positive screening results in the Finnish prostate cancer screening trial. Br J Cancer. 2010;102:469-74
56. Bangma CH, van Schaik RH, Blijenber BG. et al. on the use of prostate-specific antigen for screening of prostate cancer in European Randomised study for screening prostate cancer. Eur J Cancer. 2010;46(17):3109-19
57. Carter HB, Albertsen PC, Barry MJ, Etzioni R, Freedland SJ, Greene KL. et al. Early detection of Prostate cancer: AUA guideline. Am Urol Assoc. 2013:1-28 https://www.auanet.org/education/guidelines/prostate-cancer-detection.cfm
58. Albin RJ. The great prostate mistake. New York Times. March 9. 2010 http://www.nytimes.com/2010/03/10/opinion/10Ablin.html?_r=0
59. Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA. et al. DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999;59:5975-9
60. Hessels D, Verhaegh GW, Schalken JA, Witjes JA. Applicability of biomarkers in the early diagnosis of prostate cancer. Expert Rev Mol Diagn. 2004;4:513-26
61. de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW. et al. DD3 (PCA3), a very sensitive and specific marker to detect prostate tumours. Cancer Res. 2002;62:2695-8
62. Hessels D, Gunnewiek KJM, van Oort I, Karthaus HF, van Leenders GJ, van Balken B. et al. DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur Urol. 2003;44:8-15
63. Ruiz-Aragon J, Marquez-Pelaez S. Assessment of the PCA3 test for prostate cancer diagnosis. A systematic review and meta-analysis. Actas Urol Esp. 2010;34:346-55
64. Marks LS, Fradet Y, Deras IL, Blase A, Mathis J, Aubin SM. et al. PCA3 molecular urine assay for prostate cancer in men undergoing repeat biopsy. Urology. 2007;69:532-5
65. Deras IL, Aubin SM, Blase A, Day JR, Koo S, Partin AW. et al. PCA3: a molecular urine assay for predicting prostate biopsy outcome. J Urol. 2008;179:1587-92
66. Haese A, de la Taille A, van Poppel H, Marberger M, Stenzl A, Mulders PF. et al. Clinical utility of the PCA3 urine assay in European men scheduled for repeat biopsy. Eur Urol. 2008;54:1081-8
67. van Gils MP, Hessels D, van Hooij O, Jannink SA, Peelen WP, Hanssen SL. et al. The time resolved fluorescence-based PCA3 test on urinary sediments after digital rectal examination; a Dutch multicentre validation of the diagnostic performance. Clin Cancer Res. 2007;13:939-43
68. Groskopf J, Aubin SM, Deras IL, Blase A, Bodrug S, Clark C. et al. APTIMA PCA3 molecular urine test: development of a method to aid in the diagnosis of prostate cancer. Clin Chem. 2006;52:1089-95
69. Sokoll LJ, Ellis W, Lange P, Noteboom J, Elliott DJ, Deras IL. et al. A multicenter evaluation of the PCA3 molecular urine test: pre-analytical effects, analytical performance, and diagnostic accuracy. Clin Chim Acta. 2008;389:1-6
70. Nakanishi H, Groskopf J, Fritsche HA, Bhadkamkar V, Blasé A, Kumar SV. et al. PCA3 molecular urine assay correlates with prostate cancer tumour volume: implication in selecting candidates for active surveillance. J Urol. 2008;179:1804-9
71. Schalken JA. Towards early and more specific diagnosis of prostate cancer? Beyond PSA: New biomarkers ready for prime time. Eur Assoc Urol. 2009;8:97-102
72. Cottrez F, Auriault C, Capron A, Groux H. Quantitative PCR: validation of the use of a multispecific internal control. Nucleic Acids Res. 1994;22:2712-3
73. Totze G, Sachinidis A, Vettre H, Ko Y. Competitive reverse transcription/polymerase chain reaction for the quantification of p53 and mdm2 mRNA expression. Mol Cell Probes. 1996;10:427-33
74. Schalken J, Tombal B, Van Poppel H. Prostate cancer antigen 3. Ismar Healthcare. 2015
75. Baugh R, Hill AA, Brown EL, Hunter CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res. 2001;29:E29
76. Ferdinandusse S, Denis S, IJIst L, Dacremont G, Waterham HR, Wanders RJ. Subcellular localization and physiological role of a-methylacyl-CoA racemase. J Lipid Res. 2000;41:1890-6
77. Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG. et al. Alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. J Am Med Assoc. 2002;287:1662-70
78. Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ. et al. Alpha-Methylacyl-CoA racemase: a new molecular marker for prostate cancer. Cancer Res. 2002;62:2220-6
79. Xu J, Stolk JA, Zhang X, Silva SJ, Houghton RL, Matsumura M. et al. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer Res. 2000;60:1677-82
80. Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427-33
81. DiGiuseppe JA, Sauvageot J, Epstein JI. Increasing incidence of minimal residual cancer in radical prostatectomy specimens. Am J Surg Pathol. 1997;21:174-8
82. Epstein JI. Diagnostic criteria of limited adenocarcinoma of the prostate on needle biopsy. Hum Pathol. 1995;26:223-9
83. Epstein JI, Potter SR. The pathological interpretation and significance of prostate needle biopsy findings: implications and current controversies. J Urol. 2001;166:402-10
84. Jiang Z, Wu CL, Woda BA, Dresser K, Xu J, Fanger GR. et al. P504S/alpha-methylacyl-CoA racemase: a useful marker for diagnosis of small foci of prostatic carcinoma on needle biopsy. Am J Surg Pathol. 2002;26:1169-74
85. Beach R, Gown AM, De Peralta-Venturina MN, Folpe AL, Yaziji H, Salles PG. et al. P504S immunohistochemical detection in 405 prostatic specimens including 376 18-gauge needle biopsies. Am J Surg Pathol. 2002;26:1588-96
86. Magi-Galluzzi C, Luo J, Isaacs WB, Hicks JL, de Marzo AM, Epstein JL. Alpha-methylacyl-CoA racemase: a variably sensitive immunohistochemical marker for the diagnosis of small prostate cancer foci on needle biopsy. Am J Surg Pathol. 2003;27:1128-33
87. Jiang Z, Woda BA, Rock KL, Xu Y, Savas L, Khan A. et al. P504S: a new molecular marker for the detection of prostate carcinoma. Am J Surg Pathol. 2001;25:1397-404
88. Yang XJ, Wu CL, Woda BA, Dresser K, Tretiakova M, Fanger GR. et al. Expression of alphamethylacyl-CoA racemase (P504S) in atypical adenomatous hyperplasia of the prostate. Am J Surg Pathol. 2002;26:921-5
89. Jiang Z, Li C, Fischer A, Dresser K, Woda BA. Using an MACR(P504S)/34Βe12/p63 cocktail for the detection of small focal prostate carcinoma in needle biopsy specimens. Amer J Clin Path. 2005;123:231-6
90. Rubin MA, Bismar TA, Andren O, Mucci L, Kim R, Shen R. et al. Decreased α-methylacyl CoA racemase expression in localised prostate cancer is associated with an increased rate of biochemical recurrence and cancer-specific death. Cancer Epidemiol Biomarkers Prev. 2005;14:1424-32
91. Kuefer R, Varambally S, Zhou M, Lucas PC, Loeffler M, Wolter H. et al. Alpah-Methylacyl-CoA racemase: expression levels of this novel cancer biomarker depend on tumour differentiation. Am J Pathol. 2002;161:841-8
92. Kumaresan K, Kaklar N, Verma A, Mandal AK, Singh SK, Joshi K. Diagnostic utility of α-methylacyl CoA racemase (P504S) and HMWCK in morphologically difficult prostate cancer. Diagnos Path. 2010;22:83
93. Rubin MA, Zerkowski MP, Camp RL, Kuefer R, Hofer MD, Chinnaiyan AM. et al. Quantitative determination of expression of the prostate cancer protein a-methylacyl-CoA racemase using automated quantitative analysis (AQUA): a novel paradigm for automated and continuous biomarker measurements. Am J Pathol. 2004;164:831-40
94. Ferrari M, Cancer nanotechnology. opportunities and challenges. Nat Rev Cancer. 2005;5:161-71
95. Kingsmore SF. Multiplexed protein measurement: technologies and applications of protein and antibody arrays. Nat Rev Drug Discov. 2006;5:310-20
96. Ludwig JA, Weinstein JN. Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer. 2005;5:845-56
97. Rusling JR, Kumar VC, Gutkind JS, Patel V. Measurement of biomarker proteins for point-of-care early detection and monitoring of cancer. Analyst. 2010;135:2496-511
98. Mani V, Chikkaveeraiah BV, Patel V, Gutkind JS, Rusling JF. Ultrasensitive immunosensor for cancer biomarker proteins using gold nanoparticle film electrodes and Multienzyme-particle amplification. ACS Nano. 2009;3:585-94
99. Chikkaveeraiah BV, Mani V Patel V, Gutkind JS Rusling JF. Microfluidic electrochemical immunoarray for ultrasensitive detection of two cancer biomarker proteins in serum. Biosen Bioelectron. 2011;26:4477-83
100. Sardesai NP, Kadimisetty K, Faria R, Rusling JF. A microfluidic electrochemiluminscent device for detecting cancer biomarker proteins. Anal Bioanal Chem. 2013;405:3831-8
Author contact
![]() Corresponding author: p.kanyongac.uk.
Corresponding author: p.kanyongac.uk.