Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(14):1926-1938. doi:10.7150/jca.15603 This issue Cite
Research Paper
Selection, Analysis and Improvement of Anti-Angiogenesis Compounds Identified by an Anti-HIF-1α Screening and Validation System
Chikezie Madu, Liyuan Li, Yi Lu ![]()
Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Memphis, TN 38016, USA.
Received 2016-3-21; Accepted 2016-5-23; Published 2016-9-27
Abstract
Cancer cells resort to activating hypoxia-inducible factor-1 (HIF-1) as one of several responses to hypoxic conditions. Overexpression of HIF-1, the transcriptional regulator for a group of malignant-pathway related genes including vascular endothelial growth factor (VEGF), is associated with increased tumor growth, vascularization, and metastasis. HIF-1 is composed of an inducible subunit, HIF-1α and a constitutively expressed subunit, HIF-1ß. HIF-1 activity is mainly dependent on the level of HIF-1α protein, the inducible and regulatory subunit of the HIF-1 heterodimer complex; thus, identification of novel anti-HIF-1α agents will lead to effective blockage of the HIF-1 (HIF-1α)-mediated “switch-on” function for those malignant-pathway related genes and suppression of the HIF-1α/VEGF-mediated signaling pathway that promotes cancer progression and metastasis.
While there is an extremely large number of small molecule compounds in the database (compound libraries), the currently existing screening system is inefficient and time-consuming; or, at best, the application of the existing screening system is very limited as it is usually not coupled with biological validation processes. The further development of potential drugs is partly hindered due to the cumbersome steps in between the primary screen and consequent validation: the slow, exhausted and sometimes lack of a linked biological validation process contributes to the dismal fate of scant compounds uncovered in the primary screen.
To improve upon the status quo, we developed a prototype screening system that is coupled anti-HIF-1α primary screen with secondary anti-VEGF/anti-angiogenesis validation screens. We used breast cancer cells as the model to select potent anti-HIF-1α small-molecule compounds by their abilities to inhibit transactivation of a VEGF promoter fused to a luciferase reporter gene under hypoxia. Positive compounds were then validated by a series of assays that confirm compounds' anti-HIF-1α activities including measurement of their effects on HIF-1α downstream VEGF gene expression and angiogenic ability of breast cancer cells. Moreover, we demonstrated that we could further improve the compound's potency of anti-HIF-1α and anti-angiogenesis by modifying the identified lead to synthesize a superior (novel) drug.
Keywords: hypoxia-inducible factor-1, anti-angiogenesis
Introduction
Regions of hypoxia commonly develop within many solid tumors including breast cancer (BCa), due to the fact that the rate of oxygen consumption by these tumors often surpasses the supply. The degree of tumor hypoxia plays a profound role in determining the biological behavior, response to therapy and prognosis of many types of human cancers (1). Cancer cells respond to this hypoxic condition by activating a number of cellular pathways that foster continuous growth, specifically through the increase in glycolysis, proliferation, and angiogenesis (2). One of the critical steps that cancer cells resort to in achieving this goal is the activation of the transcription factor HIF-1 (3). Activation of the HIF-1 transcriptional complex represents the primary molecular mechanism by which oxygen regulates gene expression. In general, HIF-1 activation promotes hypoxic adaptation and survival by increasing oxygen delivery, decreasing oxygen consumption, expressing growth factors for autocrine signaling, suppressing cell death, and promoting angiogenesis (34) and metastasis (4,5).
HIF, the master transcriptional factor of cells responding to hypoxia, is a heterodimer composed of one of the three alpha subunits (HIF-1α, HIF-2α or HIF-3α) and one constitutively expressed HIF-1ß subunit. In the cancer setting, at least in the case for hypoxia induction, HIF-1α was found to play a predominant role in regulating the transcriptional targets of HIF-1 (6,7). Once translated in the cytoplasm, under normal oxygen conditions, the HIF-1α protein becomes hydroxylated and ubiquinated, leading them to proteasomal degradation. Under hypoxic conditions, HIF-1α is stable and translocates to the nucleus, where it forms a transcriptionally active heterodimer complex (HIF-1) with the HIF-1β subunit. HIF-1 then binds to a consensus DNA-binding site (hypoxia-response element or HRE) of the target genes including VEGF and transcribes/regulates the gene expression (8). Thus, the biologic activity of HIF-1 depends on the presence of HIF-1α (9,10), the inducible and regulatory subunit. As the master regulator of the hypoxic transcriptional response, HIF-1 plays a central role in tumor growth and angiogenesis (1,4). The level of available HIF-1α directly affects VEGF activity and its mediated tumor angiogenesis and malignant progression (11). For example, HIF-1α expression has been reported to contribute to angiogenesis in xenograft tumors and metastases of human BCa cells to the bone (36,38), one of the most common sites of metastases in BCa. Overexpression of HIF-1α has been reported in many primary human cancers and their metastases. Clinical studies indicate that HIF-1α protein expression correlates not only with tumor aggressiveness, increased vascularity, metastasis, and advanced disease stages, but also with treatment resistance to chemotherapy and radiotherapy, and poor prognosis in cancer patients (2,3,5,13,14,16). Therefore, the discovery of agents that target and inhibit the activity of HIF-1α should provide an effective strategy to suppress tumor growth, angiogenesis, and metastasis, as well as improve existing cancer treatments.
The aim of this study was to establish a prototype system (that can later be developed into a high-throughput screening system with automated validation) that can identify novel anticancer compounds based on anti-HIF-1α biochemical activity and anti-angiogenesis biological validation. This system is capable of rapidly screening and selecting potential leads from a large number of small molecule compounds. The method employs human breast cancer cells expressing VEGF promoter-luciferase reporter gene treated with or without candidate compounds under hypoxia incubation. The compounds that exhibit the strongest anti-HIF-1α activity (i.e., lowest luciferase readout) would be further screened and validated by their abilities to inhibit the HIF-1α/VEGF signaling pathway including HIF-1α expression, VEGF secretion, and angiogenesis. In this report, we demonstrate that we can rapidly screen and identify potential anti-HIF-1α compounds using this approach. Furthermore, synthetic derivatives of the most potent hits were designed, screened and analyzed in order to generate more potent, novel compounds targeting HIF-1α/VEGF signaling pathway and angiogenesis.
Materials and methods
Cell lines, medium and culture conditions
RPM1-1640 medium and Dulbecco's Modified Eagle medium (D-MEM) were purchased from Gibco BRL (Gaithersburg, MD), fetal bovine serum (FBS) from Hyclone Laboratories (Logan, UT), and Endothelial Cell Basal Medium-2 (EBM-2) from Lonza (Walkersville, MD). Human breast cancer (BCa) cell line MDA-MB-231 (American Type Culture Collection) and murine mammary carcinoma cell line JygMC(A) (15) (a generous gift from Dr. H. Azuma, Osaka Medical College, Osaka, Japan) were grown in RPM1-1640 medium with 10% FBS and D-MEM with 10% FBS, respectively. Human microvascular endothelial cells (HMEC) were grown in EBM-2 medium containing 10% FBS. All cell lines were grown in medium containing 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2. Cells were incubated at 37°C under normoxic conditions (5% CO2, 21% O2). Whenever necessary, breast cancer cells were also incubated at 37°C under hypoxic conditions (5% CO2, 0.5% O2, balanced with N2).
Western blot analysis of HIF-1α and HIF-1α-inducible protein expression
For detecting HIF-1α expression induced by hypoxia, BCa lines MDA-MB-231 and JygMC(A) cells were cultured under hypoxic conditions for hours (h) as indicated. The cells were lysed in lysis buffer containing 10 mM Tris-HCl (pH 8.0), 0.25 M Sucrose, 0.05 mM CaCl2, 0.02% Azide, 0.5% NP-40, 1x protease inhibitor cocktail (Sigma, St. Louis, MO), and 1x phosphatase inhibitor cocktail I and II (Sigma), and processed for gel electrophoresis. Protein concentration was determined by using Coomassie Plus Protein Assay Reagent (Pierce, Rockford, IL). Protein extracts were loaded on a precast 4-12% gradient polyacrylamide gel (NuPAGE gel, Invitrogen, Carlsbad, CA) and subject to SDS-gel electrophoresis, followed by transferring to a polyvinylidene fluoride (PVDF) membrane (Immobilon-P Transfer Membrane, Millipore). The membrane was incubated with blocking solution (5% nonfat milk and 0.02% sodium azide in phosphate-buffered saline) overnight at 4°C. The membrane was incubated for 16 h at 4°C with mouse anti-human HIF-1α antibody (BD Biosciences). The membrane was then incubated for 1 h at room temperature with corresponding secondary antibody coupled to peroxidase, followed by detection with chemiluminescent reagents (ECL kit, Amersham). For detecting HIF-1α inducible protein expression by hypoxia, the same blots were immunoblotted with antibody against GLUT1 (glucose transporter 1) and VEGF as the primary antibody (Santa Cruz Biotech, Santa Cruz, CA), respectively. The same blots were also immunoblotted with anti-actin antibody (Millipore, Temecula, CA) as the internal control.
ELISA assay to detect secreted VEGF concentration
Cells were plated in six-well plates and cultured to 80-90% confluence. The medium was replaced and cultures were treated with indicated concentrations of compounds for 48 h, followed by incubation under normoxic or hypoxic conditions for another 24 h. Secreted VEGF in extracellular medium was quantified (R&D Systems, Minneapolis, MN) by comparison with a series of concentration-known VEGF standard samples. VEGF levels were measured in duplicate in each of two independent experiments.
Sources of compounds and anti-HIF-1α screening
By a contract between the two universities, we (UTHSC) are able to access a large small-molecule-compound library containing approximate 350,000 drug-like small molecule compounds that are housed in the Drug Discovery Center at University of Cincinnati (DDCUC). YC-1, the known anti-HIF-1α compound (18), was purchased from Alexis Biochemicals Corp (San Diego, CA). PG-928310 (G3) (benzamide, 2-iodo-N-[1-(phenyl methyl)-1H-benzimidazol-2-yl]), a compound which was originally identified from DDCUC compound library and subsequently purchased from Ryan Scientific, Inc (Mt. Pleasant, SC.). CJ-III-60 (CJ-3-60) was a synthetic derivative of G3. All compounds were dissolved in dimethyl sulfoxide to 100 mM stock solution and stored at -80°C. The culture medium was used for all the subsequent dilution of compounds in the testing to reach the desired concentrations.
MDA-MB-231 cells transfected with a VEGF promoter-luciferase reporter construct (pVEGF/Luc) (24) were used for compound screening in a 96-well plate platform by incubating with various concentrations of candidate compound under hypoxic conditions as described previously (19). The inhibitory effect of compounds on the cells' VEGF gene transactivation was measured by the luciferase readout (Luc readout); compounds with higher anti-HIF-1α potency exhibit lower Luc readout. YC-1 was included as a positive control in all assays for comparison.
In vitro angiogenesis assay for evaluating potential anti-angiogenesis compounds
To validate whether potential compounds inhibit tumor cell-induced angiogenesis, culture supernatants from MDA-MB-231 cells treated with compounds (10 μM for 48 h under normoxic conditions and another 24 h under hypoxic conditions) were used as the conditioned medium for culturing HMEC cells for the tubular network formation assay. The HMEC model for tubular network formation assay was performed as we previously described (20). Briefly, 200 µl ice-cold matrigel (BD Biosciences) was pipetted onto a 48-well plate and let polymerize for 1 h at 37°C. HMEC cells incubated in EBM-2 containing 10% FBS for 24 h were harvested after trypsin treatment, resuspended in culture supernatants (conditioned medium) collected from MDA-MB-231 cells (see above), and then replated onto the layer of above-mentioned matrigel coated well at a density of 3x104 cells in 300 µl per well. The matrigel cultures of HMEC were incubated at 37°C, and at indicated time points (2, 5, 8, 11 and 24 h after HMEC planting onto the matrigel), the tube-like structure of the cells was photographed to record network-like structure. The tubular connections (i.e., tubular network formation) of five randomly selected fields at each time point were counted as vessel crossing points (20,32) for quantitive comparison.
Statistical analysis
All experiments, unless otherwise indicated, were repeated at least three times and the statistical analysis was by one-way analysis of variance (ANOVA) followed by Student's t-test, Graph Pad Prism®. P-values less than 0.05 were considered significant.
Results
Hypoxia is associated with an increased expression of HIF-1α and its targeted protein
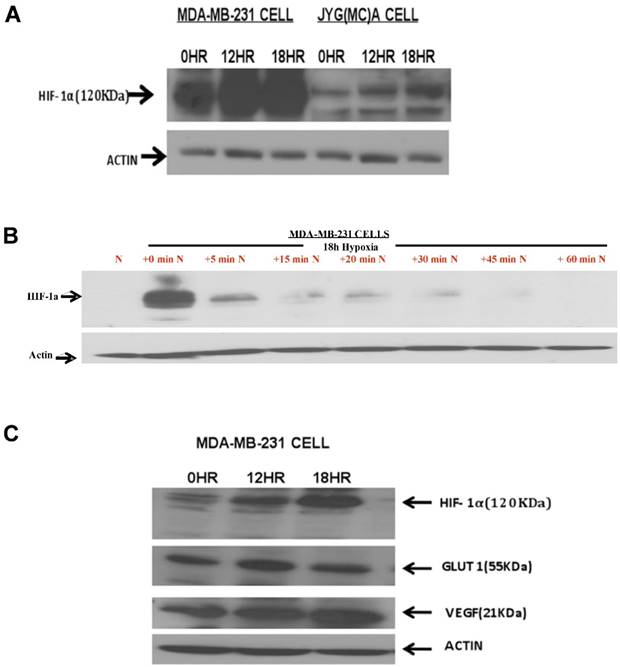
We used human BCa line MDA-MB-231 and a mouse mammary carcinoma line JygMC(A) (21) for the experiments in this project. We initially set out to confirm by Western blot analysis that hypoxia induces HIF-1α and its targeted protein in the BCa lines. Indeed, as we demonstrated, hypoxia condition (0.5% O2, 5% CO2) induced a time-dependent increased expression of HIF-1α in both MDA-MB-231 and JygMC(A) BCa cells (Fig. 1A). The induction of HIF-1α protein in MDA-MB-231 line reached its highest level at 18 h in hypoxia (Fig. 1A), as examined by our previous time-course study (19). This increased expression of HIF-1α by hypoxia is reversible, as cells returned to normoxic condition, the induced HIF-1α protein degraded rapidly, with a half-life of less than 5 minutes (min) (Figure 1B). The effect of hypoxia on the expression of two HIF-1α-driven downstream proteins, GLUT1 and VEGF, was also examined by Western blot analysis, and we observed that hypoxia also led to increased expression of these proteins in MDA-MB-231 cells (Fig. 1C) and JygMC(A) cells (not shown). While hypoxia-induced GLUT1 protein increase reached a peak level around 12 h, VEGF protein had a time-dependent increase up to 18 h (Fig. 1C).
Effects of hypoxia on HIF-1α and HIF-1α-inducible genes. Breast cancer cell lines MDA-MB-231 and JygMC(A) incubated at either normoxia (0 h) or hypoxia (0.5% O2) for 12 h or 18 h were harvested and proceeded for Western blot analysis for expression of (A) HIF-1α protein and (C) HIF-1α-targeted proteins of VEGF and Glucose transporter 1 (GLUT1). (B) The time-course degradation of HIF-1α protein under normoxic condition was also presented: after 18 hr incubation in a hypoxic environment, MDA-MB-231 cells were reoxygenenated (20% O2) for different periods of time (5, 15, 20, 30, 45, and 60 min) and cell extracts were harvested for Western blot analysis. The actin levels were used as the internal controls in all blots. *HR, hours; min, minutes.
Compound screening for anti-HIF-1α activity
HIF-1α is a transcriptional activator of VEGF gene promoter (21-23,27). We have previously demonstrated that a chimeric construct, pVEGF/Luc, where a 2.4-kb VEGF promoter was fused to a luciferase reporter gene, can be used to measure the functional transcriptional activity of HIF-1α (24). MDA-MB-231 line is a suitable cell model for the anti-HIF-1α screen because the cells express a very low basal level of endogenous HIF-1α protein in normoxia but an inducible high expression of HIF-1α under hypoxia, with a peak at 18 h (Fig. 1) (19). Therefore, we used MDA-MB-231 cells transfected with pVEGF/Luc to measure the inhibitory effect of compounds on the cells' VEGF gene transactivation (luciferase readout) under 18 h hypoxia, in order to select compounds with higher anti-HIF-1α (lower Luc readout) potency.
We sought to establish a high-throughput screening system by conducting a biochemically/biologically-coupled screen on the library of small compounds to rapidly identify functional novel inhibitors of HIF-1α. To this aim, a known HIF-1α inhibitor, YC-1 (18), was used to validate our screening method. YC-1 [3-(5'-hydroxymethyl-2'-furyl)-1-benzylindazole] (Fig. 2A insert), a pharmacologic compound, was originally developed for circulatory disorders through inhibition of platelet aggregation and vascular contraction (25) by activating soluble guanylyl cyclase (28). YC-1 was later discovered to be able to inhibit HIF-1α activity in vitro; it completely blocks HIF-1α expression at the post-transcriptional level and consequently inhibits HIF-1α transcriptional activity including hypoxia-induced VEGF expression in hepatoma cells (26). As VEGF is critical for tumor angiogenesis (21), which in turn is essential for the growth and metastasis of solid tumors (29), YC-1 halts tumor growth by blocking angiogenesis (40). As a HIF-1α inhibitor, YC-1 has shown effective anti-tumor effects in vitro (39,40) and in animal models (40). However, the severe side effects of YC-1 including increased bleeding time, and causes for hypotension and penile erection significantly limit its clinical applications (41).
Anti-HIF-1α compound YC-1 decreased hypoxia-stimulated HIF-1α-mediated VEGF transactivation and VEGF secretion from breast cancer cells. MDA-MB-231 cells expressing VEGF-Luc in 96-well platform were treated with a serial indicated concentrations of YC-1 (a known HIF-1α inhibitor, Fig. 2A insert) at 37°C for 24 h, with the last 18 h at hypoxic conditions (0.5% O2, 5% CO2, and balanced with N2). The cells were then harvested for analysis of luciferase activity. The normalized data (as luciferase activity: RLU per µg cell extracts) is presented. RLU stands for “relative light units.” Data shown are means and standard deviations (S.D.) from at least two experiments performed in duplicate. *The p values of differences between the control and the compound-treated groups are statistically significant (p <0.05). (B) MDA-MD-231 cells were treated with YC-1 at 0, 10 and 100 µM either under normoxia for 72 h or hypoxia (i.e., the cells were incubated under normoxia for 48 h and switched to hypoxia conditions for another 24 h). VEGF levels in the conditioned media (supernatant) were then quantified by using the ELISA assay. The normalized results based on the equal amount of cells are presented here. Data shown are means and S.D. from two experiments performed in duplicate. *The p values of differences between the corresponding hypoxic group and the normoxic group, and between the control and YC-1-treated samples, respectively, are statistically significant (p<0.05).
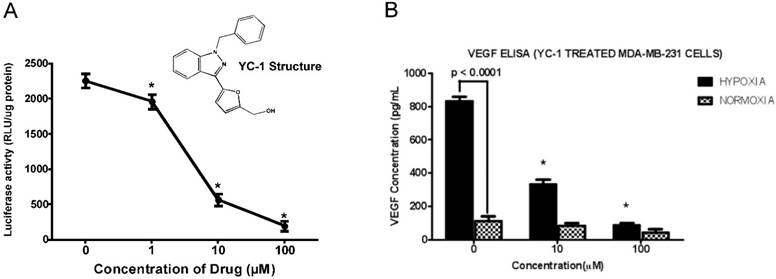
As shown in Fig. 2A, YC-1 showed a dose-dependent inhibition of VEGF-Luc transactivation as detected by our screening system. We also measured VEGF secretion from MDA-MB-231 cells under normoxia and hypoxia conditions with or without YC-1 treatment, we found that secreted VEGF proteins were elevated under hypoxia when compared with normoxia, and YC-1 inhibited VEGF secretion in both normoxia and hypoxia (Fig. 2B). These results confirmed that our screen system is capable of identifying compounds that inhibit HIF-1α activity and expression of HIF-1α downstream target protein (VEGF). Moreover, by using mutant pVEGF/Luc construct with abolished HRE sequence (where HIF-1α binds to the VEGF promoter), we demonstrated previously that it is indeed HIF-1α which is mainly responsible for the transactivation of VEGF promoter and YC-1-mediated inhibition of transactivation under hypoxic conditions under our screening system (19).
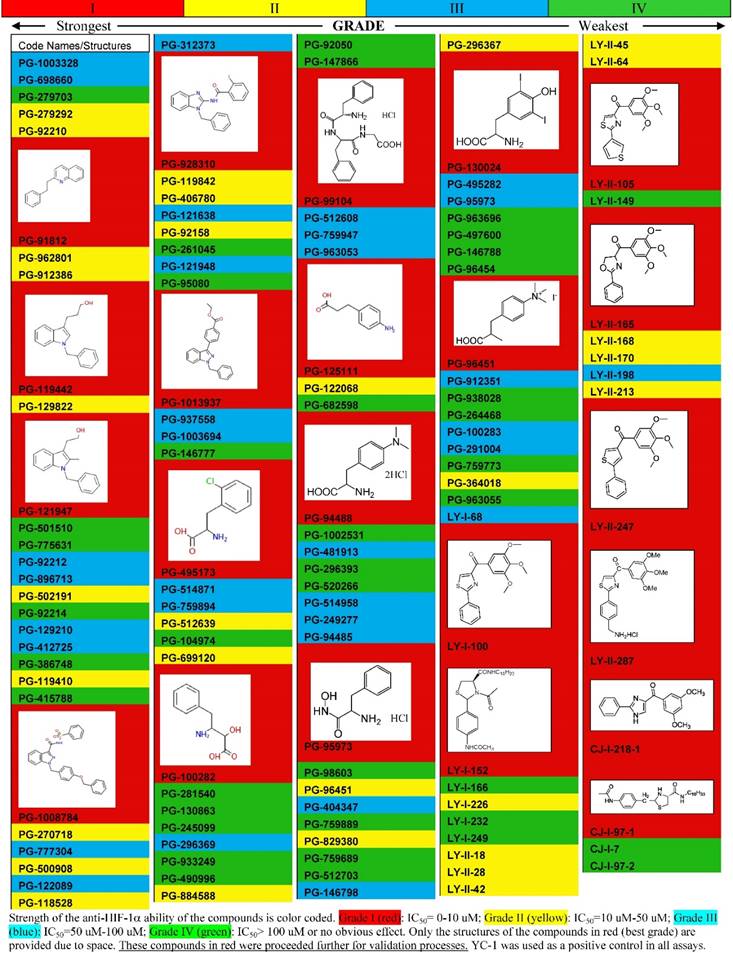
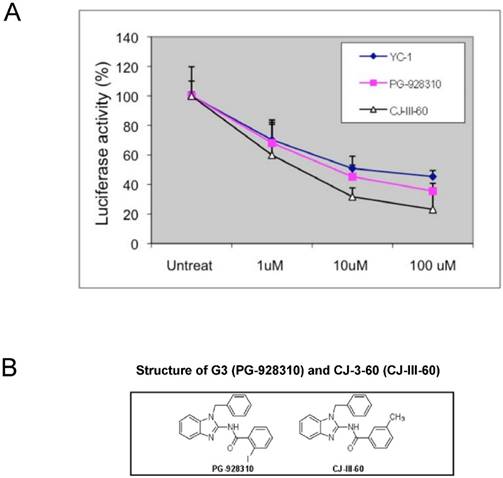
Both targeted and nontargeted screen approaches were used in our screening strategy. In our targeted screening strategy, we initially focused on compounds of the library that shared structural motif with YC-1. Over several hundreds of compounds were screened by the designed 96-well platform in the MDA-MB-231 cells using VEGF promoter-luciferase based reporter assay for HIF-1α inhibitory activities. We categorized and ranked the compounds based on their anti-HIF-1α abilities: a partial list of these screened results is illustrated in Table 1 as a representative of the screening. For example, compounds that have IC50 (the concentration that reaches the half maximal inhibition) below 10 µM were coded as red color (the best category), the second best category of compounds that give IC50 between 10-50 µM was coded as yellow color, and so on (Table 1). Only those compounds in the red category (IC50≤10 µM) were further screened by validation assays for confirmation of their anti-HIF-1α and anti-VEGF abilities (HIF-1α protein analysis and VEGF ELISA assays, respectively). Those compounds showing active (inhibitory effects) in both validation assays were further analyzed by in vitro angiogenic assay for their anti-angiogenesis (biological) effects. The compounds that were scrutinized through all these selection criteria and showed the overall best potency were selected as lead compounds (hits). Several such compounds stood out as potential lead compounds from our initial screenings: not only were they the most potent ones among the compounds screened and validated, but they also showed more anti-HIF-1α potency than YC-1 (with lower IC50 concentration than YC-1). One such lead compound, PG-928310 (also called G3 in this paper) (Fig. 3B), displayed superior anti-HIF-1α activity compared to that of YC-1, in terms of exhibiting a more potent concentration-dependent inhibition of luciferase expression than YC-1 (Fig. 3A).
Partial List of Compounds by the Pilot High Throughput Screenings Based on Anti-HIF-1α
Screening for anti-HIF-1α compounds and chemical modification of the lead compound G3. (A) MDA-MB-231 cells expressing VEGF-Luc in 96-well platform were treated with a serial dilution of potential anti-HIF-1α compounds including YC-1 at 37°C for 24 h, with the last 18 h at hypoxic conditions (0.5% O2, 5% CO2, and balanced with N2). The cells were then harvested for analysis of luciferase activity. The normalized data is presented as luciferase activity (RLU per µg cell extracts). (B) Structure of G3 (PG-928310) and CJ-3-60 (CJ-III-60). CJ-3-60 is a chemically-modified compound based on G3 that shown even higher anti-HIF-1α ability.
Modification and improvement of existing anti-HIF-1α lead compound
To improve the anti-HIF-1α potency further, the lead compound PG928310 (G3) was chosen for further modification in an attempt to generate novel compounds that may have even greater potency. Under a structure-activity relationship analysis, a dozen of analogs (derivatives) were synthesized after chemical modification. These synthesized novel analogs (not available in the compound database) were screened again by our screening and validation system. We found that one analog among them, CJ-3-60 (CJ-III-60) (Fig. 3B), demonstrated an even better anti-HIF-1α potency than its parental lead compound PG-928310 (G3) (Fig. 3A).
Effects of compounds on hypoxia-mediated expression of HIF-1α and its target protein VEGF
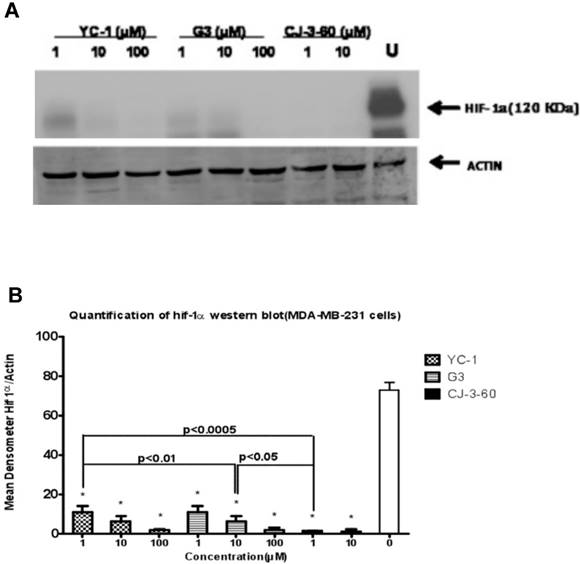
We analyzed the effect of YC-1, G3, and CJ-3-60 on hypoxic induction of HIF-1α protein in BCa cells by Western blot analysis. MDA-MB-231 and JygMC(A) cells were cultured under hypoxic conditions and treated in the absence or presence of YC-1, G3, and CJ-3-60 at various concentrations (0, 1, 10, and 100 µM). Both G3 and CJ-3-60 inhibited HIF-1α protein expression under hypoxia in a dose-dependent manner, and more potently than YC-1 did. Greater than 60% or more HIF-1α protein reduction in MDA-MB-231 cells (Fig. 4B) and 50% or more HIF-1α protein reduction in JygMC(A) cells (not shown) were observed between the untreated control and cells treated with anti-HIF-1α compounds (YC-1, G3 and CJ-3-60). In terms of inhibiting HIF-1α expression capability, CJ-3-60 is the most effective compound, followed by G3, while YC-1 is the weakest among the three (Fig. 4A). For example, at a concentration of 1 µM, YC-1 inhibited hypoxia-induced HIF-1α expression in MDA-MB-231 cells at just below 60% compared to the untreated control, while G3 inhibited at above 60%, and CJ-3-60 inhibited at nearly 70% (Fig. 4B).
Inhibition of HIF-1α protein by lead anti-HIF-1α compounds. (A) MDA-MB-231 cells were incubated for 72 h under hypoxia (0.5% O2) in the absence (U for untreated) or presence of the YC-1, G3, and CJ-3-60 (at various concentrations of 1, 10, and 100 µM), respectively. Cell extracts were then harvested for Western blot analysis using primary anti-HIF-1α antibody. The same blot was immunoblotted with anti-actin antibody as the internal control. (B) Quantification of HIF-1α expression data in the immunoblot. The signal bands in the Western blot (A) were densitometized (NIH Imager software) and the density ratios of HIF-1α and actin were presented as the normalized quantified data (means±S.D.). *The p values of differences between the untreated control and all treated samples are statistically significant (p<0.05).
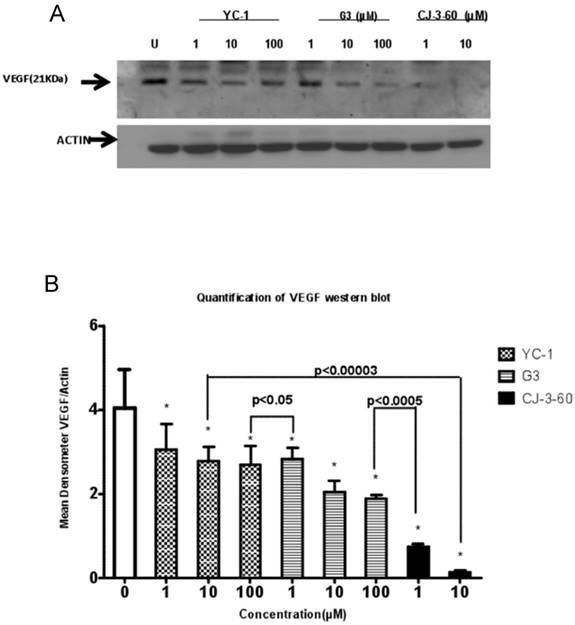
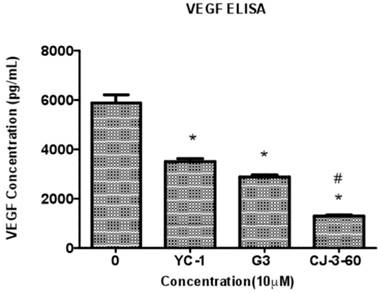
Given that HIF-1α is the main transcriptional regulator of the VEGF gene and VEGF is a prominent angiogenic factor for tumor angiogenesis and malignant progression, we analyzed the effect of these anti-HIF-1α compounds on VEGF production under hypoxia in MDA-MB-231 cells both by Western blot analysis (for the level of VEGF cellular expression, Fig. 5) and by an enzyme-linked immunosorbent assay (ELISA) (for the amount of VEGF extracellular secretion, Fig. 6). We observed a dose-dependent reduction of VEGF protein expression by these anti-HIF-1α compounds (Fig. 5A). The level of VEGF was apparently lower in G3-treated cells than in YC-1-treated cells at the same concentration, and this reduction became even more markedly significant in CJ-3-60-treated cells (Fig. 5B). Correspondingly, ELISA assay revealed that MDA-MB-231 cells had reduced hypoxia-induced VEGF secretion into the medium by these anti-HIF-1α compounds, with YC-1, G3 and CJ-3-60 (all at 10 µM) at inhibition of 41%, 51% and 78%, respectively (Fig. 6).
Inhibition of cellular VEGF proteins by lead anti-HIF-1α compounds. (A) MDA-MB-231 cells were incubated for 72 h under hypoxia (0.5% O2) in the absence (U for untreated) or presence of the YC-1, G3, and CJ-3-60 (at various concentrations of 1, 10, and 100 µM), respectively. Cell extracts were then harvested for Western blot analysis using primary anti-VEGF antibody. The same blot was immunoblotted with anti-actin antibody as the internal control. (B): Quantification of VEGF expression data in the immunoblot. The signal bands in the Western blot (A) were densitometized (NIH Imager software) and the density ratios of VEGF and actin were presented as the normalized quantified data (means±S.D.). *The p values of differences between the untreated control and all treated samples are statistically significant (p<0.05).
Inhibition of VEGF secretion from breast cancer cells by lead anti-HIF-1α compounds. MDA-MD-231 cells were untreated or treated with 10 µM compound (YC-1, G3, and CJ-3-60, respectively) under normoxia for 48 h and then switched to hypoxia conditions for another 24 h. The cell medium (supernatant) was collected for VEGF determination by ELISA assay. The normalized results based on the equal amount of cells are presented here. Data shown are means±S.D. (error bars) from two independent experiments performed in duplicate. *The p values of differences between the untreated control group and all compound-treated samples are statistically significant (p<0.05). #The p values of differences between the CJ-3-60 group and YC-1 (or G3) samples are statistically significant (p<0.05), respectively.
Compounds inhibit BCa-induced angiogenesis
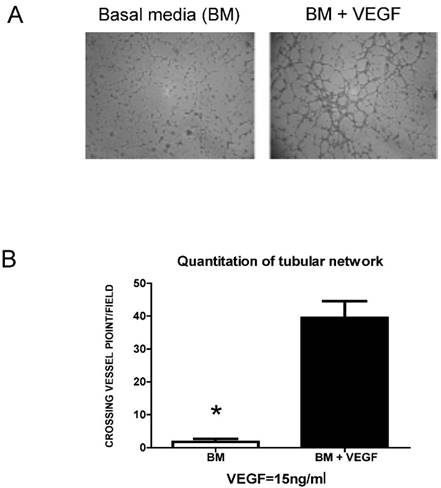
To further explore if the inhibition of VEGF protein production by anti-HIF-1α compounds would parallel their capabilities of inhibiting BCa cell-induced angiogenesis, a HMEC-based in vitro angiogenesis assay, a classic tubular network formation study that quantifies the degree of angiogenesis in vitro, was used. In the absence of required angiogenic factor (i.e., basal medium with no fetal bovine serum and any growth factors), HMEC cells failed to form meaningful tubular network at 8 h after incubation (Fig. 7A, Basal Media or BM). However, the addition of the angiogenic factor VEGF into basal medium (to the final concentration of 15 ng/ml) stimulated the HMEC cells to form network-like structures (Fig. 7A, BM + VEGF). The tubular connections (i.e., vessel crossing points) of five randomly selected fields were counted for quantitive comparison (30,32) of angiogenic ability (Fig. 7B). These results confirm that the HMEC-based tubular formation assay can be used to measure the in vitro angiogenic ability induced by growth factors such as VEGF.
HMEC-based in vitro angiogenesis assay for measuring VEGF-mediated tubular network formation. HMEC cells were planted onto matrigel-coated wells and cultured in the basal media (BM) (i.e., RPMI-1640 medium with no FBS) or BM containing 15 ng/ml VEGF (BM+VEGF). (A) The tubular network formation was recorded at 8 h after planting HMEC cells onto the matrigel-coated wells in the indicated media. (B) The vessel crossing points of five randomly selected fields were counted (30,32) as angiogenic ability for quantitation comparison. *The p values of difference between the untreated control and the VEGF-treated samples are statistically significant (p<0.05).
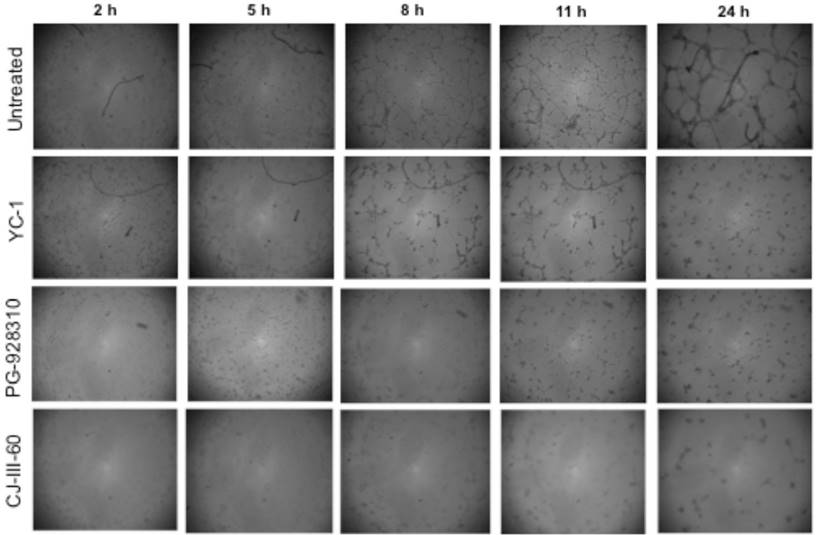
When the HMEC cells grew in conditioned medium (i.e., the culture supernatant) isolated from the untreated MDA-MB-231 cells under hypoxia, the cells tended to form tubular structures on the matrigel due to the growth factors including VEGF secreted from BCa cells; at 5 h, the cells started to aggregate together to form a pre-tubular structure (Fig. 8, top row). The apparent tubular structure was observed at 8 h, and reached the mature, well-developed tubular network by 11 h and 24 h. These results suggested that there were some growth factors (including VEGF) secreted from MDA-MB-231 cells that were able to stimulate angiogenesis of HMEC cells. Our previous publication demonstrated that VEGF is the major growth factor stimulating the tubular network formation in this case: MDA-MB-231 cells that were cultured in conditioned medium containing neutralized anti-VEGF antibody failed to form the tubular network structure (30). However, when HMEC cells grew in conditioned medium isolated from YC-1-treated or PG-928310 (G3)-treated MDA-MB-231 counterparts, their abilities to form the tubular network were significantly reduced, with more inhibition on this angiogenesis by CJ-III-60 (CJ-3-60) (compare the top 3 rows, Fig. 8). For example, at 11 h, by counting the cross-points as quantitation for angiogenic ability (32), YC-1 and PG-928310 (G3) had 70.8% and 82.3% inhibition (Fig. 8, middle two rows at 11 h time point), respectively, compared to the untreated, while CJ-III-60 (CJ-3-60) had the highest (93.1%) inhibitory effect on HMEC angiogenesis among all compounds (Fig. 8, bottom row).
Anti-HIF-1α compounds inhibited BCa-induced angiogenesis. HMEC cells were planted onto matrigel-coated wells in the conditioned medium collected from untreated or 10 µM-compound treated (YC-1, G3, and CJ-3-60, respectively) MDA-MB-231 cells as described in Fig. 6 legend. The development of the tubular network of HMEC cells were recorded at indicated time points after HMEC cells seeding onto the matrigel. The degrees of effect of conditioned medium of compound-treated MDA-MB-231 cells that inhibited HMEC tubular network formation are, in order of effectiveness, CJ-3-60>G3>YC-1.
Discussion
Angiogenesis, as the growth of new blood vessels from pre-existing vessels, is an essential prerequisite for aggressive tumor proliferation and spreading (31) and it requires several angiogenic factors during the malignant transformation (12). VEGF is a prominent angiogenic factor that plays a pivotal role in promoting tumor angiogenesis (17,21,31,33), which leads to malignant progression of BCa including metastasis - the major cause of BCa patient death. As a result of its role in angiogenesis, VEGF and its receptors have been main targets for antiangiogenic therapy. Until recently, the most famous and commonly used anti-angiogenesis agent in clinical is Avastin, a VEGF-neutralizing monoclonal antibody; but the severe side effects made FDA withdrew the approval of Avastin for treating BCa in December 2010.
Searches for other effective anti-angiogenesis drugs for cancer treatment naturally enforce the identification of small-molecule inhibitors targeting HIF-1α activity, which has a different anti-angiogenesis mechanism from Avastin's direct VEGF ligand-targeting and neutralization approach. Since HIF-1 is a hypoxia-activated transcription factor that can regulate VEGF synthesis, and HIF-1 activity is dependent upon the level of available HIF-1α, a rationale is therefore provided for developing agents aimed at targeting HIF-1-dependent responses in solid tumors. The discovery and development of novel agents targeting hypoxia-induced HIF-1α/VEGF signaling pathways has become a crucial area of developmental therapeutics to effectively combat cancer. Importantly, in the case of treating BCa, anti-HIF-1α therapy is also effective for the triple-negative breast cancer (TNBC) (35), the more aggressive and most treatment-resistant subtype BCa. Inhibition of HIF-1 activity in mice after orthotopic transplantation of TNBC cells has a dramatic effect on primary tumor growth as well as metastasis to lymph nodes and lungs (37).
Although several anti-HIF-1α compounds (4) were identified including YC-1 (18), these identified compounds are not clinically applicable due to their severe side effects or insufficient potency to inhibit tumor angiogenesis. For example, YC-1 prolongs bleeding time due to its inhibition of platelet aggregation (28) and thus undermines its potential for clinical use. At present, few small-molecule HIF-1α inhibitors have completed evaluation in the clinic and proven particularly effective in BCa clinical trial (32). Hence, little success has occurred to date in developing specific and effective small molecules that can be regarded as “gold standard” for HIF-1α inhibition (32). The stagnant advance in this field is partly because there is no reliable mass screening method available that is coupled with automated validation processes in order to rapidly identify promising leads. This project will fill this gap by linking a novel high-throughput system prototype, that allows for highly efficient screening of new anti-cancer drugs based on the anti-HIF-1α mechanism with validation screens (which have the potential to be developed into automated processes) for the compounds' inhibitory effect on target (HIF-1α) expression, its downstream target gene (VEGF) and relevant biological effects (angiogenesis).
Summary of our results in this report include: (A) We have established a reliable screening system coupled with secondary/counter screens to validate the compounds' anti-HIF-1α/anti-VEGF property. (B) By using this system to screen several hundreds of small molecule compounds, we identified several lead compounds including G3 (PG-928310) that is an effective and more potent HIF-1α inhibitor than the known anti-HIF-1α agent YC-1, as validated by anti-VEGF transactivation, inhibition of HIF-1α expression, VEGF secretion and BCa-induced angiogenesis. (C) We can improve the compound's anti-HIF-1α potency by chemically modifying the identified lead (G3) in order to make an even better drug (e.g., CJ-3-60). We are currently investigating some of these lead compounds for their abilities on other anti-cancer properties including anti-proliferation, induction of apoptosis, and anti-cell migration. We hope that our prototype screening system would provide an efficient screen/validation system that is capable of rapidly identifying, modifying, and developing better anti-HIF-1α/anti-angiogenesis drugs that give maximal anti-BCa efficacy with no or minimal side effect.
Acknowledgements
This research project was partially supported by NIH grant CA107162 (YL) and UTHSC research funding (YL). We thank Jordan Masters and Andrew Lu for their participation in the screening of anti- HIF-1α compounds. We also thank Jianjun Chen and Dr. Wei Li for providing CJ-3-60 compound.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Harris AL. Hypoxia - a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
2. Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408-16
3. Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med. 2002;8:S62-67
4. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
5. Zhong H, De Marzo AM, Laughner E. et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830-5
6. Sowter HM, Raval RR, Moore JW. et al. Predominant role of hypoxia-inducible transcription factor (Hif)-1α versus Hif-2α in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63:6130-4
7. Carroll VA, Ashcroft M. Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res. 2006;66:6264-70
8. Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007;80:51-60
9. Le QT, Denko NC, Giaccia AJ. Hypoxic gene expression and metastasis. Cancer Metastasis Rev. 2004;23:293-310
10. Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230-7
11. Blagosklonny MV. Hypoxia-inducible factor: Achilles' heel of antiangiogenic cancer therapy. Int J Oncol. 2001;19:257-62
12. Brem SS, Gullino PM, Medina D. Angiogenesis, a marker for neoplastic transformation of mammary papillary hyperplasia. Science. 1997;195:880-1
13. Birner P, Schindl M, Obermair A. et al. Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res. 2000;60:4693-6
14. Bos R, Zhong H, Hanrahan CF. et al. Levels of hypoxia-inducible factor-1α during breast carcinogenesis. J Natl Cancer Inst. 2001;93:309-14
15. Azuma H, Takahara S, Ichimaru N. et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62:1410-9
16. Brizel DM, Scully SP, Harrelson JM. et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941-3
17. Risau W. What, if anything, is an angiogenic factor?. Cancer Metastasis Rev 15: 149-151. 1996
18. Yeo EJ, Chun Y-S, Cho Y-S. et al. YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst. 2003;95:516-25
19. Lu Y, Madu C, Masters J, Lu A, Li L. Development of a novel anti-HIF-1α screening system coupled with biochemical and biological validation for rapidly selecting anti-cancer compounds. J Cancer. 2014;5:417-24
20. Zhang J, Lu A, Beech D, Jiang B, Lu Y. Suppression of breast cancer metastasis through the inhibition of VEGF-mediated tumor angiogenesis. Cancer Ther. 2007;5:273-86
21. Grunstein J, Roberts WG, Mathieu-Costello O. et al. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999;59:1592-8
22. Michel G, Minet E, Mottet D. et al. Site-directed mutagenesis studies of the hypoxia-inducible factor-1alpha DNA-binding domain. Biochim Biophys Acta. 2002;1578:73-83
23. Burroughs KD, Oh J, Barrett JC, DiAugustine RP. Phosphatidylinositol 3-kinase and mek1/2 are necessary for insulin-like growth factor-I-induced vascular endothelial growth factor synthesis in prostate epithelial cells: a role for hypoxia-inducible factor-1? Mol Cancer Res. 2003;1:312-22
24. Zhang J, Lu A, Li L, Yue J, Lu Y. p16 modulates VEGF expression via its interaction with HIF-1α in breast cancer cells. Cancer Invest. 2010;28:588-97
25. Teng CM, Wu CC, Ko FN. et al. YC-1, a nitric oxide-independent activator of soluble guanylate cyclase, inhibits platelet-rich thrombosis in mice. Eur J Pharmacol. 1997;320:161-6
26. Chun YS, Yeo EJ, Choi E. et al. Inhibitory effect of YC-1 on the hypoxic induction of erythropoietin and vascular endothelial growth factor in Hep3B cells. Biochem Pharmacol. 2001;61:947-54
27. Forsythe JA, Jiang BH, Iyer NV. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604-13
28. Ko FN, Wu CC, Kuo SC. et al. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226-33
29. Folkman J. Tumor angiogenesis, therapeutic implications. N Engl J Med. 1971;285:1182-6
30. Zhang J, Lu A, Beech D, Jiang B, Lu Y. Suppression of breast cancer metastasis through the inhibition of VEGF-mediated tumor angiogenesis. Cancer Ther 5: 273-286. 2007
31. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13: 9-22. 1999
32. Kim YM, Kim YM, Lee YM. et al. TNF-related activation-induced cytokine (TRANCE) induces angiogenesis through the activation of Src and phospholipase C (PLC) in human endothelial cells. J Biol Chem. 2002;277:6799-805
33. Detmar M, Velasco P, Richard L. et al. Expression of vascular endothelial growth factor induces an invasive phenotype in human squamous cell carcinomas. Am J Pathol. 2000;156:159-67 2000
34. Hashimoto T, Shibasaki F. Hypoxia-inducible factor as an angiogenic master switch. Front Pediatr. 2015;3:33
35. Yehia L, Boulos F, Jabbour M. et al. Expression of HIF-1α and markers of angiogenesis are not significantly different in triple negative breast cancer compared to other breast cancer molecular subtypes: implications for future therapy. PLoS One. 2015;10(6):e0129356
36. Liu Z, Semenza GL, Zhang H-F. Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang Univ Sci B. 2015;16:32-43
37. Zhang H, Wong CC, Wei H. et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757-70
38. Hiraga T, Kizaka-Kondoh S, Hirota K. et al. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157-63
39. Feng Y, Zhu H, Ling T. et al. Effects of YC-1 targeting hypoxia-inducible factor 1 alpha in oesophageal squamous carcinoma cell line Eca109 cells. Cell Biol Int. 2011;35:491-7
40. Pan SL, Guh JH, Peng CY. et al. YC-1 [3-(5'-hydroxymethyl-2'-furyl)-1-benzyl indazole] inhibits endothelial cell functions induced by angiogenic factors in vitro and angiogenesis in vivo models. J Pharmacol Exp Ther. 2005;314:35-42
41. Chun YS, Yeo EJ, Park JW. Versatile pharmacological actions of YC-1: anti-platelet to anticancer. Cancer Lett. 2004;207:1-7
Author contact
![]() Corresponding author: Yi Lu, Ph.D. Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 218, 19 South Manassas Street, Memphis, TN 38163, USA. Tel: (901) 448-5436 Fax: (901) 448-5496 e-mail: yluedu.
Corresponding author: Yi Lu, Ph.D. Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 218, 19 South Manassas Street, Memphis, TN 38163, USA. Tel: (901) 448-5436 Fax: (901) 448-5496 e-mail: yluedu.