Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(4):537-554. doi:10.7150/jca.15989 This issue Cite
Research Paper
Differentiation by NK cells is a prerequisite for effective targeting of cancer stem cells/poorly differentiated tumors by chemopreventive and chemotherapeutic drugs
Anna Karolina Kozlowska1,2*, Paytsar Topchyan1*, Kawaljit Kaur1, Han-Ching Tseng1, Antonia Teruel1, Toru Hiraga3, Anahid Jewett1 ![]()
1. The Jane and Jerry Weintraub Center for Reconstructive Biotechnology, The Jonsson Comprehensive Cancer Center, Dental Research Institute, Division of Oral Biology and Oral Medicine. UCLA School of Dentistry, Los Angeles, CA 90095, USA.
2. Department of Tumor Immunology, Chair of Medical Biotechnology, Poznan University of Medical Sciences, Poznan, Poland.
3. Department of Histology and Cell Biology Matsumoto Dental University, Gobara-Hirooka, Shiojiri, Nagano, Japan.
* These authors contributed equally to the manuscript.
Received 2016-4-27; Accepted 2016-7-24; Published 2017-2-11
Abstract
Natural Killer (NK) cells target oral, pancreatic, lung, breast, glioblastoma and melanoma stem-like/poorly differentiated tumors. Differentiation of the abovementioned tumors with supernatants from split-anergized NK cells decreases their susceptibility to NK cells, but increases their sensitivity to cisplatin (CDDP)-mediated cell death. Breast and melanoma tumor cells with CD44 knockdown display enhanced susceptibility to NK cell-mediated lysis, potentially due to decreased differentiation. We also demonstrate that sulindac, a non-steroidal anti-inflammatory drug and a chemopreventive agent, not only limits the growth of oral tumor cells, but also aids in cancer cell elimination by NK cells. Treatment of oral tumors with sulindac, but not adriamycin inversely modulates the expression and function of NFκB and JNK, resulting in a significant down-regulation of IL-6, and VEGF secretion by oral tumor cells. In addition, increased secretion of IL-6 and VEGF is blocked by sulindac during interaction of oral tumors with NK cells. Sulindac treatment prevents synergistic induction of VEGF secretion by the tumor cells after their co-culture with untreated NK cells since non-activated NK cells lack the ability to efficiently kill tumor cells. Moreover, sulindac is able to profoundly reduce VEGF secretion by tumor cells cultured with IL-2 activated NK cells, which are able to significantly lyse the tumor cells. Based on the data presented in this study, we propose the following combinatorial approach for the treatment of stem-like/ poorly differentiated tumors in cancer patients with metastatic disease. Stem-like/ poorly differentiated tumor cells may in part undergo lysis or differentiation after NK cell immunotherapy, followed by treatment of differentiated tumors with chemotherapy and chemopreventive agents to eliminate the bulk of the tumor. This dual approach should limit tumor growth and prevent metastasis.
Keywords: cancer stem-like cells, NFκB, VEGF, IL-6, NSAIDs, sulindac, NK cells
Introduction
Despite advances in our understanding of cancer biology, the development of effective treatment strategies remains a therapeutic challenge. One of the main reasons for the failure to control certain types of cancers is the lack of strategies targeting cancer stem-like cells (CSCs)/poorly differentiated tumors. CSCs have been found in a wide variety of human tumors including leukemia, breast, head and neck, pancreatic and prostate cancers as well as glioblastoma (GBM) and melanoma. CSCs may arise from stem cells, progenitor cells, or other differentiated cancer cells [1]. Although CSCs comprise only a small population in the tumor bulk, they are capable of self-renewal in both primary tumors and their metastatic variants, thus maintaining the bulk of the tumor through cycles of self-renewal, differentiation, and de-differentiation [2]. Several markers that identify CSCs have been described previously, including CD44, ALDH activity and CD133 [3, 4]. There is growing evidence that CSCs are resistant to chemotherapy or radiation, and their increased numbers within the tumor microenvironment predict poor prognosis [5-7].
Lack or lower expression of MHC class I are hallmarks of poorly differentiated cells, including mesenchymal stem cells, hematopoietic stem cells, embryonic stem cells, induced pluripotent stem cells, and CSCs in various tumor types [8, 9]. On the other hand, such cells are targets of NK cells since NK cells are known to kill cells expressing decreased MHC class I. Although, higher levels of circulating NK cells are associated with better prognosis [10], NK cell cytotoxic activity in peripheral blood of cancer patients is usually reduced, and the expression of NK cell activating receptors is diminished even at the early stages of cancer, as well as in advanced disease [11, 12]. It is known that the cytotoxic function of NK cells is suppressed after their interaction with stem cells [13-15]. We have previously shown that poorly differentiated oral squamous carcinoma stem cells (OSCSCs) were excellent targets for NK cell-mediated cytotoxicity and triggered significant IFN-γ secretion by the NK cells, whereas their differentiated counterparts, oral squamous carcinoma cells (OSCCs), were significantly more resistant [16]. OSCSCs expressed CD44high, EpCAMhigh, CD26high, CD166low, and CD338+ oral stem cell markers [17, 18]. Furthermore, de-differentiation of tumors by either blocking NFκB or genetically knocking out COX2 results in an increased NK cell-mediated cytotoxicity [17, 19-21]. We have recently demonstrated that induction of split anergy in NK cells results in differentiation of CSCs by cytokines secreted from such NK cells. Consequently, NK differentiated tumors become resistant to NK cell-mediated cytotoxicity and are unable to trigger secretion of cytokines or chemokines [22].
Anti-carcinogenic and chemopreventive effects of non-steroidal anti-inflammatory drugs (NSAIDs) have been well documented [23-27]. In addition to their well-known preventive effect against colon and gastric cancers, NSAIDs mediate a protective effect against bladder cancer [28], breast cancer [29], prostate cancer [30], and lung cancer [31]. Aspirin and sodium salicylates have previously been shown to suppress the activation of Nuclear Factor κB (NFκB) pathway [32-34]. Inhibition of NFκB by an IκB super-repressor in tumors or decreased function of NFκB in oral tumors led to enhanced activation and survival of NK and T cells [35, 36]. We have also shown that the inhibition of NFκB activation in HEp2 tumor cells rendered these cells susceptible to NK and CD8+ T cell-mediated cytotoxicity, and prevented NK and CD8+ T cells from undergoing inactivation and cell death mediated by tumor cells [35, 37]. Thus, the protective effect of NSAIDs against a variety of cancers could relate to their ability to decrease NFκB and increase the cytotoxic function of effector cells in order to clear newly arising cancer cells. Indeed, as shown here and elsewhere, treatment of HEp2 cells with sulindac, an NSAIDs compound, increased the susceptibility of HEp2 cells to NK cell-mediated cytotoxicity [35].
NFκB is also shown to be an important regulator of interleukin-6 (IL-6) gene expression [38, 39]. IL-6 is a pleiotropic cytokine, produced during a variety of inflammatory conditions, and is found to modulate several physiological processes such as cell proliferation, differentiation, survival, and apoptosis [40]. Various cancer types, including melanoma, prostate cancer [40], basal cell carcinomas [41], and head and neck squamous cell carcinomas (HNSCC) [42] produce increased amounts of IL-6 which acts as an autocrine and/or paracrine growth factor for tumor cells [40]. In addition to its role in tumor proliferation, IL-6 is also known to play a key role in immune cell inactivation [36, 43]. IL-6 has been shown to decrease the expression of the co-stimulatory molecule CD80 in a murine model of oral squamous cell carcinomas [44]. Therefore, NFκB expression in tumor cells could negatively regulate immune function by increasing IL-6 secretion.
C-Jun N-terminal kinase (JNK) belongs to the family of mitogen-activated protein kinases (MAPKs), which are responsible for phosphorylation and mediation of signal transduction from extracellular stimuli. The role of JNK in cell growth, differentiation, and cell death remains controversial. Studies have shown that activation of JNK generally either promotes cell growth or activates cell death [45, 46]. The induction of strong and prolonged JNK activity in response to a variety of stresses including UV light, ionizing irradiation, and hydrogen peroxide, have been shown to induce cell death [47, 48]. Moreover, we have previously shown that the induction of JNK in the absence of NFκB was found to trigger significant cell death of HEp2 cells after 2-hydroxyethyl methacrylate (HEMA) treatment [49]. Similarly, studies conducted by several groups implicated the significance of inverse regulation of JNK and NFκB in the induction of cell death [23, 50-53]. Therefore, identification of novel pharmacological compounds, which could target signaling pathways that mediate cell death, is of significance. Sulindac is a prodrug, structurally related to a methylated indole derivative, indomethacin, which exhibits antipyretic, analgesic and anti-inflammatory functions. It differs from indomethacin by its ability to bind IKKβ and inhibit NFκB activation in response to TNF-α [39, 54].
In this paper, we demonstrate that the differentiation stage of cancer cells is predictive of their susceptibility to sulindac and CDDP-mediated cell death. NK cells, in addition to their well-known ability to lyse CSCs/undifferentiated tumors, are also able to differentiate tumors, resulting in their enhanced susceptibility to drug-mediated cell death. Moreover, we show that the protective effect of sulindac against cancer is likely mediated by the inverse regulation of NFκB and JNK which results in the decrease of IL-6, IL-8 and vascular endothelial growth factor (VEGF) secretion favoring NK cell activation.
Materials and Methods
Cell Lines, reagents, and antibodies
Oral squamous carcinoma cells (OSCCs) and oral squamous cancer stem-like cells (OSCSCs) were isolated from oral cancer patient tongue tumors at UCLA. These cells were cultured as described previously [17].
MIA PaCa-2 (MP2), PL12 and Capan-1 human pancreatic cell lines were provided by Drs. Guido Eibl and Nicholas Cacalano (UCLA David Geffen School of Medicine). MP2 and PL12 cells were cultured in DMEM supplemented with 10% FBS and 2% penicillin-streptomycin (Gemini Bio-Products, CA, USA). Capan-1 was cultured in RPMI 1640 medium supplemented with 10% FBS and 2% penicillin-streptomycin.
X0GB2 stem-like glioblastoma (GBM) cells were isolated from freshly resected human tumor tissues, and were characterized and cultured as described previously [55-57].
MDA-MB231 and A375 cells were generously provided by Dr. Toru Hiraga (Department of Histology and Cell Biology, Matsumoto Dental University). MDA-MB231 and A375 cells modified with vectors containing CD44 and luciferase (LUC) shRNA were prepared by Dr. Hiraga's group [58]. MDA-MB231 and A375 cells were cultured in DMEM (Gemini Bio-Products, CA, USA) supplemented with 10% FBS and 100 mg/mL kanamycin sulfate (Life Technologies, MA, USA).
A549 (catalog # CCL-185) cell line, human acinar adenocarcinoma with gland formation, was purchased from ATCC (Manassas, VA, USA). A549 cells were cultured in DMEM supplemented with 3% FBS, 1.4% antibiotic-antimycotic, 1% sodium pyruvate, 1.4% non-essential amino acids, and 1% L-glutamine.
HEp2 cancer cell line (catalog # CCL-23) was obtained from ATCC (Manassas, VA, USA) and was maintained in DMEM supplemented with 10% FBS. RPMI 1640 supplemented with 10% FBS was used for the cultures of NK cells and PBMCs.
OSCSCs and OSCCs were irradiated with a single dose of 10 or 20 Gy with MARK-I Irradiator (J.L. Shepherd), and the cell viability was assessed using propidium iodide (PI) at different time points followed by flow cytometric analysis.
Recombinant human IL-2 was obtained from Hoffman La Roche (NJ, USA). Recombinant human IFN-γ was obtained from Biolegend (San Diego, CA, USA). Antibodies to TNF-α and IFN-γ were prepared in our laboratory. The antibodies against p65 subunit of NFκB, actin and β-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p-JNK and JNK were purchased from Cell Signaling Technology (Danvers, MA, USA). Cisplatin (CDDP) and adriamycin were obtained through Ronald Reagan Pharmacy at UCLA. Sulindac and DMSO were purchased from Sigma Aldrich (Buffalo, NY, USA).
Surface staining and cell death assays
As previously described, staining was performed by labeling cells with antibodies or PI [59-61]. Flow cytometry analysis was performed using Beckman Coulter Epics XL cytometer (Brea, CA, USA) and results were analyzed in FlowJo vX software (Tree Star, Ashland, OR, USA).
Purification of NK cells
Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated as described before [60]. Briefly, peripheral blood lymphocytes were obtained after Ficoll-hypaque centrifugation and NK cells were negatively selected using NK cell isolation kits (Miltenyi Biotec (Auburn, CA, USA) and Stem Cell Technologies (Vancouver, Canada). The purity of the NK cell population was found to be greater than 95% based on flow cytometric analysis of anti-CD16 antibody stained cells. The levels of contaminating CD3+ T cells remained low, at 2.4%±1%, similar to that obtained by the non-specific staining using isotype control antibody throughout the experimental procedures. Written informed consents approved by UCLA Institutional Review Board (IRB) were obtained from the blood donors and all the procedures were approved by the UCLA-IRB.
Stem cell differentiation with NK cell supernatant
Human NK cells were purified from healthy donors' PBMCs as described above. NK cells were left untreated or treated with a combination of IL-2 (1000 units/mL) and anti-CD16mAb (3 μg/mL), (a condition to induce split anergy in NK cells) for 18- 24 hours before the supernatants were removed and used in differentiation experiments. The amounts of IFN-γ produced by activated split-anergized NK cells were assessed with IFN-γ ELISA (Biolegend, San Diego, CA, USA). Differentiation of OSCSCs and MP2 cells was conducted with an average total of 1500-2000 pg and 4000-7000 pg of IFN-γ from IFN-γ containing supernatants, respectively, over 4-5 days as described previously [18]. Differentiation of A549 and A375 cells was conducted with an average total of 6000 pg and 15000 pg of IFN-γ containing supernatants, respectively over the course of 5 days. Differentiation of X02GB required 35000 pg of IFN-γ containing supernatants as described previously [57]. Initially, 1X106 tumor cells were cultured and treated with split-anergized NK supernatants for differentiation. Afterward, target cells were rinsed with 1X PBS, detached and used for experiments.
Luciferase reporter assay
Vector alone and IκB(S32AS36A) super-repressor transfected HEp2 tumor cells were generated in our laboratory. Transfections were performed using an NF-κB Luciferase reporter vector and Lipofectamine 2000 reagent in Opti-MEM media (Invitrogen, CA, USA) after which HEp2 tumor cells were treated with different agents as described in the results section. The cells were then lysed with lysis buffer and the relative luciferase activity was measured using the luciferase assay reagent kit obtained from Promega (Madison, WI, USA). NFκB knockdown HEp2 tumor cells (HEp2-IκB(S32AS36A)) were used as negative controls for luciferase assay.
Preparation of the nuclear extracts
Treated and untreated samples were incubated for 15 minutes at 37°C, after which they were washed in Buffer A (l0 mM HEPES, 1.5 mM MgCl, 10 mM KCL, 0.5 mM DTT and 0.5 mM PMSF) without Nonidet P-40. The pellets were resuspended in 300 uL of Buffer A with 0.1 % Nonidet P-40. The incubation was continued for 5 minutes on ice. The insoluble fraction was washed in Buffer A without NP-40 once and Buffer C (25% Glycerol, 20 mM HEPES, 0.6 M KCl, 1.5 mM MgCl and 0.2mM EDTA) was added to the pellets for 30 minutes. The pellets were centrifuged at 14000 rpm for 10 minutes in cold and the recovered supernatants were frozen in -80°C until used.
Western blot
Treated and untreated cells were lysed in a lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40 (v/v), 1 mM sodium orthovanadate, 0.5 mM EDTA, 10 mM NaF, 2 mM PMSF, 10 ug/mL leupeptin, and 2 U/mL aprotinin for 15 minutes on ice. The samples were then sonicated for 3 seconds. The cell lysates were centrifuged at 14000 rpm for 10 minutes, supernatants were removed and the levels of protein were quantified by Bradford method. The cell lysates were denatured by boiling in 5X SDS sample buffer. Equal amounts of cell lysates were loaded onto 10% SDS-PAGE and transferred onto Immobilon-P membranes (Sigma Aldrich, MO, USA). The membranes were blocked with 5% non-fat milk in PBS plus 0.1% Tween-20 for 1 hour. Primary antibodies at the predetermined dilution were added for 1 hour at room temperature. Membranes were then incubated with 1:1000 dilution of horseradish peroxidase-conjugated secondary antibody. Blots were developed by enhanced chemiluminescence (ECL).
ELISA and multiplex cytokine array kit
Single ELISAs were performed as described previously [60]. Fluorokine MAP cytokine multiplex kits were purchased from R&D Systems (Minneapolis, MN, USA) and the procedures were conducted as suggested by the manufacturer. JNK and pJNK levels were analyzed using the multiplex array kit from EMD Millipore, according to manufacturer's protocol (Billerica, MA, USA). To analyze and obtain the cytokine and chemokine concentrations, standard curves were generated using either two or three-fold dilution of recombinant cytokines provided by the manufacturer. Analysis was performed using the software provided by Luminex.
51Cr release cytotoxicity assay
The 51Cr release assay was performed as described previously [35]. Briefly, different numbers of purified NK cells were incubated with 51Cr-labeled tumor target cells. After a 4-hour incubation period, the supernatants were harvested from each sample and counted for released radioactivity using the gamma counter. The percentage specific cytotoxicity was calculated as follows:
% Cytotoxicity = Experimental cpm - spontaneous cpm / Total cpm - spontaneous cpm
LU (lytic unit) 30/106 is calculated by using the inverse of the number of effector cells needed to lyse 30% of tumor target cells X100.
Results
Differentiation of oral cancer stem-like cells by split-anergized NK cell supernatants resulted in upregulation of differentiation antigens and loss of susceptibility to NK cell-mediated cytotoxicity
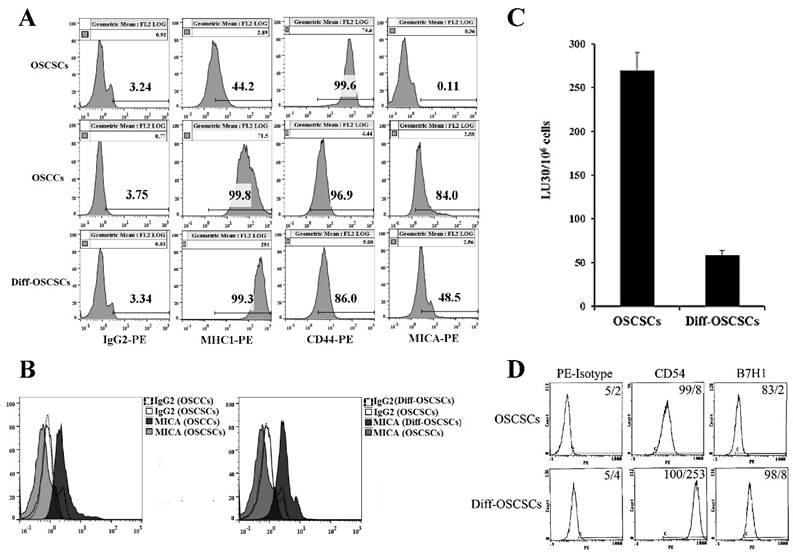
As shown previously [17, 18, 60], NK cells mediated much higher cytotoxicity against oral squamous carcinoma stem-like cells (OSCSCs) when compared to their differentiated counterpart, oral squamous carcinoma cells (OSCCs). OCSCSs displayed higher CD44 stem cell marker expression and lower MHC-I, MICA and CD54 levels (Fig 1A-1B and [22]), whereas the reverse profile was seen on their differentiated counterpart OSCCs (Fig. 1A-1B). To determine the effects of supernatants from split-anergized NK cells on OSCSCs, highly purified NK cells were treated with IL-2 and anti-CD16mAb for 18-24 hours before their supernatants were removed and added to OSCSCs. The treatment of OSCSCs with split-anergized NK cell supernatants significantly decreased their sensitivity to IL-2-treated NK cell-mediated lysis (Fig. 1C) and mediated upregulation of MHC-I (100 fold), CD54 (32 fold), B7H1 (4 fold) and decreased CD44 (15 fold) expression as compared to untreated OSCSCs (Fig. 1A and 1D). The levels of MICA were 4.6 fold higher on OSCCs and 5.3 fold higher on NK differentiated as compared to untreated OSCSCs (Fig. 1A-1B).
Differentiation stage of oral tumor cells correlates with sensitivity to NK cell-mediated lysis. OSCSCs were differentiated as described in the Materials and Methods section. The surface expression of CD44, MHC-I, and MICA on OSCSCs, OSCCs and split-anergized NK cell supernatant-differentiated OSCSCs was assessed using flow cytometric analysis after staining with respective PE-conjugated antibodies. Isotype control antibodies were used as controls (A). Histograms from isotype control and MICA staining for OSCSCs and OSCCs or OSCSCs and split-anergized NK cell supernatant-differentiated OSCSCs (diff-OSCSCs) are overlayed in this figure (B) The cytotoxicity against untreated and diff-OSCSCs using IL-2-treated NK cells was assessed in a 4-hour 51Cr release assay. Percent cytotoxicity was obtained at different effector to target ratio and the lytic units 30/106 cells were determined as described in the Materials and Methods (C). OSCSCs were differentiated with split-anergized NK cell supernatants as described in the Materials and Methods section and the surface expression of CD54 and B7H1 was assessed by flow cytometric analysis after staining with the respective PE-conjugated antibodies. Isotype control antibodies were used as controls. The numbers on the right-hand corner are the percentages and the mean channel fluorescence intensities in each histogram (D).
Differentiation stage of pancreatic cancer cells correlated directly with the resistance to NK cell-mediated cytotoxicity and expression of key surface antigens
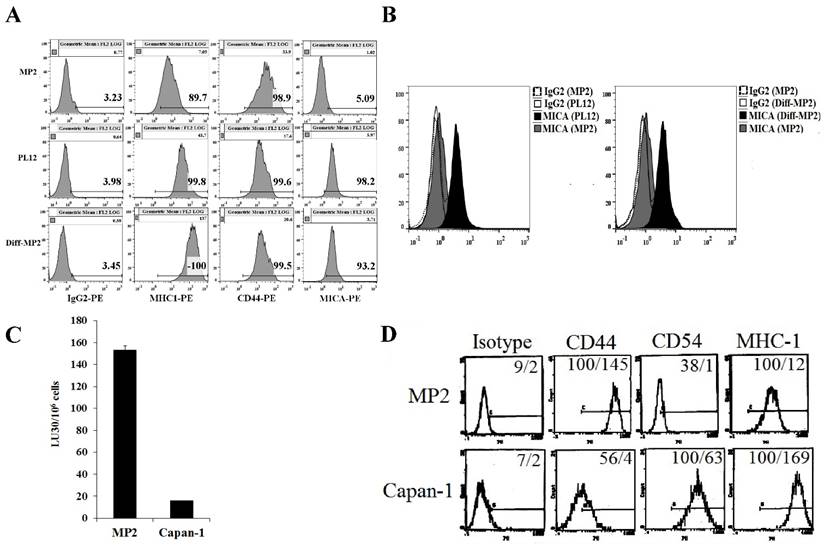
Six pancreatic tumor cells were screened for surface receptor expression of select markers, susceptibility to NK cell-mediated cytotoxicity, and secretion of cytokines and chemokines to determine the relationship between the differentiation stage of each tumor cell line and their susceptibility to NK cells (manuscript in preparation). In this study, we focused on poorly differentiated pancreatic tumor cell line, MP2, and highly differentiated pancreatic tumor cell lines, Capan-1 and PL-12 (Fig. 2). Similar to the profiles observed in OSCSCs and OSCCs, a direct correlation between the stage of differentiation, surface expression of differentiation antigens and susceptibility to NK cell-mediated cytotoxicity was observed in pancreatic tumor cells (Fig. 2). Stem-like/undifferentiated pancreatic tumor cells, MP2 had higher levels of CD44 and lower levels of CD54, MHC-I and MICA (Fig. 2A, 2B and 2D), and they were excellent targets for NK cell-mediated lysis (Fig. 2C) in comparison to more differentiated pancreatic tumor cell lines, Capan-1 and PL-12. Two differentiated pancreatic tumor cells were used for comparison with stem-like/undifferentiated tumors to demonstrate reproducibility. We have previously shown that the addition of supernatants from split-anergized NK cells to MP2 tumor cells also lowered sensitivity to NK cell-mediated lysis, upregulated B7H1 and MHC-I and downregulated CD44 expression [18]. Our recent studies also demonstrated that differentiated pancreatic tumor cells, both patient-derived and those differentiated by NK cells, did not grow or metastasize to vital organs in NSG mice after orthotopic injection of tumors in the pancreas (manuscript in preparation).
Stem-like pancreatic MP2 tumor cells differentiated with split-anergized NK cell supernatants, as well as patient-derived, well-differentiated pancreatic tumor cells, PL12 and Capan-1 were resistant to NK cell-mediated lysis and had higher surface expression of differentiation antigens. MP2 cells were differentiated as described in the Materials and Methods section. The surface expression of CD44, MHC-I, and MICA on MP2, PL12 and split-anergized NK cell supernatant-differentiated MP2 tumor cells was assessed using flow cytometric analysis after staining with respective PE-conjugated antibodies. Isotype control antibodies were used as controls (A). Histograms from isotype control and MICA staining for MP2 and PL-12 or MP2 and split-anergized NK cell supernatant-differentiated MP2 (diff-MP2) cells are overlayed in this figure (B). Freshly isolated NK cells were treated with IL-2 (1000 units/mL) for 18-24 hours before they were added to 51Cr labeled MP2 and Capan-1 cells. NK cell-mediated cytotoxicity was determined using a 4-hour 51Cr release assay and the lytic units 30/106 cells were determined as described in the Materials and Methods (C). Surface expression of CD44, CD54, and MHC-I was determined on MP2 and Capan-1 cells by flow cytometric analysis after staining with the respective PE-conjugated antibodies. Isotype control antibodies were used as controls. The numbers on the right hand corner are the percentages and the mean channel fluorescence intensities in each histogram (D).
Differentiation of glioblastoma cancer stem-like cells with supernatants from split-anergized NK cells resulted in decreased susceptibility to NK cell lysis and upregulation of CD54 and MHC-I surface expression
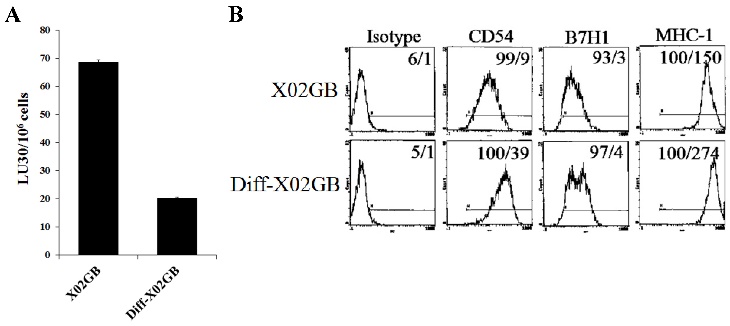
We have recently demonstrated that IL-2-treated NK cells-mediated higher cytotoxicity against X02GB, which is a CD44-high glioblastoma stem-like tumor, as compared to more differentiated U87 glioblastoma cells with lower CD44 surface expression [56, 57]. Differentiation of X02GB with supernatants from split-anergized NK cells caused a 3.5 fold inhibition in their susceptibility to lysis by IL-2-treated NK cells (Fig. 3A) and significantly upregulated CD54 (4.3 fold) and MHC-I (1.8 fold) expression when compared to undifferentiated X02GB (Fig. 3B and [57]).
Differentiation of stem-like X02GB tumors with split-anergized NK cell supernatants resulted in their decreased susceptibility to NK cell-mediated cytotoxicity and upregulation of CD54 and MHC-I surface expression. Differentiation of X02GB was carried out as described in the Materials and Methods section. Afterwards, untreated and NK supernatant differentiated X02GB (diff-X02GB) were labeled with 51Cr and used as target cells against IL-2-treated NK cells in a 4-hour 51Cr release cytotoxicity assay. Percent cytotoxicity was obtained at different effector to target ratios and the lytic units 30/106 cells were determined as described in the Materials and Methods (A). Surface expression of CD54, B7H1, and MHC-I on untreated and diff-X02GB was assessed by flow cytometric analysis after staining with the respective PE-conjugated antibodies. Isotype control antibodies were used as control. The numbers on the right-hand corner are the percentages and the mean channel fluorescence intensities in each histogram (B).
CD44 knockdown in breast cancer and melanoma cell lines resulted in an increased sensitivity to NK cell-mediated lysis
Since increased CD44 correlated with stem-like/undifferentiated phenotype of tumor cells, we used CD44 knockdown tumor cells to assess their susceptibility to NK cell-mediated cytotoxicity. CD44 was knocked down in human breast cancer cell line, MDA-MB231 (MDA-MB231shCD44) and human melanoma cell line, A375 (A375shCD44) as described [58]. Cells stably expressing shRNA against firefly luciferase were used as controls (MDA-MB231shLUC and A375shLUC). CD44 knockdown and control cell lines were used as target cells against IL-2-treated NK cells (Fig. 4A). Surprisingly, MDA-MB23shCD44 cells were more susceptible to NK cell-mediated lysis than MDA-MB231shLUC (Figs. 4A-4B). CD44 knockdown in MDA-MB231 also resulted in a downmodulation of CD54, B7H1 and MHC-I (Fig. 4C).
Increased susceptibility of CD44 knockdown MDA-MB231 and A375 tumor cells to NK cell-mediated cytotoxicity, and the induction of resistance after treatment with supernatants from split-anergized NK cells or IFN-γ. NK cells were treated with IL-2 (1000 units/mL) for 18-24 hours before they were added to 51Cr labeled MDA-MB231shLUC or MDA-MB231shCD44 tumor cells. NK cell-mediated cytotoxicity was determined using a 4-hour 51Cr release assay (A). MDA-MB231shLUC and MDA-MB231shCD44 tumor cells were treated with recombinant human IFN-γ (50 units/mL) for 24 hours. Afterwards, the cells were detached from the tissue culture plates and labeled with 51Cr and used in a 4-hour 51Cr release assay against IL-2-treated NK cells. Percent cytotoxicity was obtained at different effector to target ratio and the lytic units 30/106 cells were determined as described in the Materials and Methods (B). The surface expression of CD44, CD54, B7H1, and MHC-I on MDA-MB231shLUC and MDA-MB231shCD44 was assessed with flow cytometric analysis after staining with the respective PE-conjugated antibodies. Isotype control antibodies were used as controls. The numbers on the right hand corner are the percentages and the mean channel fluorescence intensities in each histogram (C). A375shLUC and A375shCD44 cells were left untreated or treated with recombinant human IFN-γ (50 units/mL) for 24 hours. Afterwards, the cells were detached from the tissue culture plates and labeled with 51Cr and used in a 4-hour 51Cr release assay against untreated (D), IL-2-treated (1000 units/mL) (E), and IL-2 (1000 units/mL) and anti-CD16mAb (3μg/mL) treated (F) NK cells. Pre-treatment of NK cells with IL-2 or IL-2+anti-CD16mAb was carried out for 18-24 hours. Percent cytotoxicity was obtained at different effector to target ratio and the lytic units 30/106 cells were determined as described in the Materials and Methods. Differentiation of A375shLUC and A375shCD44 cells with split-anergized NK cell supernatants was carried out as described in the Materials and Methods section. The cytotoxicity against untreated, diff- A375shLUC, and diff- A375shCD44 with untreated (G) and IL-2 (1000 units/ml) treated (H) NK cells was assessed using 4-hour 51Cr release assay. Percent cytotoxicity was obtained at different effector to target ratio and the lytic units 30/106 cells were determined as described in the Materials and Methods. A375shLUC and A375shCD44 were treated with split-anergized NK cell supernatants as described in the Materials and Methods section and the surface expression of CD44, CD54, MHC-I and MICA was assessed with flow cytometric analysis after staining with the respective PE-conjugated antibodies (I, J). Isotype control antibodies were used as controls. The numbers on the right-hand corner are the percentages and the mean channel fluorescence intensities for each histogram in Fig. (I) as well as mean channel fluorescence intensities in Fig. (J) with percentages shown above the cursors.
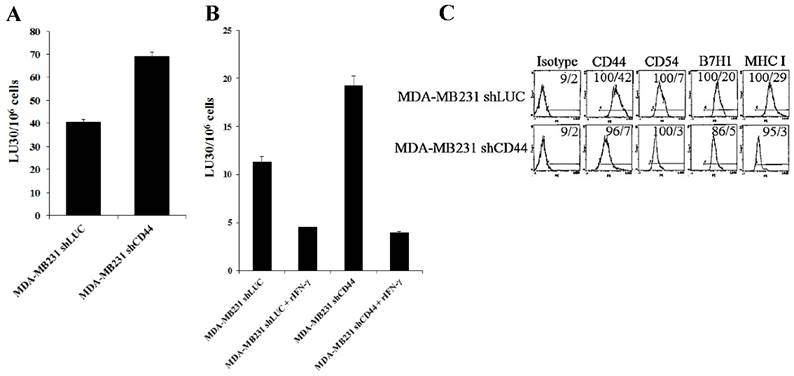
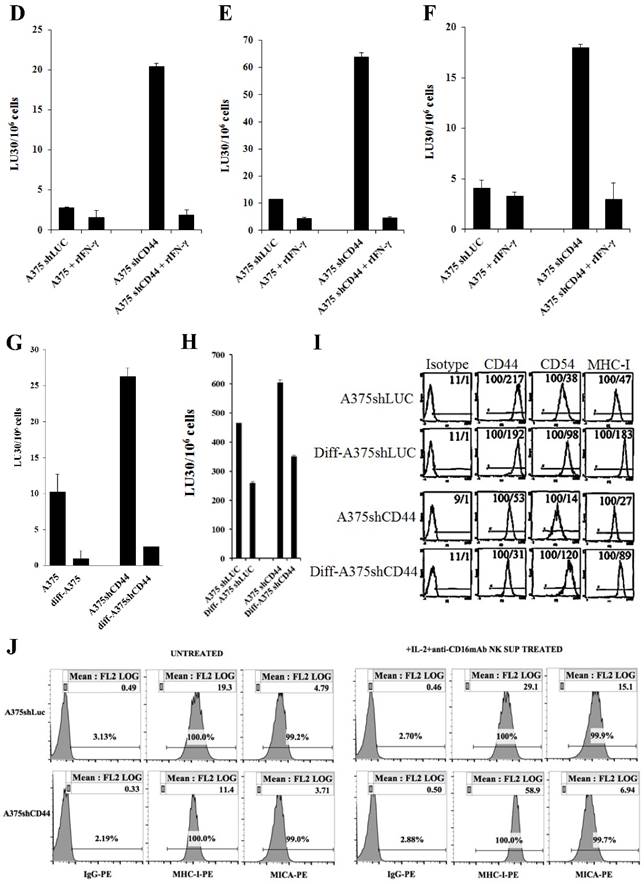
In our previous studies and in here, we demonstrated that IFN-γ secreted by split-anergized NK cells induced differentiation in OSCSCs and caused resistance against NK cell-mediated cytotoxicity [18]. As shown in Fig. 4B, the fold increase in resistance against IL-2-treated NK cell-mediated lysis by the addition of rIFN-γ to the cells was greater in MDA-MB231shCD44 (5 fold) than MDA-MB231shLUC (3 fold) (Fig. 4B).
Similarly, A375 shCD44 melanoma cells were more susceptible to untreated, IL-2 or IL-2+anti-CD16mAb-treated NK cells when compared to A375shLUC tumor cells (Fig. 4D-4F). A decrease in NK cell susceptibility was observed when untreated (Fig. 4D), IL-2-treated (Fig. 4E), or IL-2+anti-CD16mAb treated (Fig. 4F) NK cells were added to A375shLUC or A375 shCD44 cells pre-treated with rIFN-γ, and the fold increase in resistance compared to untreated tumors was much greater in A375shCD44 cells treated with IFN-γ than in IFN-γ-treated A375shLUC. The addition of rIFN-γ to A375shCD44 cells also induced higher fold increase in the surface expression of CD54, B7H1 and MHC-I in comparison with A375shLUC treated with rIFN-γ (data not shown).
Treatment of A375shCD44 and A375shLUC with split-anergized NK cell supernatants (diff-A375shCD44 and diff-A375shLUC) decreased substantially cytotoxicity of both untreated (Fig. 4G) or IL-2-treated (Fig. 4H) NK cells. The increase in resistance against NK cell cytotoxicity observed in diff-A375shCD44 and diff-A375shLUC directly correlated with an increase in CD54, MHC-I and MICA expression (Fig. 4I and 4J).
The resistance of lung cancer cells to NK cell-mediated cytotoxicity after differentiation with split-anergized NK cell supernatants was mediated by the combination of TNF-α and IFN-γ
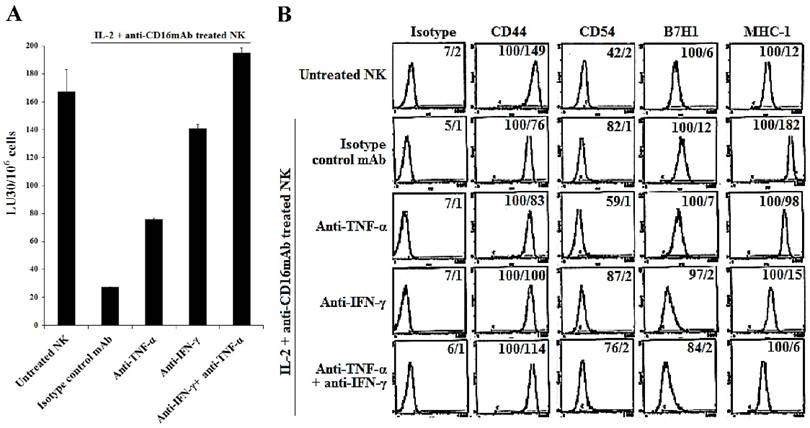
We have previously shown that resistance of OSCSCs or MP2 pancreatic cancer cells to NK cell-mediated cytotoxicity after differentiation by the supernatants from split-anergized NK cells was caused by the combination of TNF-α and IFN-γ function [18]. We aimed to determine whether similar pattern of resistance to cytotoxicity could be seen when A549, a lung tumor line was also differentiated with split-anergized NK cell supernatants, and if the resistance was mediated by the dual function of TNF-α and IFN-γ. Treatment of A549 cells with split-anergized NK cell supernatants significantly decreased NK cell-mediated lysis by IL-2-treated NK cells (Fig. 5A). To examine the mechanism by which A549 cells become resistant to NK cells, we determined NK cell-mediated cytotoxicity after the addition of each of TNF-α and IFN-γ antibodies alone or in combination with split-anergized NK cell supernatant to A549 tumor cells (Fig. 5A). The addition of each of the antibodies to TNF-α and IFN-γ alone had inhibitory effect on the induction of resistance to NK cells. The combination of anti-TNF-α and anti-IFN-γ completely abrogated NK cell resistance of A549 tumor cells differentiated with split-anergized NK cell supernatants (Fig. 5A). Decreased susceptibility of A549 to NK cell-mediated cytotoxicity induced by split-anergized NK supernatants directly correlated with the increase in CD54, B7H1 and MHC-I surface expression, and the addition of the combination of anti-IFN-γ and anti-TNF-α antibodies completely abrogated the upregulation of the above-mentioned surface antigens (Figs. 5A and 5B). Anti-IFN-γ had a more pronounced effect on restoration of NK cytotoxicity and modulation of surface receptor expression in comparison with TNF-α (Fig. 5B).
The induction of NK cell resistance in split-anergized NK supernatant-differentiated A549 was mediated by TNF-α and IFN-γ. Supernatants from untreated and split-anergized NK cells were prepared as described in the Materials and Methods section, and then added to A549 cells in the presence or absence of anti-TNF-α (1:100) and/or anti-IFN-γ (1:100). The cytotoxicity against untreated and split-anergized NK supernatant-differentiated A549 in the presence of antibodies to TNF-α and IFN-γ using IL-2 treated NK cells was assessed in a 4-hour 51Cr release assay. Percent cytotoxicity was obtained at different effector to target ratio and the lytic units 30/106 cells were determined as described in the Materials and Methods (A). The surface expression of CD44, CD54, B7H1, and MHC-I on untreated and split-anergized NK supernatant-treated A549 tumor cells in the presence and absence of anti-TNF-α (1:100) and/or anti-IFN-γ (1:100) was assessed by flow cytometric analysis after PE-conjugated antibody staining. Isotype control antibodies were used as controls. The numbers on the right hand corner are the percentages and the mean channel fluorescence intensities in each histogram (B).
Radiation or treatment with chemotherapeutic agent cisplatin (CDDP) induced significant cell death in OSCCs as well as OSCSCs, MP2 and A375shCD44 cells differentiated with split-anergized NK cell supernatant
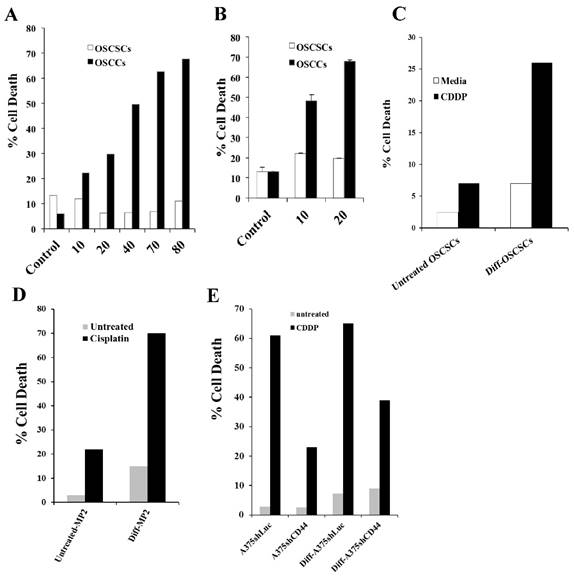
OSCSCs and OSCCs were treated with increasing concentrations of CDDP and the percent cell death was determined using propidium iodide (PI) staining followed by flow cytometric analysis (Fig. 6A). CDDP induced significant cell death in OSCCs as compared to OSCSCs (Fig. 6A). Similarly, OSCCs were highly susceptible to radiation treatment, whereas OSCSCs did not undergo cell death after radiation (Fig. 6B). Differentiation of OSCSCs (Fig. 6C) or MP2 cells (Fig. 6D) with supernatants from split-anergized NK cells resulted in a significant increase in their susceptibility to CDDP, whereas untreated OSCSCs or MP2 cells remained relatively resistant to CDDP (Fig. 6C and 6D). A375shLUC melanoma cells demonstrated higher differentiation antigens, and were significantly more susceptible to CDDP-mediated death when compared with A375shCD44. Differentiation with split-anergized NK cell supernatant did not change the levels of A375shLUC cell death but it decreased viability of A375shCD44 cells (Fig. 6E).
Cisplatin induced significant death in OSCCs, A375shLUC and diff-OSCSCs, diff-MP2 and diff-A375shCD44 but not in stem-like/undifferentiated OSCSCs, MP2 and A375shCD44 cells. OSCSCs and OSCCs were seeded in 24-well plates at 1X105 cells/well for 18-24 hours before the cells were treated with CDDP at 10, 20, 40, 70, and 80 μg/mL for 24 hours. The viability of the cells was determined using propidium iodide staining and analyzed by flow cytometry (A). OSCSCs and OSCCs were seeded in 24-well plates at 1X105 cells/well for 18-24 hours before the cells were irradiated at 10 or 20 Gy. Afterwards, the viability of the cells was determined using propidium iodide staining and analyzed by flow cytometry (B). OSCSCs, MP2, A375shLUC and A375shCD44 cells were left undifferentiated or differentiated with split-anergized NK cell supernatants as described in the Materials and Methods section. Untreated or Diff-OSCSCs (C), Diff-MP2 (D), Diff- A375shLUC and diff-A375shCD44 (E) were seeded in 24-well plates at 1X105 cells/well for 18-24 hours before the cells were treated with CDDP at 60 μg/mL for OSCSCs and MP2 cells, and 20 μg/mL for A375shLUC and A375shCD44. After an overnight treatment period, the viability of tumors was determined using propidium iodide staining followed by flow cytometric analysis.
Decreased VEGF secretion in co-cultures of NK cells with sulindac-treated oral cancer stem-like cells and their differentiated counterparts
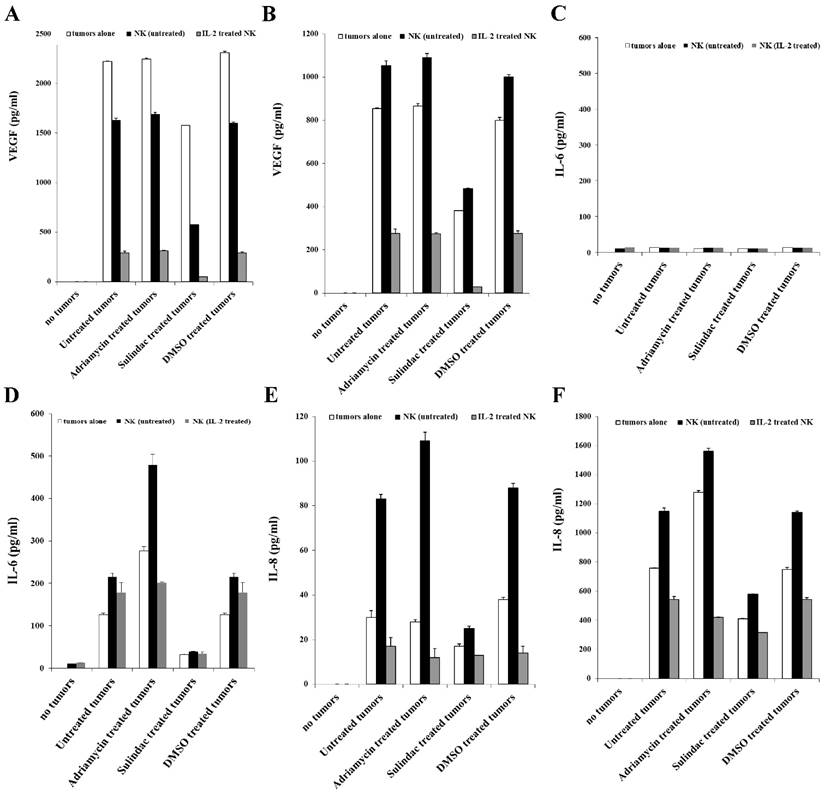
We next determined the levels of VEGF secretion in the co-cultures of NK cells with OSCSCs or OSCCs treated with or without adriamycin or sulindac (Figs. 7A and 7B). No significant release of VEGF could be observed from untreated or IL-2-treated NK cells (Figs. 7A and 7B). In contrast, OSCSCs and OSCCs oral tumors secreted high levels of VEGF (Table 1 and Fig. 7A and 7B). Synergistic induction of VEGF release was observed when untreated NK cells (Table 1 and Fig. 7B) were co-cultured with OSCCs, whereas there was a decrease in VEGF secretion in co-cultures of untreated NK cells and stem-like OSCSCs in comparison to VEGF levels secreted by tumor cells alone (Fig. 7A). Secretion of VEGF in the supernatants obtained from the co-cultures of IL-2-treated NK cells with tumor cells was significantly decreased for both cell lines (Figs. 7A and 7B), due to an increased lysis of tumor cells by IL-2-treated NK cells (Table 1). Sulindac treatment of oral tumor cells cultured in the absence or presence of untreated and IL-2-treated NK cells decreased VEGF secretion (Figs. 7A and 7B). Moreover, VEGF production in OSCCs in the presence of untreated NK cells was suppressed when tumor cells were treated with sulindac, but not with adriamycin (Figs. 7A and 7B).
Decreased VEGF, IL-6, and IL-8 secretion in NK cells co-cultures with sulindac-treated OSCSCs or OSCCs. OSCSCs (A, C, E) and OSCCs (B, D, F) were left untreated or treated with sulindac (200μM), adriamycin (10 μg/mL), or DMSO control. After an overnight incubation, tumor cells were washed twice and co-cultured with untreated or IL-2-treated NK cells at an effector to target ratio of 5:1. Treatment of NK cells with IL-2 (500 units/mL) was carried out for 8-12 hours before their co-culture with the tumor cells. The concentrations of VEGF (A and B), IL-6 (C and D) and IL-8 (E and F) in the cell culture supernatants were measured with a multiplex cytokine array kit after an overnight incubation.
Inability of untreated NK cells to lyse OSCCs oral tumors resulted in synergistic induction of VEGF secretion by the tumor cells. NK cells were left untreated or treated with IL-2 (500units/mL) for 8-12 hours before they were added to OSCCs primary oral tumors at an effector to target ratio of 5:1. The levels of VEGF secretion in each sample were determined using a multiplex ELISA array kit specific for VEGF. * Values were normalized to the levels of VEGF secretion obtained in oral tumors in the absence of immune effectors. NK cell cytotoxicity was determined using a standard 51Cr release assay and the lytic units 30/106 were determined using inverse number of effectors required to lyse 30% of the tumor cells X 100.
| LU30/106 | VEGF (pg/mL) | %VEGF* | |
|---|---|---|---|
| NK control | --- | 5.1 | 1.3 |
| NK IL-2 | ---- | 4.7 | 1.2 |
| OSCCs tumor alone | ---- | 367.4±14 | 100 |
| + NK control | 3.3±0.0 | 713.4±43 | 197 |
| + NK IL-2 | 27.8±0.8 | 61±22 | 17 |
Decreased secretion of IL-6 in sulindac-treated oral cancer cells with and without NK cells
OSCSCs secreted very low levels of IL-6, and sulindac treatment and/or their co-culture with NK cells did not increase secretion of IL-6 (Fig. 7C). In contrast, OSCCs secreted IL-6 and their co-cultures with NK cells increased IL-6 production. As shown in Fig. 7D, severalfold increase in IL-6 secretion could be observed in supernatants obtained from the co-cultures of NK cells with adriamycin-treated OSCCs. OSCCs treatment with sulindac was able to significantly decrease IL-6 secretion in the absence and presence of co-cultures with NK cells when compared to those, which were left, untreated or treated with DMSO or adriamycin (Fig. 7D). On the other hand, IL-6 secretion in OSCSCs that were co-cultured with NK cells showed very low levels under all conditions (Fig. 7C).
Decreased IL-8 secretion in sulindac-treated oral tumor cells with and without NK cells
Co-cultures of untreated or IL-2-activated NK cells with sulindac-treated OSCCs or OSCSCs were analyzed for IL-8 secretion. In the absence of tumor cells, both untreated and IL-2-activated NK cells showed no IL-8 secretion (Figs. 7E and 7F). Synergistic induction of IL-8 secretion was observed when untreated NK cells were co-cultured with untreated, DMSO- or adriamycin-treated oral tumor cells. In contrast, IL-8 levels obtained from IL-2 activated NK cell co-cultures with untreated, DMSO- and adriamycin-treated oral tumor cells were significantly decreased. This could once again be due to increased lysis of tumor cells by IL-2 activated NK cells. Decrease in IL-8 secretion was observed in both stem-like and differentiated sulindac-treated tumor cells with or without NK cells (Figs. 7E and 7F).
Increased NK cell cytotoxicity against sulindac-treated HEp2 cells
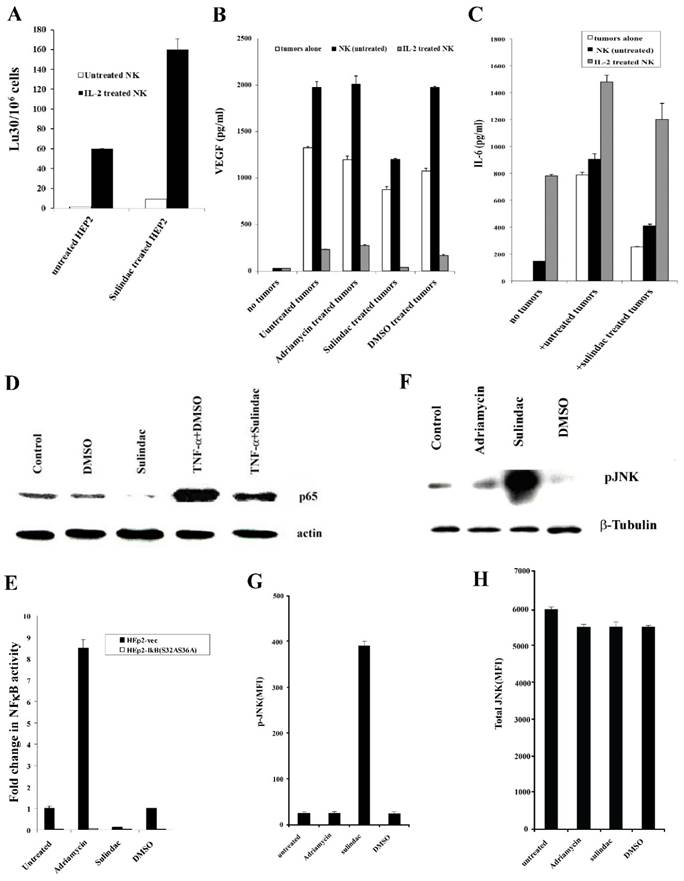
HEp2 cells were treated with sulindac at a concentration that was not cytotoxic before their co-culture with NK cells. Untreated and IL-2-treated NK cells mediated increased lysis of sulindac-treated HEp2 cells when compared to untreated HEp2 cells (Fig. 8A).
Treatment of HEp2 cells with sulindac resulted in increased susceptibility to NK cell -mediated cytotoxicity, decreased VEGF and IL-6 secretion in the co-cultures with NK cells, decreased NFκB nuclear expression and increased p-JNK. HEp2 cells (4X106) were left untreated or treated with sulindac at 200 μM for 4 hours. The cells were then washed and labeled with 51Cr and used as target cells against untreated or IL-2 (500 units/mL) pre-treated NK cells in 4-hour 51Cr release assay (A). HEp2 tumors were left untreated or treated with sulindac (200 μM), adriamycin (10 μg/mL), and DMSO for 12-18 hours. Afterwards, the tumor cells were washed twice and co-cultured alone or with either untreated or IL-2-treated (500 units/mL) NK cells at an effector to target ratio of 5:1. After an overnight incubation, the supernatants were removed from the co-cultures and the levels of VEGF (B) and IL-6 (C) secretion were determined using specific ELISAs. HEp2 tumor cells were treated with sulindac (200 μM), TNF-α (20 ng/mL), and DMSO alone or in combination, as indicated in the figure. After an overnight incubation, the nuclear extracts were prepared from each sample and run on a polyacrylamide gel. The levels of p65 NFκB sub-unit were determined in each sample. Actin expression for the nuclear extracts was used as the loading control (D). HEp2-vec and HEp2-IκB(S32AS36A) tumor cells were transfected with NFκB Luciferase reporter vector, as described in the Materials and Methods section, after which the transfected tumors were treated with and without sulindac (200 μM), adriamycin (10 μg/mL), and DMSO for 12-18 hours before they were lysed and the relative luciferase activity was measured over untreated tumors (E). HEp2 cells were treated with adriamycin (10μg/mL), sulindac (200 μM), or DMSO. After an overnight incubation, the nuclear extracts were prepared from each sample and were run on a polyacrylamide gel to determine the expression of phospho-JNK (p-JNK). β-tubulin expression was used as the loading control in the western blot (F). The levels of p-JNK (G) and total JNK (H) were determined using multiplex array kit. The Median Fluorescence Intensity (MFI) was measured with the Luminex system. The figures represent the average and standard deviation of three replicate wells.
Decreased VEGF secretion in co-cultures of NK cells with sulindac-treated HEp2 cells
We determined VEGF secretion in the co-cultures of NK cells with sulindac-treated HEp2 cells. No significant induction of VEGF release could be observed from untreated or IL-2-treated NK cells in the absence of HEp2 cells. Synergistic induction of VEGF release was observed when untreated NK cells were co-cultured with HEp2 cells (Fig. 8B). Supernatants obtained from the co-cultures of IL-2-treated NK cells with HEp2 demonstrated a significant decrease in VEGF levels (Fig. 8B), which is likely due to the increased lysis of tumor cells by IL-2-treated NK cells (Fig. 8B). Sulindac treatment decreased VEGF secretion by tumor cells in the presence and absence of NK cells when compared with untreated, DMSO- and adriamycin-treated tumor cells (Fig. 8B).
Decreased secretion of IL-6 in co-cultures of NK cells with sulindac-treated HEp2 cells
Since sulindac-treated OSCCs triggered lower IL-6 secretion, both alone or in NK co-cultures, we aimed to determine whether a decreased IL-6 secretion could also be seen in HEp2 cells cultured with untreated or IL-2-treated NK cells. HEp2 cells treated with sulindac in the presence or absence of untreated NK cells decreased IL-6 secretion (Fig. 8C). Please note that NK cells treated with IL-2 secreted higher levels of IL-6 in the absence of tumor cells, and in the presence of untreated and sulindac-treated tumor cells secretion of IL-6 rose, indicating that IL-6 secretion is likely produced by IL-2 treated NK cells and not by HEp2 tumors. Moreover, the levels of IL-6 secretion were moderately decreased in sulindac-treated tumor co-cultures with IL-2 treated NK cells in comparison to those cultured with untreated tumor cells (Fig. 8C).
Downmodulation of NFκB by sulindac
We have previously shown that NFκB knockdown in HEp2 cells resulted in their increased susceptibility to NK cell-mediated cytotoxicity [36]. Therefore, we wanted to determine the effect of sulindac on NFκB expression. Nuclear extracts obtained from DMSO, sulindac, TNF-α and the combination of TNF-α and sulindac-treated HEp2 cells were used in western blot analysis to analyze the levels of nuclear NFκB (Fig. 8D). As shown in Figs. 8D and 8E, decreased levels of nuclear NFκB were observed in sulindac-treated HEp2 cells as compared to either untreated or DMSO-treated HEp2 tumor cells. The concentration of DMSO used to solubilize sulindac did not affect the function of the cells (data not shown). Similarly, TNF-α-mediated increase in NFκB induction was inhibited when HEp2 tumor cells were treated with the combination of TNF-α and sulindac (Fig. 8D).
Upregulation of p-JNK in HEp2 tumor cells by sulindac
Protein extracts to measure p-JNK were obtained from Hep2 tumor cells treated with adriamycin, sulindac, or DMSO, and analyzed using western blot and multiplex protein arrays. Increased levels of p-JNK were observed in sulindac-treated Hep2 cells compared to untreated or DMSO-treated tumor cells in western blot and multiplex analysis (Figs. 8F and 8G). Although total JNK levels were comparable throughout all treatments, p-JNK levels in sulindac-treated HEp2 tumor cells were higher (Figs. 8G and 8H).
Discussion
We have recently shown in both in vitro and in vivo studies that COX2 deletion in myeloid cells substantially boosts NK cell activation [21]. In addition, we and others have demonstrated that deletion of NFκB in tumor cells significantly increases NK cell-mediated cytotoxicity and IFN-γ secretion [35, 36], and causes autoimmunity and inflammation in vivo [62, 63]. Moreover, conditional STAT3 knockout in hematopoietic cells was found to induce colitis in mice due to chronic gut inflammation [64, 65]. It has been described that various gene knockouts augment NK cell function, possibly due to the fundamental function of NK cells targeting cells with impaired capability to differentiate [21]. The list of candidate genes that activate NK cells and/or other key immune effectors when knocked down in the target cells have been increasing steadily [21]. In this paper, we also demonstrate that CD44 knockdown, which is a well-established marker for cancer stem-like cells of different origin, increases NK cells' cytotoxic function in both mammary tumors and in melanoma. It is counter-intuitive to suggest that CD44 knockdown may serve to rise NK cell cytotoxicity due to de-differentiation of the cells since CD44 is not only a marker of stemness but also its role in adhesion and cell-cell contact has been demonstrated in previous studies. Indeed, initially the rationale for the use of CD44 knockdown tumors was to demonstrate their resistance to NK cell-mediated cytotoxicity, since differentiated cells exhibited less CD44 and were significantly more resistant to NK cell-mediated cytotoxicity. However, to our surprise, we obtained the opposite result potentially strengthening the hypothesis that knockdown of important cellular genes including CD44 may result in de-differentiation of the cells and greater susceptibility to NK cell-mediated cytotoxicity.
We have previously shown that NK cells target poorly differentiated cells or stem cells with lower expression of key differentiation antigens [17-19]. In our studies we characterized the link between the stage of maturation and differentiation of tumors and their sensitivity to NK cell-mediated lysis. In this regard, we have shown that stem-like/poorly differentiated oral and pancreatic tumor cells were significantly more susceptible to NK cell-mediated cytotoxicity whereas, their differentiated counterparts were significantly more resistant [17]. Based on these results, we have proposed, that NK cells play a significant role in differentiation of cells by providing critical signals via secreted cytokines as well as direct cell-cell contact [18]. In addition, we have demonstrated that human monocytes promote significant split anergy in NK cells [16, 19, 21, 66-68]. Induction of split anergy in NK cell effector function is thought to ultimately aid in driving differentiation of healthy, as well as transformed stem-like cells [19, 66-69]. Therefore, it appears from our studies that the loss of key molecules involved in differentiation in target cells may activate NK cells in order to drive both selection and differentiation of interacting cells. Furthermore, we demonstrated that differentiated oral, pancreatic and glioblastoma tumor cells isolated from patients, and cancer stem cells/poorly differentiated tumor cells differentiated in vitro with supernatants from split-anergized NK cells became resistant to NK cell-mediated cytotoxicity. Unlike the CSCs/poorly differentiated tumor cells, both patient-derived differentiated tumor cells and split-anergized NK supernatant-differentiated tumor cells exhibited upregulated CD54, B7H1, and MHC class I surface expression, and demonstrated decreased CD44 expression. Tumor differentiation was largely mediated by both IFN-γ and TNF-α secreted by activated NK cells, since the addition of the combination of anti-TNF-α and anti-IFN-γ retained the OSCSCs, MP2 pancreatic [18] and A549 lung cancer cells (Fig. 5) in a non-differentiated stage as assessed by susceptibility to NK cell-mediated lysis and decreased B7H1 and MHC class I expression. Addition of IFN-γ augumented differentiation in A375 melanoma and MBA-MB231 breast cancer cells and upregulated CD54, B7H1, MHC class I and MICA surface expression similar to the effect mediated by split-anergized NK cell supernatants. Therefore, we demonstrated that differentiation of oral, pancreatic, glioblastoma, lung, melanoma and breast cancer cells either by split-anergized NK supernatants or addition of IFN-γ rendered the tumor cells resistant to NK cell-mediated cytotoxicity, whereas their stem-like/poorly differentiated counterparts remained susceptible to NK cell-mediated cytotoxicity. Additionally, expression of MICA was higher on differentiated OSCCs and PL12 when compared to undifferentiated OSCSCs and MP2 cell lines, and their levels rose when OSCSCs or MP2, A374shLUC or A375shCD44 cells were differentiated with supernatants from split-anergized NK cells, indicating that differentiation is the mechanism involved in upregulation of MICA expression in cancer cells.
Although stem-like oral and pancreatic tumor cells are highly susceptible to NK cell-mediated cytotoxicity, they are quite resistant to either CDDP-mediated or radiation-induced cell death, whereas their differentiated counterparts are killed efficiently by either treatment. Differentiation with split- anergized NK cell supernatants made the tumor cells susceptible to CDDP-mediated cell death, indicating that differentiation of CSCs by NK cells is a crucial pre-conditioning step for the success of chemotherapy. Interestingly, A375shCD44 cells were quite resistant to CDDP-mediated cell death whereas A375shLUC cells were significantly more susceptible. Differentiation with split-anergized NK cell supernatants increased susceptibility to CDDP in A375shCD44 tumor cells. This data suggests that knockdown of cellular genes, and their reversion to a less differentiated phenotype may activate NK cell mediated cytotoxicity but it may also lead to resistance of those cells to chemotherapeutic agents. Thus, stage of differentiation is a clear determinant of tumor susceptibility to NK cell mediated cytotoxicity as well as their response to chemotherapeutic drugs. Similarly, sulindac, a chemopreventive agent, inhibited only 29% of VEGF secreted by OSCSCs whereas it decreased 55% of secretion by OSCCs, demonstrating direct effect on VEGF inhibition. However, in the presence of NK cells, sulindac inhibited 65% of VEGF secreted by OSCSCs since OSCSCs are susceptible to NK cell-mediated cytotoxicity whereas no significant change in VEGF level could be seen in OSCCs in the presence of untreated NK cells. This data indicates that tumor susceptibility to chemotherapheutics and NK cells operates in a differential manner. In addition, untreated NK cells synergize with sulindac to inhibit VEGF secretion in stem-like but not in differentiated cancer cells. Furthermore, in the absence of sulindac, IL-2-treated NK cells inhibited 87% of VEGF secreted by OSCSCs whereas they decreased 67% of VEGF secretion by OSCCs. Although the trend for IL-8 secretion remained somewhat similar, lower secretion in OSCSCs co-cultures with the NK cells and potential contribution of both tumor cells and NK cells to IL-8 secretion made it difficult to draw conclusions based on the results. OSCSCs did not secrete IL-6 and no increase in IL-6 secretion could be observed in co-cultures with NK cells, whereas OSCCs secreted IL-6 and the secreted levels were modulated in the presence of NK cells. Although there were important differences in VEGF secretion, depending on the differentiation status of oral tumor cells cultured with or without NK cells, overall, sulindac inhibited VEGF, IL-8 and IL-6 secretion in both cell types, emphasizing the key function of this agent in chemoprevention.
Sulindac as well as other NSAIDs, such as aspirin, have been demonstrated to inhibit NFκB activation [32, 33, 70, 71]. In agreement with those studies, we also show that sulindac treatment significantly inhibits nuclear NFκB expression, as well as TNF-α-mediated NFκB upregulation in HEp2 tumor cells. NFκB downmodulation by sulindac is shown to be caused by specific inhibition of Iκκ kinase activity, which prevents degradation of IκBα and results in a decreased translocation of NFκB [70]. We have previously shown that targeted downmodulation of NFκB in HEp2 tumor cells auguments IFN-γ secretion by the NK and T cells, prevents depletion of immune cells and results in functional activation of NK and T cells [35]. Thus, increased function of NK cells co-cultured with sulindac-treated tumor cells may be due to downmodulation of NFκB nuclear function.
The antineoplastic effect of sulindac is well established in gastrointestinal tract cancers, in particular in colon cancer [72]. Sulindac, sulindac sulfone and sulindac sulfide were found to induce apoptosis in HEpG2 cells. Sulindac inhibited proliferation and increased caspase-3 activation in a dose-dependent manner in HepG2 cells [73]. Similar results were obtained in a study using human gastric cancer cells [74]. Thus, these studies suggested that inhibition of NFκB might be one of the mechanisms by which sulindac-treated tumor cells are signaled to undergo cell death. In addition, we and others have shown previously, and demonstrated in this report, that decrease in NFκB in the presence of JNK activation could be the potential signal for cells to undergo cell death [50]. Therefore, sulindac may exert its suppressive effect through induction of cell death in tumors.
Tumor growth is generally associated with immunosupression. Signaling abnormalities, spontaneous apoptosis and reduced proliferation of NK, T cells and dendritic cells (DCs) have all been observed in patients with oral cancer [44, 75]. Furthermore, cytotoxic function of NK and T cells is inhibited by a variety of tumor target cells [60, 61]. Thus, treatment with sulindac may be one way to reduce inactivation and death of immune effectors in oral cancer patients. Sulindac may not only inhibit the survival and function of tumor cells, but it may also augment survival and function of cytotoxic immune effectors resulting in further lysis of tumor cells.
Increased IL-6 levels have been demonstrated in oral cancer patients. Furthermore, squamous cell carcinomas of head and neck (HNSCC) are shown to secrete significant levels of IL-6 [44, 76-78]. IL-6 has been shown to impair the development of DCs [75] as well as the NK function in athymic mice [43]. Thus, use of sulindac to control IL-6 secretion by tumor cells may rescue NK cells from undergoing functional inactivation as a result of interaction with tumor cells. Therefore, the use of NSAIDs, such as sulindac, as an adjunct therapy in several digestive tract and oral neoplasias are well warranted and should limit the tumor burden as well as enhance cytotoxic immune cell function.
In this study, we conclude that conventional regimens that target differentiated tumor cells should be supplemented with immunotherapy that targets and eliminates poorly differentiated/ cancer stem-like cells. Since cancer stem cells resist chemotherapy and radiotherapy but can be targeted by NK cells, whereas differentiated oral tumor cells are resistant to NK cell-mediated cytotoxicity but are more sensitive to chemotherapeutic drugs, a tailored therapeutic scheme should improve elimination of both undifferentiated and differentiated cancer cells present in the tumor nest. In addition, for those cancer patients whose NK cells have lost ability to kill and/or differentiate tumor cells repeated allogeneic NK cell transplantation should support elimination of cancer stem cells.
Acknowledgements
Anna K. Kozlowska was supported by the Polish Ministry of Sciences and Higher Education and Mobility Plus award.
We are grateful to Omid Nikjeh, Nick Ohanian, Jessica Cook and Jessica Chiang, for their technical help and critical review of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806-23
2. Rodriguez-Iglesias J, Maranon E, Castrillon L, Riestra P, Sastre H. Life cycle analysis of Municipal Solid Waste management possibilities in Asturias, Spain. Waste Manag Res. 2003;21:535-48
3. Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Mol Cancer Ther. 2010;9:2450-7
4. Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15:338-44
5. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P. et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nature medicine. 2011;17:1086-93
6. Rich JN, Bao S. Chemotherapy and cancer stem cells. Cell Stem Cell. 2007;1:353-5
7. Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980-4
8. Cabrera CM, Nieto A, Cortes JL, Montes RM, Catalina P, Cobo F. et al. The low rate of HLA class I molecules on the human embryonic stem cell line HS293 is associated with the APM components' expression level. Cell Biology International. 2007;31:1072-8
9. Suarez-Alvarez B, Rodriguez RM, Calvanese V, Blanco-Gelaz MA, Suhr ST, Ortega F. et al. Epigenetic Mechanisms Regulate MHC and Antigen Processing Molecules in Human Embryonic and Induced Pluripotent Stem Cells. Plos One. 2010:5
10. Qin J, Han B, Pang J. [The relationship between TIL from human primary hepatic carcinoma and prognosis]. Zhonghua Yi Xue Za Zhi. 1997;77:167-70 [Article in Chinese]
11. Aparicio-Pages MN, Verspaget HW, Pena AS, Lamers CB. Natural killer cell activity in patients with adenocarcinoma in the upper gastrointestinal tract. Journal of clinical & laboratory immunology. 1991;35:27-32
12. Rocca YS, Roberti MP, Arriaga JM, Amat M, Bruno L, Pampena MB. et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun. 2013;19:76-85
13. Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815-22
14. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L. et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4(+)CD25(high)FOXP3(+) regulatory T cells. Stem Cells. 2008;26:212-22
15. Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111:1327-33
16. Jewett A, Man Y-G, Tseng H-C. Dual Functions of Natural Killer Cells in Selection and Differentiation of Stem Cells; Role in Regulation of Inflammation and Regeneration of Tissues. Journal of Cancer. 2013;4:12-24
17. Tseng H-C, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A. et al. Increased Lysis of Stem Cells but Not Their Differentiated Cells by Natural Killer Cells; De-Differentiation or Reprogramming Activates NK Cells. Plos One. 2010:5
18. Tseng HC, Bui V, Man YG, Cacalano N, Jewett A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Frontiers in immunology. 2014;5:269
19. Jewett A, Arasteh A, Tseng HC, Behel A, Arasteh H, Yang W. et al. Strategies to rescue mesenchymal stem cells (MSCs) and dental pulp stem cells (DPSCs) from NK cell mediated cytotoxicity. PLoS One. 2010;5:e9874
20. Jewett A, Cacalano NA, Head C, Teruel A. Coengagement of CD16 and CD94 receptors mediates secretion of chemokines and induces apoptotic death of naive natural killer cells. Clinical Cancer Research. 2006;12:1994-2003
21. Tseng HC, Arasteh A, Kaur K, Kozlowska A, Topchyan P, Jewett A. Differential Cytotoxicity but Augmented IFN-gamma Secretion by NK Cells after Interaction with Monocytes from Humans, and Those from Wild Type and Myeloid-Specific COX-2 Knockout Mice. Front Immunol. 2015;6:259
22. Tseng HC, Cacalano N, Jewett A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget. 2015;6:8947-59
23. Giovannucci E, Rimm EB, Stampfer MJ, Colditz GA, Ascherio A, Willett WC. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann Intern Med. 1994;121:241-6
24. Giovannucci E, Egan KM, Hunter DJ, Stampfer MJ, Colditz GA, Willett WC. et al. Aspirin and the risk of colorectal cancer in women. N Engl J Med. 1995;333:609-14
25. Thun MJ, Namboodiri MM, Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N Engl J Med. 1991;325:1593-6
26. Thun MJ. Aspirin, NSAIDs, and digestive tract cancers. Cancer Metastasis Rev. 1994;13:269-77
27. Rosenberg L, Louik C, Shapiro S. Nonsteroidal antiinflammatory drug use and reduced risk of large bowel carcinoma. Cancer. 1998;82:2326-33
28. Castelao JE, Yuan JM, Gago-Dominguez M, Yu MC, Ross RK. Non-steroidal anti-inflammatory drugs and bladder cancer prevention. Br J Cancer. 2000;82:1364-9
29. Sotiriou C, Lacroix M, Lagneaux L, Berchem G, Body JJ. The aspirin metabolite salicylate inhibits breast cancer cells growth and their synthesis of the osteolytic cytokines interleukins-6 and -11. Anticancer Res. 1999;19:2997-3006
30. Nelson JE, Harris RE. Inverse association of prostate cancer and non-steroidal anti-inflammatory drugs (NSAIDs): results of a case-control study. Oncol Rep. 2000;7:169-70
31. Castonguay A, Rioux N, Duperron C, Jalbert G. Inhibition of lung tumorigenesis by NSAIDS: a working hypothesis. Exp Lung Res. 1998;24:605-15
32. Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science. 1996;274:1383-5
33. Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956-9
34. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77-80
35. Jewett A, Wang MY, Teruel A, Poupak Z, Bostanian Z, Park NH. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: effect on the function of natural killer cells. Hum Immunol. 2003;64:505-20
36. Jewett A, Cacalano NA, Teruel A, Romero M, Rashedi M, Wang M. et al. Inhibition of nuclear factor kappa B (NFkappaB) activity in oral tumor cells prevents depletion of NK cells and increases their functional activation. Cancer Immunol Immunother. 2006;55:1052-63
37. Jewett A. et al. Dedifferentiation of epithelial tumors enhances citotoxicity, survival, and expansion of allogenic CD8+ T cells and natural killer cells. J Carcinogen Mutagen. 2012 S1:007
38. Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327-34
39. Shimizu H, Mitomo K, Watanabe T, Okamoto S, Yamamoto K. Involvement of a NF-kappa B-like transcription factor in the activation of the interleukin-6 gene by inflammatory lymphokines. Mol Cell Biol. 1990;10:561-8
40. Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1-38
41. Jee SH, Shen SC, Chiu HC, Tsai WL, Kuo ML. Overexpression of interleukin-6 in human basal cell carcinoma cell lines increases anti-apoptotic activity and tumorigenic potency. Oncogene. 2001;20:198-208
42. Chen Z, Malhotra PS, Thomas GR, Ondrey FG, Duffey DC, Smith CW. et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369-79
43. Tanner J, Tosato G. Impairment of natural killer functions by interleukin 6 increases lymphoblastoid cell tumorigenicity in athymic mice. J Clin Invest. 1991;88:239-47
44. Thomas GR, Chen Z, Leukinova E, Van Waes C, Wen J. Cytokines IL-1 alpha, IL-6, and GM-CSF constitutively secreted by oral squamous carcinoma induce down-regulation of CD80 costimulatory molecule expression: restoration by interferon gamma. Cancer Immunol Immunother. 2004;53:33-40
45. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326-31
46. Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567-77
47. Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L. et al. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr Biol. 1996;6:606-13
48. Butterfield L, Storey B, Maas L, Heasley LE. c-Jun NH2-terminal kinase regulation of the apoptotic response of small cell lung cancer cells to ultraviolet radiation. J Biol Chem. 1997;272:10110-6
49. Paranjpe A, Cacalano NA, Hume WR, Jewett A. Mechanisms of N-acetyl cysteine-mediated protection from 2-hydroxyethyl methacrylate-induced apoptosis. J Endod. 2008;34:1191-7
50. Jewett A. Activation of c-Jun N-terminal kinase in the absence of NFkappaB function prior to induction of NK cell death triggered by a combination of anti-class I and anti-CD16 antibodies. Hum Immunol. 2001;62:320-31
51. Arsura M, Panta GR, Bilyeu JD, Cavin LG, Sovak MA, Oliver AA. et al. Transient activation of NF-kappaB through a TAK1/IKK kinase pathway by TGF-beta1 inhibits AP-1/SMAD signaling and apoptosis: implications in liver tumor formation. Oncogene. 2003;22:412-25
52. Ting HC, Christian SL, Burgess AE, Gold MR. Activation and phosphatidylinositol 3-kinase-dependent phosphorylation of protein kinase C-epsilon by the B cell antigen receptor. Immunol Lett. 2002;82:205-15
53. Castrillo A, Traves PG, Martin-Sanz P, Parkinson S, Parker PJ, Bosca L. Potentiation of protein kinase C zeta activity by 15-deoxy-delta(12,14)-prostaglandin J(2) induces an imbalance between mitogen-activated protein kinases and NF-kappa B that promotes apoptosis in macrophages. Mol Cell Biol. 2003;23:1196-208
54. Jewett A, Bonavida B. TARGET-INDUCED ANERGY OF NATURAL-KILLER CYTOTOXIC FUNCTION IS RESTRICTED TO THE NK-TARGET CONJUGATE SUBSET. Cellular Immunology. 1995;160:91-7
55. Soeda A, Inagaki A, Oka N, Ikegame Y, Aoki H, Yoshimura S. et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283:10958-66
56. Tseng HC, Inagaki A, Bui VT, Cacalano N, Kasahara N, Man Yg, Jewett A. Differential Targeting of Stem Cells and Differentiated Glioblastomas by NK Cells. J Cancer. 2015;6(9):866-876
57. Kozlowska AK, Tseng HC, Kaur K, Topchyan P, Inagaki A, Bui VT. et al. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-gamma after differentiation of glioblastoma by human natural killer cells. Cancer Immunol Immunother. 2016
58. Hiraga T, Ito S, Nakamura H. Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res. 2013;73:4112-22
59. Jewett A, Bonavida B. Interferon-alpha activates cytotoxic function but inhibits interleukin-2-mediated proliferation and tumor necrosis factor-alpha secretion by immature human natural killer cells. J Clin Immunol. 1995;15:35-44
60. Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. Journal of Immunology. 1996;156:907-15
61. Jewett A, Cavalcanti M, Bonavida B. Pivotal role of endogenous TNF-alpha in the induction of functional inactivation and apoptosis in NK cells. Journal of Immunology. 1997;159:4815-22
62. Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A. et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861-6
63. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301-10
64. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S. et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature medicine. 2005;11:1314-21
65. Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z. et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1879-84
66. Jewett A, Tseng HC. Potential rescue, survival and differentiation of cancer stem cells and primary non-transformed stem cells by monocyte-induced split anergy in natural killer cells. Cancer Immunol Immunother. 2012;61:265-74
67. Jewett A, Tseng HC, Arasteh A, Saadat S, Christensen RE, Cacalano NA. Natural killer cells preferentially target cancer stem cells; role of monocytes in protection against NK cell mediated lysis of cancer stem cells. Curr Drug Deliv. 2012;9:5-16
68. Jewett A, Tseng HC. Tumor Microenvironment may Shape the Function and Phenotype of NK Cells Through the Induction of Split Anergy and Generation of Regulatory NK Cells. In: (ed.) Shurin M, Umansky V, Malyguine A. The Tumor Immunoenvironment. Dordrecht: Springer. 2013:361-84
69. Jewett A, Man YG, Tseng HC. Dual functions of natural killer cells in selection and differentiation of stem cells; role in regulation of inflammation and regeneration of tissues. J Cancer. 2013;4:12-24
70. Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulindac inhibits activation of the NF-kappaB pathway. The Journal of biological chemistry. 1999;274:27307-14
71. Berman KS, Verma UN, Harburg G, Minna JD, Cobb MH, Gaynor RB. Sulindac enhances tumor necrosis factor-alpha-mediated apoptosis of lung cancer cell lines by inhibition of nuclear factor-kappaB. Clin Cancer Res. 2002;8:354-60
72. Giardiello FM, Spannhake EW, DuBois RN, Hylind LM, Robinson CR, Hubbard WC. et al. Prostaglandin levels in human colorectal mucosa: effects of sulindac in patients with familial adenomatous polyposis. Dig Dis Sci. 1998;43:311-6
73. Liu JJ, Wang JY, Hertervig E, Nilsson A, Duan RD. Sulindac induces apoptosis, inhibits proliferation and activates caspase-3 in Hep G2 cells. Anticancer Res. 2002;22:263-6
74. Wu YL, Sun B, Zhang XJ, Wang SN, He HY, Qiao MM. et al. Growth inhibition and apoptosis induction of Sulindac on Human gastric cancer cells. World J Gastroenterol. 2001;7:796-800
75. Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B. et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood. 2002;100:230-7
76. Mann EA, Spiro JD, Chen LL, Kreutzer DL. Cytokine expression by head and neck squamous cell carcinomas. Am J Surg. 1992;164:567-73
77. Chen F, Castranova V, Shi X, Demers LM. New insights into the role of nuclear factor-kappaB, a ubiquitous transcription factor in the initiation of diseases. Clin Chem. 1999;45:7-17
78. Wang YF, Chang SY, Tai SK, Li WY, Wang LS. Clinical significance of interleukin-6 and interleukin-6 receptor expressions in oral squamous cell carcinoma. Head & neck. 2002;24:850-8
Author contact
![]() Corresponding author: Address: 10833 Le Conte Ave, UCLA School of Dentistry, Los Angeles, CA 90095, USA. Telephone: (310) 206-3970; Fax: (310) 794-7109; E-mail: ajewettedu.
Corresponding author: Address: 10833 Le Conte Ave, UCLA School of Dentistry, Los Angeles, CA 90095, USA. Telephone: (310) 206-3970; Fax: (310) 794-7109; E-mail: ajewettedu.