Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(18):3225-3235. doi:10.7150/jca.26052 This issue Cite
Research Paper
Molecular Characterization of HBV DNA Integration in Patients with Hepatitis and Hepatocellular Carcinoma
Liu Yang1*, Song Ye3*, Xinyi Zhao4*, Liyan Ji8, Yinxin Zhang8, Pingyu Zhou7, Jun Sun7, Yanfang Guan8, Yingxin Han2,8, Chao Ni1, Xiaoge Hu1, Weilong Liu6, Haiyan Wang6, Boping Zhou5 ![]() , Jian Huang2,5,6
, Jian Huang2,5,6 ![]()
1. Key Laboratory of Tumor Molecular Diagnosis and Individualized Medicine of Zhejiang Province and Key Laboratory of Gastroenterology of Zhejiang Province, Zhejiang Provincial People's Hospital, People's Hospital of Hangzhou Medical Colleg, Shang Tang Road 158, Hangzhou 310014, Zhejiang, P. R China.
2. Key Laboratory of Systems Biomedicine (Ministry of Education) and Collaborative Innovation Center of Systems Biomedicine, Shanghai Center for Systems Biomedicine, Chinese National Human Genome Center at Shanghai. Shanghai Jiao Tong University, Shanghai, 200240, China.
3. Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, Qing Chun Road 79, Hangzhou 310003, Zhejiang, P. R. China.
4. State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Zhejiang University School of Medicine, Hangzhou, 310003, Zhejiang, P. R. China.
5. Shenzhen People's Hospital, Second Clinical Medical College of Jinan University. Shenzhen, 518109, China.
6. Shenzhen Key Laboratory of Infection and Immunity, Shenzhen Third People's Hospital, Guangdong Medical College, Shenzhen, 518112, China.
7. STD Institute, Shanghai Skin Disease Hospital, Tong Ji University, Shanghai, China.
8. Binhai Genomics Institute, BGI-Tianjin, Tianjin, 300308, China.
*Liu Yang, Song Ye and Xinyi Zhao contributed equally to this work.
Received 2018-3-14; Accepted 2018-7-6; Published 2018-9-7
Abstract
Infection by chronic hepatitis B virus (HBV) is one of the major causes of liver cirrhosis and primary hepatocellular carcinoma (HCC). Viral DNA integration into the host cell genome is a key mechanism of hepatocarcinogenesis. However, the molecular characterization and the potential clinical implications of HBV DNA integration into patients suffering from different hepatitis and HCC remain unclear. In this study, we analyzed HBV integrations in patients with hepatitis B and HCC using HBV probe-based capturing and next-generation sequencing. The results revealed that the sizes of the HBV integrations ranged from 28 bp to 3215 bp, including the full-length HBV DNA sequence. The integration breakpoints were preferentially distributed in the viral enhancer, X protein, and core protein regions of the HBV genome. The number of HBV integrations followed an increasing trend from hepatitis to HCC, which was positively correlated with the HBV virus load in patients with hepatitis. The number of HBV integrations in the HBeAg positive chronic hepatitis B group was significantly greater than that in the other hepatitis B groups (P < 0.05). However, the relative abundance of HBV integrations was significantly higher in HCC tissues than in the adjacent liver tissues. Interestingly, 61.6% (8/13) of HBV-human DNA integration fragments could be detected at the RNA level. Our results also showed that HBV integration-targeted genes (ITGs) were significantly enriched in many cancer-related pathways, such as MAPK, extracellular matrix (ECM)-receptor interaction, and the hedgehog signaling pathway. Individuals with HBV integrations exhibited shorter disease-free survival (DFS) and overall survival (OS) than those without HBV integrations in some ITGs including LINC00293 (long intergenic non-protein coding RNA 293; DFS P = 0.008, OS P = 0.009), FSHB (follicle stimulating hormone beta subunit; DFS P = 0.05, OS P = 0.186), and LPHN3 (latrophilin-3; DFS P = 0.493, OS P = 0.033). This study determined the underlying mechanism of HBV DNA integration in liver diseases and laid the foundation for future studies on the pathogenesis of liver cancer.
Keywords: Chronic hepatitis B virus, Hepatocellular carcinoma, Capture sequencing, HBV integration.
1. Introduction
Hepatocellular carcinoma (HCC) is the most common subtype of liver cancer and the third leading cause of cancer death worldwide [1]. Infection with hepatitis B virus (HBV) greatly increases the incidence of HCC, because it causes chronic hepatitis, liver cirrhosis, and ultimately, HCC. The estimated risk of developing HCC was observed to be 25-37-fold higher in hepatitis B surface antigen (HBsAg) carriers compared with that in non-infected patients [2, 3]. HBV infection is one of the most important pathogeneses of HCC.
Several major molecular mechanisms of hepatocarcinogenesis are caused by HBV infection. First, the HBV genome is altered by integration breakpoints. The expression of hepatitis B virus X protein (HBx) is involved in many intracellular signal transduction pathways associated with cell proliferation and cell apoptosis, and both the C-terminal truncated HBx and the C-terminal region of HBx play important roles in hepatocarcinogenesis [4]. Second, the integration of HBV DNA into the host genome alters the expression and function of important genes that are mostly associated with cell proliferation, differentiation, and survival, and the induction of chromosomal instability. Several recurrent HBV integration genes, including TERT (telomerase reverse transcriptase, FAR2 (fatty acyl-coA reductase 2), ITPR1 (inositol 1,4,5-trisphosphate receptor type 1, also known as IP3R1), IRAK2 (interleukin 1 receptor associated kinase 2), MAPK1 (mitogen-activated protein kinase 1), MLL2 (myeloid/lymphoid or mixed-lineage leukemia 2), and MLL4, have been identified in recent years [5-7]. Additionally, the accumulation of genetic damage caused by chronic inflammation mediated by regulatory T cells contributes to the development of HCC. Traditional technologies such as PCR and northern blotting focus on known genes and small-scale sample analysis to investigate HBV integration in HCC. With the development of next-generation sequencing (NGS), many studies have achieved genome-wide surveys of HBV integration in HCC, and a great deal of data on the HBV pathogen in HCC has been obtained [8-11]. However, no studies have yet explored HBV integration in hepatitis samples or performed a comparison of hepatitis and HCC based on HBV sequence capture sequencing.
In this study, we conducted HBV probe-based capture assays, followed by NGS for 18 samples obtained from patients with different types of hepatitis who underwent liver biopsy, in addition to 54 HBV-positive HCCs and adjacent normal tissues, with the aim of surveying and comparing HBV integration in the disease trilogy from hepatitis to HCC. We used HBV probe-based capture technology to analyze the HBV integration profile from hepatitis to HCC. The results of the present study will increase our understanding the pathogenesis of liver cancer.
2. Materials and methods
2.1 Tissue samples
Fifty-four HCC tissues and adjacent liver tissues were obtained via surgical excision from the livers of patients with HCC at the First Affiliated Hospital of the School of Medicine of Zhejiang University, between November 2010 and April 2012. At the same time, we collected 18 samples from patients with different types of hepatitis who underwent liver biopsy at Shenzhen People's Hospital at the Second Clinical Medical College of Jinan University (Table S1). The diagnoses of HCC and hepatitis were confirmed based on clinical and serological characteristics, ultrasonography (US), computed tomography (CT), magnetic resonance imaging (MRI), and pathological examination, and were consistent with the Hepatitis and Primary Liver Cancer Clinical Diagnosis and Staging Criteria. In addition, normal liver tissues from five patients were resected surgically because of hemangioma in the liver at the First Affiliated Hospital of the School of Medicine of Zhejiang University. The samples were obtained from the part of the liver that was unaffected by hemangioma and were immediately frozen in liquid nitrogen. The samples were subsequently sectioned and confirmed by histological technology. All laboratory data related to the assessment of hepatic function were within normal ranges, including serum alanine aminotransferase, aspartate aminotransferase, g-glutamyltranspeptidase, alkaline phosphatase, total bilirubin, and albumin levels, prothrombin activity, and glucose, cholesterol, and triglyceride levels. Serological tests for hepatitis B surface antigen, hepatitis C virus antibodies, and human immunodeficiency virus antibodies were negative. Neither heavy alcohol consumption nor the intake of chemical drugs was observed before surgical resection. This study was approved by the Ethics Committee of the First Affiliated Hospital (Hangzhou, China) and Shenzhen People's Hospital (Shenzhen, China), and informed written consent was obtained from all patients.
2.2 DNA extraction and DNA library preparation
DNA was extracted from the tissue samples using TIANamp Genomic DNA Kits (Tiangen Biotech, Beijing, China) according to the manufacturer's protocols. The amount and integrity of the DNA were assayed using a Qubit® 2.0 fluorometer (Life Technologies, Carlsbad, CA, USA) and gel electrophoresis. Each DNA sample was quantified via agarose gel electrophoresis and measured using a NanoDrop system (Thermo Fisher, Waltham, MA, USA). First, 3 µg of genomic DNA was fragmented using ultrasound. The fragments were then purified, blunt-ended, A-tailed, and ligated to adaptors. Twelve cycles of PCR were then performed. Libraries with insert sizes of 350-400 bp were constructed for sequencing following the instructions provided by Illumina. After size selection in Agarose, 10 cycles of PCR were performed. The concentrations of the libraries were then quantified using a Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA, USA).
2.3 HBV enrichment and sequencing
The amplified DNA was captured using an HBVcap kit (MyGenostics, Baltimore, MD, USA). The hybridization probes were designed to tile along all HBV subtype sequences (A, B, C, D, E, F, G, and H). The capture experiment was conducted as follows. First, 1 μg of a DNA library was mixed with Buffer BL and the HBVcap probe (MyGenostics), and the mixture was heated at 95 °C for 7 min and 65 °C for 2 min. Next, 23 μL of prewarmed (65 °C) buffer HY (MyGenostics) was added to the mixture, which was maintained at 65 °C with a heated PCR lid for 22 hours, to allow hybridization. Then, 50 µL of MyOne beads (Thermo Fisher) were washed 3 times in 500 μL of 1× binding buffer and resuspended in 80 μL of 1× binding buffer. A 64-µL aliquot of 2× binding buffer was subsequently added to the hybrid mixture, followed by transfer to a tube containing 80 μL of MyOne beads. This mixture was mixed for 1 hour on a rotator, after which the beads were washed once with WB1 buffer at room temperature for 15 minutes and three times with WB3 buffer at 65 °C for 15 minutes. The bound DNA was then eluted using Elution Buffer, and the eluted DNA was finally amplified using the following program: 98 °C for 30 s (1 cycle); 98 °C for 25 s, 65 °C for 30 s, and 72 °C for 30 s (15 cycles); followed by 72 °C for 5 min (1 cycle). The amplicons were then purified using SPRI beads (Beckman Coulter, Inc., Fullerton, CA, USA), and paired-end 100-bp read-length sequencing was performed on a HiSeq 2000 sequencer according to the manufacturer's instructions (Illumina, Sa Diego, CA, USA).
2.4 Analysis of HBV integration sites
We detected HBV integration sites using local scripts. The workflow is shown in Figure S1. The raw data were first mapped to the HBV genome (NC_003977). Reads for which at least half of the bases mapped to the HBV genome were retained and then mapped to the human genome (NCBI build 37, HG19). Reads for which at least thirty percent of the bases mapped to the human genome were retained. Chimeric reads (read sequences that were partially aligned to the human genome and partially to the HBV genome, allowing 15 percent mismatch to both genomes) and mate-pair reads (allowing 10 percent mismatch to both genomes) were analyzed to detect HBV integration breakpoints. Considering errors in the experimental procedure and bioinformatics analysis, as well as the highly heterogeneous nature of tumors, we merged breakpoints within 70-bp regions in the chimeric reads, within 500-bp regions in the mate-pair reads, and within 500-bp regions in the chimeric mate-pair reads. We finally output the leftmost integration sites. HBV breakpoints with a supporting read number ≥ 2 were regarded as highly confident HBV integration sites in the subsequent analysis. HBV integration sites were annotated using Annovar with the default parameters [12].
2.5 PCR-based Sanger re-sequencing
Aliquots of 1 μL of the supernatant from the DNA samples were used as targets in the first round of PCR amplification. PCR amplification was performed in a reaction mixture containing 0.2 μM forward and reverse primers, 10 μL of 2× PCR mix, and 1 μL of the DNA sample (water was added to bring the total volume to 20 µL). Then, 1 μL of the universal reaction was used as a template for a second round of PCR amplification. The PCR system was the same as that used in the first round of PCR. PCR primers were designed on the basis of the pairedend assembled fragment (Table S2), in which one primer was located in the human genome and the other was in the HBV genome. PCR was performed using a GeneAmp PCR System 9700 thermal cycler. For each PCR product, we performed dye terminator Sanger sequencing using an Applied Biosystems 3730 DNA analyzer.
2.6 RNA extraction and semi-quantitative RT-PCR
RNA was extracted from the tissues and matched normal tissue samples using the TRIzol reagent according to the manufacturer's instructions (Life Technologies). Quality control for the RNA samples was performed using a 2100 Bioanalyzer Instrument (Agilent Technologies) to assay the amount and integrity of the RNA. First-strand cDNA was synthesized using the PrimeScript 1st Strand cDNA Synthesis Kit (Takara, Shiga, Japan) according to the manufacturer's instructions. Semi-quantitative RT-PCR was then performed using the cDNA as the template and the primers designed for the target regions (Table S2). H2O was employed as a blank control. The PCR product was detected by electrophoresis through a 2% agarose gel.
3. Results
3.1 Sequencing of HBV DNA using HBV probe-based capture sequencing
To investigate the relationship between HBV DNA integration and hepatocarcinogenesis, we enriched potential HBV integrated fragments in 54 paired HCC and adjacent liver tissues, and in 18 liver tissues from patients with different types of hepatitis B diagnosed by the American Association for the Study of Liver Diseases (AASLD) [13] based on HBV probe capture technology [14] (Table S1). The enriched DNA was then employed for library construction and sequenced at single-base resolution using a HiSeq 2500 sequencer. We obtained a total of 497,648,760 raw reads with 100-bp-length target regions from 126 specimens, among which 70,729,110 reads completely or partially matched to the HBV genome (NC_003977). On average, 89.79% coverage with at least 10-fold read mapping was achieved for the HBV genome. The average depth of coverage was 3,188-fold (Table S3) in 126 samples. The highest depth of coverage was 16,729-fold in HCC sample 080962T, and the lowest depth of coverage was seven-fold in hepatitis sample SD090 (Table S3). After removing reads that were completely matched to the HBV genome, a total of 25,962,712 reads were mapped to the human genome reference (HG19). We finally obtained 233,339 effective reads comprising potential integration events (Figure S1 and Table S3). Thus, the coverage and read depth were adequate to reliably detect HBV DNA integration.
3.2 Identification of integration breakpoints
All 233,339 effective reads (Table S3), including chimeric reads and mate-pair reads, were used to identify integration breakpoints according to the following criteria: If some junctions supported by the chimeric reads were clustered within the range of 70bp, the only upstream junction was defined as a HBV integration breakpoint (Figure S2). The same way, if some junctions supported by the mate-pair reads were clustered within the range of 500 bp, the only upstream junction was defined as a HBV integration breakpoint (Figure S2) [14]. Our results identified a total of 7513 breakpoints in 126 samples, among which 2870 HBV integration breakpoints were identified in 54 HCC samples; 4466 breakpoints were identified in paired adjacent liver samples; and 177 breakpoints were identified in hepatitis samples (after filtering out breakpoints with only one read) (Table S3, Table S4). To confirm the results, we randomly selected 75 integration breakpoints for PCR and Sanger sequencing validation tests and successfully validated 85.3% (64/75) of these integration sites (Table S5). Our results suggested that the results of HBV integration detected by NGS are highly reliable.
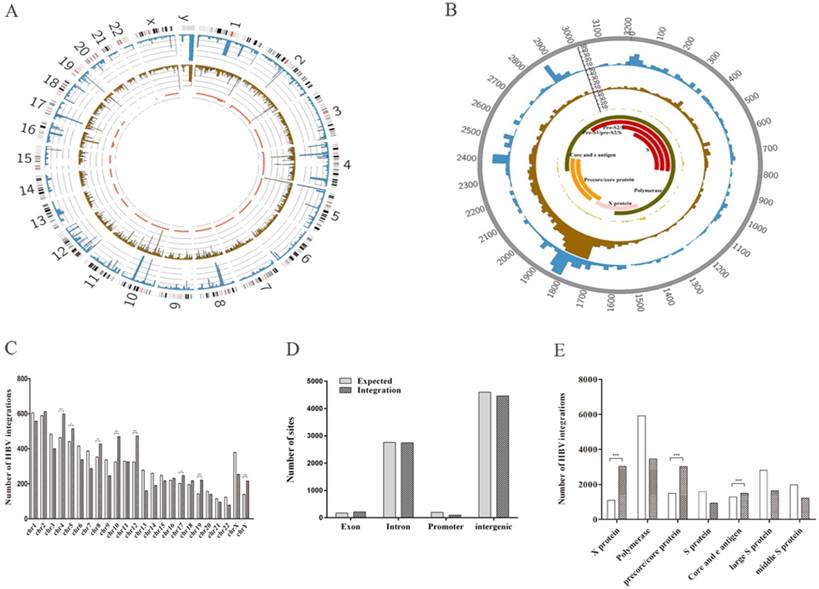
We found that the 7513 HBV integration breakpoints were distributed across the entire human genome (Figure 1A). However, HBV was preferentially integrated into chromosomes 4 (P < 0.0001), 5 (P = 0.0153), 8 (P = 0.007), 10 (P < 0.0001), 12 (P < 0.0001), 17 (P=0.0329), 19 (P < 0.0001), and Y (P < 0.0001) (experimental data compared with the expected result) (Figure 1C). In addition, we found no enrichment of HBV integration events in the gene body (Figure 1D), suggesting that HBV integration breakpoints might be randomly distributed in intragenic regions. Moreover, approximately 36.9% (2773/7513) of the breakpoints were preferentially distributed in the 1590~1840-bp region of the HBV genome that includes the viral enhancer, X protein, and core protein coding sequences [15] (Figure 1B). Clearly, more breakpoints in the X protein (P < 0.0001) and core protein (P<0.0001) were observed compared with our expectations (Figure 1E). Our results support the view that HBV breakpoints located in these regions might facilitate the formation of HBV-human fusion genes, thus initiating hepatocarcinogenesis [11].
Distribution of integration breakpoints in the human and HBV genomes in 72 samples. (A) Distribution of integration breakpoints in the human genome. Each bar represents the frequency of HBV integration breakpoints at a particular locus in the human genome (hg19). Hepatocellular carcinoma (HCC), adjacent liver tissues, and hepatitis samples with HBV integrations are shown in the outer (blue), middle (brown), and inner circles (red), respectively. The scale bar indicates the number of tumors, adjacent liver tissues, and hepatitis tissues. Chromosome numbers are shown. (B) Distribution of integration breakpoints in the HBV genome. Each histogram represents the frequency of integration breakpoints at different loci in the HBV genome. HCC, adjacent liver tissues, and hepatitis samples with HBV integrations are shown in the outer (blue), middle (brown), and inner circles (red), respectively. The locations of genes encoding HBV polymerase (green), core (orange), S (red), and X (pink) proteins are shown. Genomic positions are numbered. (C) Distribution of HBV integration breakpoints in human chromosomes. The histogram shows the number of expected and observed integrations in chromosomes. P values were calculated assuming a binomial distribution. * P < 0.05, ** P < 0.01 and ***P < 0.001. (D) Percentages of HBV integration sites located in exon, intron, promoter, and intergenic regions of the human genome. (E) The histogram shows the number of expected and observed integration breakpoints in the HBV structures, including X proteins, polymerase, precore/core proteins, S proteins, core and e antigens, large S proteins, and intermediate-sized S proteins. P values were computed assuming a binomial distribution. * P < 0.05, ** P < 0.01 and ***P < 0.001.
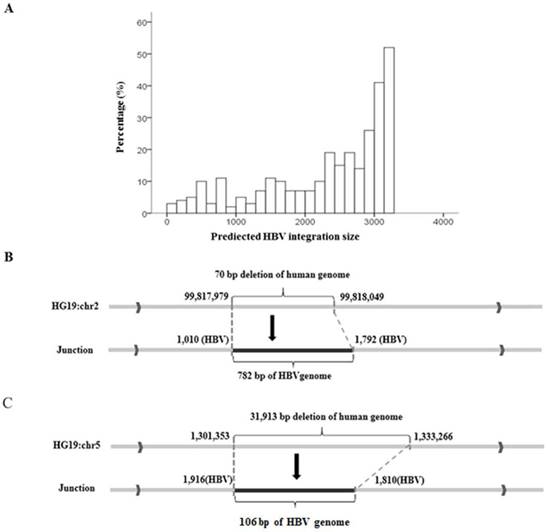
Local algorithms predicted that the integrated HBV fragments ranged in size from 28 bp to 3215 bp, and in general, long integration fragments were more frequent than short ones (Figure 2A). To confirm the predicted results, we used nested PCR to amplify the complete integrated HBV fragments in the human genome. In sample 807Ca, we found a 70-bp deletion at the integration breakpoint for the human genome and a 782-bp HBV fragment insertion in the human genome, as shown in Figure 2B. In sample 813Ca, we found a 31,913-bp deletion at the integration breakpoint for the human genome and a 106-bp HBV fragment insertion in the human genome, as shown in Figure 2C. Both of the above findings were consistent with the length of the HBV integration predicted by NGS.
Prediction and validation of integration fragment lengths. (A) Distribution of predicted lengths of HBV integration fragments. (B) Sanger validation of two HBV integration fragments. A 70-bp deletion at the integration breakpoint for the human genome and a 782-bp HBV fragment inserted in the human genome. (C) A 31,913-bp deletion at the integration breakpoint for the human genome and a 106-bp HBV fragment inserted into the human genome.
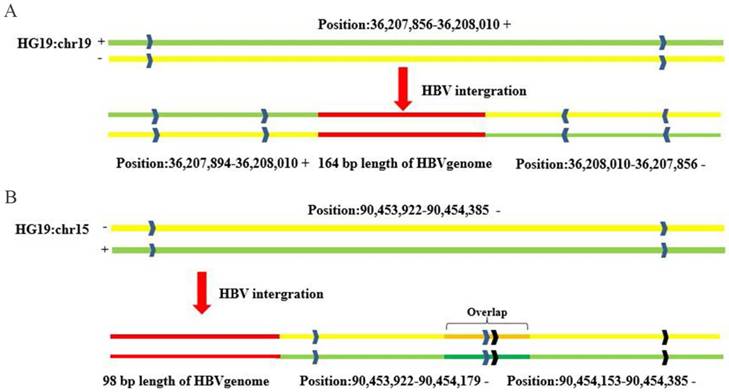
Interestingly, the results of integration breakpoint validation using PCR-based Sanger sequencing showed structural abnormity of the human genome at loci containing HBV integration events. For example, the complementary 164-bp sequence (chr19:36207856-36208010) in the left flanking HBV integration fragment was reversed and then inserted into the right flanking region of the HBV integration fragment (chr19:36208010-36207856) compared with the reference sequence for 815Ca (HCC) (Figure 3A). In another case (734Ca), there was a repeat sequence of 26 bp (chr15: 90454153-90454179) in the right flanking region of the HBV integration fragment compared with the reference sequence (Figure 3B). Our findings suggest that the insertion of viral DNA into the host genome may induce human genome instability, which would be consistent with earlier reports [8,15].
Sanger sequencing validation of host genome structural alterations induced by HBV integration. (A) The complementary sequence of 164 bp (chr19:36207856-36208010) in the left flanking HBV integration fragment was reversed and inserted into the right flanking HBV integration fragment (chr19:36208010-36207856) compared with the reference sequence. (B) Repeat sequence of 26 bp (chr15:90454153-90454179) in the right flanking HBV integration fragment, compared with the reference sequence.
3.3 HBV integration breakpoints at different disease stages
We then analyzed the distribution of the number of integration breakpoints in the follow groups: normal liver, hepatitis B, adjacent liver tissues, and HCC (Figure 4, Table S6). All hepatitis B patients in our study were accepted for standardized treatment and were divided into four groups according to the AASLD guide [13], including autoimmune hepatitis, acute hepatitis B, HBeAg negative chronic hepatitis B, and HBeAg positive chronic hepatitis B (Table S6). The results showed that the mean number of HBV integrations showed an increasing trend from hepatitis to HCC (Figure 4A). In addition, the number of HBV integrations in the HBeAg positive chronic hepatitis B group was significantly greater than the total number of integrations in all other hepatitis B groups (P < 0.05), demonstrating that the number of HBV integrations was positively correlated with disease progression (Figure 4A).
Distribution of the number of HBV integrations in hepatitis B, adjacent liver tissue and hepatocellular carcinoma (HCC) samples. (A) Distribution of the mean number of HBV integrations in each group. (B) The mean supported reads number in hepatitis B, adjacent liver tissue, and HCC samples (P < 0.05).
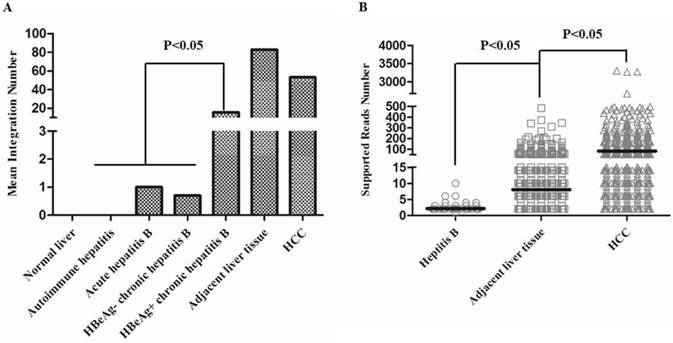
We also found that the mean number of HBV integration events in tumor samples (including HCCs and adjacent liver tissue) was greater than the mean number in the hepatitis B groups (Figure 4A). Interestingly, the mean number of HBV integrations was greater in adjacent liver tissues than in HCCs. However, the mean supported reads showed an increasing trend from hepatitis B (average, 2.2), adjacent liver tissues (average, 8.1), to HCCs (average, 82.1), suggesting that the relative abundance of HBV integration was highest in HCCs (Figure 4B).
3.4 HBV integration target genes correlate with poor prognosis
Next, we observed that the number of HBV integrations was positively associated with the titer of serum HBsAg (P < 0.0001; Figure 5A). Patients with HCC with a high number of HBV integrations were generally 40-60 years old (P = 0.035, Figure 5B), consistent with the incidence of HCC in China in relation to age [1].
Clinical correlation analysis of HBV integration. (A) The number of HBV integration sites versus serum HBV DNA levels (cut-off set at 105). P values were calculated using unpaired Student's t-tests. (B) Correlation analyses of HBV integration with the age of the affected individuals. P = 0.035, Pearson chi-square. (C, D) Kaplan-Meier survival estimates according to HBV integrations in LINC00293 (long intergenic non-protein coding RNA 293). The data are shown for the disease free survival (DFS) of patients with and without HBV integrations in LINC00293 (P = 0.008). The data are shown for the overall survival (OS) of patients with and without mutations in LINC00293 (P = 0.009); (E, F) Kaplan-Meier survival estimates according to HBV integrations in FSHB (follicle stimulating hormone beta subunit). The data are shown for the DFS of patients with and without HBV integrations in FSHB (P = 0.05). The data are shown for the OS of patients with and without mutations in FSHB (P = 0.186); (G, H) Kaplan-Meier survival estimates according to HBV integrations in LPHN3 (latrophilin-3). The data are shown for the DFS of patients with and without HBV integrations in LPHN3 (P = 0.493). The data are shown for the OS of patients with and without mutations in LPHN3 (P = 0.033).
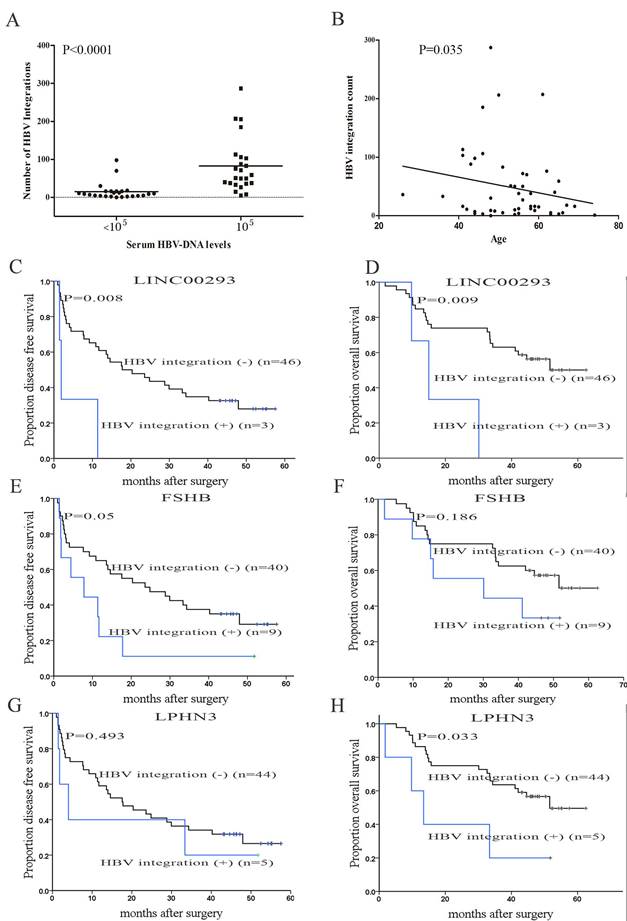
Genes with their transcription start sites (TSS) closest to the HBV integration sites (within 61 Mb from the integration site) were defined as integration targeted genes (ITGs) [15]. We then analyzed the relationship between ITGs and the prognosis of HCC patients statistically using Kaplan-Meier survival curves. The results demonstrated that individuals with HBV integrations in LINC00293 (long intergenic non-protein coding RNA 293; disease-free survival (DFS) P = 0.008, overall survival (OS) P = 0.009), FSHB (follicle stimulating hormone beta subunit; DFS P = 0.05, OS P = 0.186), and LPHN3 (latrophilin-3; DFS P = 0.493, OS P = 0.033) exhibited shorter DFS and OS than those without HBV integrations (Figure 5C-5H), suggesting that HBV integrations correlate with poor prognosis in patients with HCC. Of course, these are preliminary results, and more samples should be collected and analyzed to confirm them.
3.5 Differentially expressed HBx-human chimeric transcripts
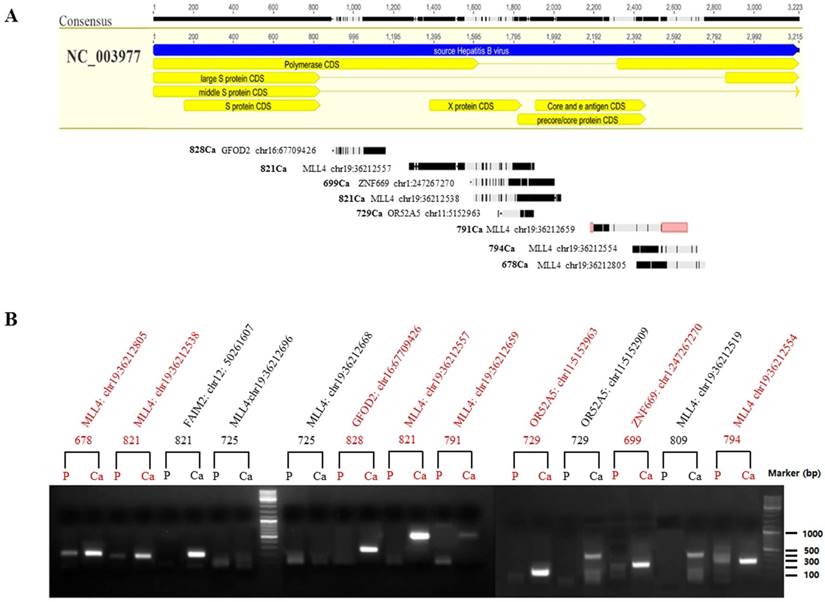
To validate the hypothesis that the integrated HBV fragments are expressed at the RNA level, we randomly selected 13 integration fragments located in exon regions of five genes (Figure 6A), which occurred only in HCC, according to the NGS analysis. RT-PCR was then performed to compare the mRNA expression between HCCs and adjacent liver tissues (Figure 6B). Among the 13 PCR assays we obtained eight high quality PCR products that were confirmed by Sanger sequencing (Figure 6B). The results revealed that 61.6% (8/13) of HBV-human DNA integration fragments were expressed at the RNA level, including four HBx integration fragments, three precore/core protein CDS, and one polymerase CDS integration fragments, and four human genes (Figure 6A). Interestingly, we found that the integration fragments were expressed at significantly higher levels in HCCs than in the adjacent liver tissues (Figure 6B). The results suggested that differentially expressed HBx-human chimeric transcripts might correlate with hepatocarcinogenesis, although more evidence is needed to confirm this hypothesis.
Location and mRNA expression of HBV-human chimeric transcripts. (A) Locations of expressed HBV-human chimeric transcripts in the HBV genome. (B) Agarose gel analysis of 13 HBV-human chimeric transcripts amplified using nested PCR technology. The PCR products were subjected to electrophoresis through a 1.3% agarose gel and visualized via ethidium bromide staining. Molecular marker lanes (m) are included. Among the 13 HBV-human DNA integration fragments, eight high quality PCR products (marked in red) were confirmed by Sanger sequencing, while the other five low quality PCR products were not validated. P indicates adjacent liver tissue and Ca indicates hepatocellular carcinoma (HCC).
4. Discussion
HBV DNA integration was first reported in 1980, which paved the way to study the mechanism of hepatocarcinogenesis at the molecular level [6]. Since then, researchers have used various methods to study HBV integration events, including Southern blotting, in situ hybridization, and PCR [17-20]. Recently, several groups have used whole-genome sequencing technology to detect HBV DNA integrations in hepatocellular tumor tissues and adjacent liver tissues [8, 21]. However, the cost of whole-genome sequencing technology is too high, the amount of data produced is too large, and too much time is required for the analysis of HBV DNA integration. Therefore, in the present study, we designed an HBV probe-based capture technology to analyze HBV integrations. We then used this technology to study changes in amount and extent of HBV integration between patients with hepatitis and those with HCC. Based on our data, we identified many more HBV integration events than were found in previous studies [8, 10, 22, 23], suggesting that HBV probe-based capture technology is highly effective, sensitive, and cost effective when applied at the genome-wide level to identify HBV integration events in the human genome [14].
Integration is not necessary for HBV replication and therefore has not routinely occurred in the natural history of HBV infection, which is quite different from the case of retroviruses [5]. It was previously thought that HBV DNA integration occurred at the chronic stage of HBV infection. However, research has shown that HBV DNA integration occurs at various stages of infection. Murakami et al. used Alu-PCR to analyze liver samples of 19 patients and 12 patients with acute and chronic hepatitis B, respectively, and HBV DNA integration was identified in three patients with acute hepatitis B and in all patients with chronic hepatitis [23]. In this study, we also found that HBV DNA integration could occur in all stages of HBV infection, including HCC (Figure 4A). In addition, the number of HBV integrations increased from hepatitis B to HCCs (Figure 4A). In the group who had experienced primary treatment failure (HBeAg positive chronic hepatitis B), there were 129 integration events in patient B9, who had the highest virus load (5.52 × 108) among the group (Table S1 and S6). Patient SD075 had the second highest number HBV integrations (22) and their virus load was 8.97 × 107. Patient SD079T, who suffered from hepatitis B cirrhosis, had a negative virus load, but had five HBV integrations. The current findings revealed that a combination of HBV integration events and HBV load might be useful to evaluate the prognosis of hepatitis B. In addition, HBV integration events might represent a novel biomarker to define the population at high risk of HCC.
Interestingly, the number of HBV integrations in tumor samples was higher than that in hepatitis B samples (Figure 4A), and the mean number of HBV integrations in HCCs was lower than that in adjacent liver tissues (Figure 4A). However, the mean supported reads was higher in HCCs (average, 82.1) than that in the adjacent liver tissues (average, 8.1) (Figure 4B), suggesting that the relative abundance of HBV integration was higher in HCCs. This finding revealed that HBV integration might play a role in hepatocellular carcinogenesis in a hit-and-run manner; however, the dosage of HBV integration might be more important in hepatocarcinogenesis [24-26].
Ding [9] and Toh [11] concluded that HBV integration preferentially occurs in human chromosomes 17 and 10. However, we found that HBV integration was significantly enriched in other chromosomes, including chromosomes 4, 5, 8, 12, 19, and Y, but not in chromosomes 10 and 17 (Figure 1C, p < 0.05), suggesting that our technology could identify more chromosomes that are enriched in HBV DNA integration events. Furthermore, Ding reported that the lengths of HBV integrations ranged from 34 to 1185 bp, based on FLX sequencing [9]. In our study, analysis of the length of the inserted HBV fragments showed that the maximum predicted length of an HBV integration was 3215 bp, except for litter fragments (Figure 2A), suggesting that almost the full length of the virus could be integrated into the host genome. We found that the HBV breakpoints were concentrated within the 1,590-1,840-bp region of the HBV genome, which was consistent with previous reports [8, 15]. The sequences located in these regions have been reported to participate in the integration of HBV into the host genome [27,28].
Microhomologies (MHs) between the human genome and the HBV genome at or near integration breakpoints were also identified in our study (Figure S3). A significant enrichment of MHs was previously reported at HPV integration sites in cervical cancer [29]. However, we did not observe significant enrichment of MHs at HBV breakpoints (compared with random integration within the human genome) (Figure S3). This disparity may have occurred because the different viruses exhibit different integration mechanisms. Further investigation will be required to clarify the underlying mechanisms.
Our results showed that 7513 HBV integrations were distributed among 4339 genes (Table S7), which we referred to as HBV integration-targeted genes (ITGs). There were 393 recurrent ITGs found in the HCC samples and 746 ITGs were found in adjacent liver tissues samples, while only one ITG (NAALADL2, encoding N-acetylated alpha-linked acidic dipeptidase like 2) was found in hepatitis samples (Table S7). In total, 4339 ITGs were used for functional annotation based on Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The results revealed that the ITGs were enriched in many cancer-related pathways, including the MAPK signaling pathway, ECM-receptor interaction, and the Hedgehog signaling pathway (P < 0.05, Figure S4A). Further KEGG analysis of the ITGs identified in hepatitis, HCC and adjacent liver tissues samples showed that the ITGs from hepatitis samples were enriched in neuroactive ligand-receptor interaction and axon guidance pathways, whereas the ITGs from HCC and adjacent liver tissues samples were enriched in cancer-related pathways (P < 0.05, Figure S4B).
Interestingly, four genes exhibited HBV integrations in more than one-third of the HCC samples, which were TERT (48.1%), MLL4 (46.3%), PXDNL (peroxidasin like; 42.6%) and SNTG1 (syntrophin gamma 1; 42.6%), accounting for 76% (41/54) of the tumor samples. Among these genes, TERT and MLL4 are well established as highly frequent ITGs in HCC [7-11]. We found 46 integration breakpoints located in the promoter region and six located in the intronic region of TERT (Table S6). HBx frequently integrated into the promoter of TERT, probably resulted in a 'cis' effect on the transcription of TERT [8, 29]. In addition, there were 17 integrations in intronic regions and 47 in exonic regions of MLL4 (Table S6). HBx frequently integrated into exon three of MLL4, which might generate fusion genes (Figure 6). Interestingly, we confirmed that the HBx-human chimeric transcripts could be expressed in HCCs using RT-PCR. As the next step of our research, we will perform additional assays to explore the relationship between HBx-human chimeric transcripts and hepatocarcinogenesis.
In summary, we systematically characterized HBV integration in patients with hepatitis and in those with HCC using HBV probe-based capture sequencing. Our findings show that HBV integrations might be a useful biomarker to monitor disease progression from hepatitis to HCC. These results provided a good foundation for future studies on the pathogenesis of liver cancer.
Abbreviations
HBV: hepatitis B virus; HCC: hepatocellular carcinoma; ITGs: HBV integration targeted genes; HBsAg: hepatitis B surface antigen; HBx: hepatitis B virus X protein; CT: computed tomography; MRI: magnetic resonance imaging; DFS: disease free survival; OS: overall survival; MHs: Microhomologies.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We gratefully acknowledge support from the China National Key Projects for Infectious Disease (2017ZX10203207); National Natural Science Foundation of China (81502463, 81272306, 81472639 and 81372227); the Science Technology Department of Zhejiang Province (2016C33116); the key project of the Health Bureau of Zhejiang Province (2018274734); Shanghai Commission for Science and Technology (15431902900 and 15JC1403000); the Program of Shenzhen Science Technology and Innovation Committee (JCYJ20160427183814675 and JCYJ20150402111430647).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Joachim Lupberger, Eberhard Hildt. Hepatitis B virus-induced oncogenesis. World Journal of Gastroenterology. 2007;13:74-81
2. Blvd. MMHBH, Hwang LY, Hatten CJ, et al. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206-13
3. Sun CA, Wu DM, Lin CC. et al. Incidence and cofactors of hepatitis C virus-related hepatocellular carcinoma: a prospective study of 12,008 men in Taiwan. American Journal of Epidemiology. 2003;157:674-82
4. Koike K, Moriya K, Iino S. et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994;19:810-9
5. Murakami Y, Saigo K, Takashima H. et al. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut. 2005;54:1162-8
6. Paterlinibréchot P, Saigo K, Murakami Y. et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911-6
7. Saigo K, Yoshida K, Ikeda R. et al. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia ( MLL4 ) gene and rearrangements of MLL4 in human hepatocellular carcinoma †. Human Mutation. 2008;29:703-8
8. Sung WK, Zheng H, Li S. et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nature Genetics. 2012;44:765-9
9. Ding D, Lou X, Hua D. et al. Recurrent Targeted Genes of Hepatitis B Virus in the Liver Cancer Genomes Identified by a Next-Generation Sequencing-Based Approach. Plos Genetics. 2011;8:1208-13
10. Jiang Z, Jhunjhunwala S, Liu J. et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Research. 2012;22:593-601
11. Toh ST. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34:787-98
12. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38:e164
13. Terrault NA, Bzowej NH, Chang KM. et al. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63:261-83
14. Li W, Zeng X, Lee NP. et al. HIVID: An efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013;102:338-44
15. Li X, Zhang J, Yang Z. et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. Journal of Hepatology. 2014;60:975-84
16. Brechot C, Pourcel C, Louise A. et al. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature. 1980;286:533-5
17. Kawai S, Yokosuka O, Imazeki F. et al. State of HBV DNA in HBsAg-negative, anti-HCV-positive hepatocellular carcinoma: Existence of HBV DNA possibly as nonintegrated form with analysis by Alu-HBV DNA PCR and conventional HBV PCR. Journal of Medical Virology. 2001;64:410-8
18. Minami M, Poussin K, Bréchot C. et al. A Novel PCR Technique Using Alu -Specific Primers to Identify Unknown Flanking Sequences from the Human Genome. Genomics. 1995;29:403-8
19. Georgigeisberger P, Berns H, Loncarevic IF. et al. Mutations on Free and Integrated Hepatitis B Virus DNA in a Hepatocellular Carcinoma: Footprints of Homologous Recombination. Oncology. 1992;49:386-95
20. Matsui H, Shiba R, Matsuzaki Y. et al. Direct detection of hepatitis B virus gene integrated in the Alexander cell using fluorescence in situ polymerase chain reaction. Cancer Letters. 1997;116:259-64
21. Fujimoto A, Totoki Y, Abe T. et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature Genetics. 2012;44:760-4
22. Jiang S, Yang Z, Li W. et al. Re-evaluation of the carcinogenic significance of hepatitis B virus integration in hepatocarcinogenesis. Plos One. 2012;7:e40363
23. Murakami Y, Minami M, Daimon Y. et al. Hepatitis B virus DNA in liver, serum, and peripheral blood mononuclear cells after the clearance of serum hepatitis B virus surface antigen. Journal of Medical Virology. 2004;72:203-14
24. Piao Z, Park C, Park J. et al. Allelotype analysis of hepatocellular carcinoma. International Journal of Cancer. 1998;75:29-33
25. Xu XR, Huang J, Xu ZG. et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proceedings of the National Academy of Sciences. 2001;98:15089-94
26. Bill CA, Summers J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11135-40
27. Guerrero RB, Roberts LR. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. Journal of Hepatology. 2005;42:760-77
28. Hu Z, Zhu D, Wang W. et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nature Genetics. 2015;47:158-63
29. Ferber MJ, Montoya DP, Yu C. et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22:3813-20
Author contact
![]() Corresponding authors: Jian Huang, E-mail: jianhuangedu.cn and Boping Zhou, zhoubopingcom
Corresponding authors: Jian Huang, E-mail: jianhuangedu.cn and Boping Zhou, zhoubopingcom