Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(18):3247-3256. doi:10.7150/jca.25921 This issue Cite
Research Paper
Scutellarin induces apoptosis and autophagy in NSCLC cells through ERK1/2 and AKT Signaling Pathways in vitro and in vivo
ChaoYue Sun1, CaiYun Li1, XiaoFeng Li2, Ying Zhu1, ZuQing Su1, XieQi Wang1, QingLian He1, GuangJuan Zheng1 ![]() , Bing Feng1
, Bing Feng1 ![]()
1. Guangdong Provincial Hospital of Chinese Medicine, The Second Clinical College of Guangzhou University of Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou 510120, China.
2. Clinical Medical College of Acupuncture and Rehabilitation, Guangzhou University of Chinese Medicine, no 232, Waihuandong Road, Guangzhou Higher Education Mega Center, Guangzhou 510006, China.
Received 2018-3-6; Accepted 2018-8-13; Published 2018-9-7
Abstract
Curative molecular therapy for non-small cell lung cancer (NSCLC) is still lacking. Scutellarin, an active flavone extracted from Erigeron breviscapus Hand-Mazz, displays anti-tumor property in diverse cancer types, yet its tumor-suppressive effect on NSCLC is not reported. In this study, we found that scutellarin significantly inhibited the proliferation of NSCLC cells, induced cell apoptosis, and triggered autophagy. Notably, inhibition of autophagy with inhibitor HCQ attenuated the anti-proliferative activity of scutellarin, indicating that scutellarin-induced autophagy is antineoplastic. In addition, HCQ treatment reduced scutellarin-induced apoptosis. Further study demonstrated that scutellarin stimulated phosphorylation of ERK1/2, and inhibition of ERK1/2 with inhibitor U0126 markedly attenuated scutellarin-induced autophagy. Similarly, scutellarin downregulated the expression of p-AKT, and AKT inhibitor MK-2206 induced autophagy. Moreover, there also existed crosstalk between ERK and AKT pathways. Finally, in vivo xenograft nude mice experiment proved that scutellarin treatment significantly reduced tumor growth and increased the levels of LC3-II and p-ERK1/2, suppressed p-AKT in mice tumors. Thus, our study for the first time uncovered the anti-cancer function of scutellarin on NSCLC cells, and might provide a potential novel therapy for treatment of patients with NSCLC.
Keywords: Scutellarin, NSCLC, apoptosis, autophagy, ERK1/2, AKT
1. Introduction
Lung cancer is still the leading cause of cancer-related mortality worldwide, at least partially due to lack of effective pharmacological treatment [1]. Non-small cell lung cancer (NSCLC) accounts for greater than 80% of lung cancer, with a low 5-year survival rate [2]. Recent treatments, mainly including surgery, chemotherapy and targeted therapy, however, have not been ultimately changed the dismal overall survival rate [3]. Therefore, understanding of the molecular mechanisms and exploiting promising therapeutic agents for NSCLC is urgently required.
Apoptosis, is the main and best-described form of programmed cell death that plays a central role in cancer therapy [4]. Autophagy is a highly conserved process that discards damaged or superfluous cytoplasmic proteins and organelles to support metabolism [5]. Autophagy plays a critical role in the pathogenesis, survival and responses to therapy in virtually all cancers, where it can suppress tumor initiation or sustain growth, survival of established tumors [6, 7]. In general, basal autophagy impairs cancer progression under stress, whereas aberrant autophagic activity was discovered to facilitate carcinoma cell survive [8]. Strikingly, mounts of studies indicate that tumors are more autophagy-dependent than normal tissues, suggesting that autophagy inhibition or activation might be a therapeutic approach for cancers [9]. As such, apoptosis and autophagy are simultaneously involved in deciding the fate of cancer cells, where exists interaction between each other. For instance, autophagy inhibitor 3-MA can abolish the apoptosis-promoting effect of anti-cancer drug, triptolide on osteosarcoma cells [10].
Extracellular signal-regulated kinase (ERK1/2) is a mitogen-activated protein kinase that transduces signals from the cytoplasm to the nucleus of the cells [11]. Once activated in response to a diverse array of stimulus, the phosphorylation of ERK1/2 is involved in directing cell proliferation, survival and apoptosis [12]. Accumulating evidence indicates that ERK is intimately linked to promote tumorigenesis as its upstream activators RAS/RAF are frequently mutated in NSCLC patients [13]. However, ERK1/2 pathway can be activated by antineoplastic agents to exert anti-tumor activity [14]. Of note, accumulated studies have demonstrated that ERK1/2 pathway positively regulates autophagy [15]. For instance, knocking down ERK1/2 by its inhibitor PD98059 suppressed autophagy in neuroblastoma cells [16]. In addition, the PI3K-AKT pathway is often hyperactivated in a variety of cancers due to loss of the tumor suppressor PTEN [17]. Similarly, the PI3K-AKT pathway can negatively regulate autophagy [18]. It is noteworthy that PI3K-AKT can crosstalk with ERK1/2 pathway [19].
Scutellarin (C21H18O12, shown in Fig 1A), an active flavone extracted from Chinese traditional medicine Erigeron breviscapus Hand-Mazz, has been demonstrated to possess a variety of bioactivities such as neuroprotective [20], anti-oxidant [21] and anti-inflammatory [22]. Recent researches have exhibited evidence that scutellarin has a potent anti-cancer effect on various types of tumors, including hepatocellular carcinoma [23], colorectal cancer [24], and tongue squamous carcinoma [25]. However, so far, the anti-tumor effect of scutellarin on NSCLC has not been reported. In this study, we analyzed the anti-tumor effect of scutellarin on NSCLC cells, and determined whether scutellarin could induce apoptosis and autophagy through ERK1/2 and AKT signaling pathways.
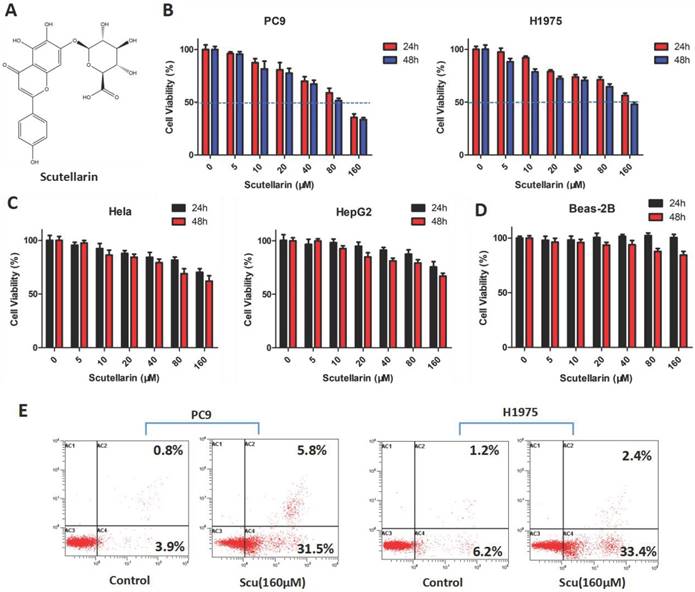
Effects of scutellarin on the proliferation and apoptosis on NSCLC cell lines. (A) Chemical structure of scutellarin. (B) PC-9 and H1975 cells were treated with various concentrations of scutellarin (0, 5, 10, 20, 40, 80, 160 μM) for 24 or 48 hours. After treatment, cell viability was measured by MTT assay. (C) Hela and HepG2 cells were treated with increased concentrations of scutellarin (0, 5, 10, 20, 40, 80, 160 μM) for 24 or 48 hours, and cell viability was measured by MTT assay. (D) Human normal lung epithelial Beas-2B cells were exposed to scutellarin (0, 5, 10, 20, 40, 80, 160 μM) for 24 or 48 hours, and cell viability was measured by MTT assay. (E) PC-9 and H1975 cells were incubated with 160 μM scutellarin for 48 hours, the cell apoptosis was detected by flow cytometry. Data are representative of three independent experiments.
2. Materials and Methods
2.1 Chemicals and Reagents
Scutellarin (purity ≥ 98%) was purchased from Sigma Aldrich (St. Louis, MO, USA), and dissolved in PBS (pH 7.4). Autophagy inhibitor hydroxychloroquine (HCQ), ERK1/2 inhibitor U0126 and AKT inihbitor MK-2206 were obtained from Selleck Chemicals LLC ( Houston, TX, USA ). The primary antibodies against LC3, p62, ERK1/2, p-ERK1/2, AKT, p-AKT, β-actin and HRP-conjugated secondary antibodies were purchased from Cell Signaling Technology (Boston, MA, USA).
2.2 Cell Lines and Cell Culture
Human NSCLC cell lines, including PC-9 and H1975, human cervical cancer cell line Hela, human hepatocellular carcinoma cell line HepG2, and human normal lung epithelial cell line Beas-2B were provided by Shanghai Life Sciences Research Institute Cell Resources Center. All cell lines were maintained in RPMI (Roswell Park Memorial Institute medium) 1640 containing 10% FBS (fetal bovine serum), and incubated at 37 ℃ in a humidified atmosphere with 5% CO2.
2.3 Cell Viability Assay
Cell viability was examined by the MTT assay. In brief, PC-9, H1975, HepG2, Hela and Beas-2B cells were seeded in 96-well plates (4×103cells/well), and cultured overnight. After 24 or 48 hours treatment of scutellarin (0, 5, 10, 20, 40, 80, 160 μM), cells were subjected to 20 μL of MTT solution (5 mg/mL), followed by addition with 150 μL of dimethyl sulfoxide (DMSO). The optical density (OD) at wavelength of 490 nm was detected by a Multiskan Ascent Revelation Plate Reader (Thermo, USA).
2.4 Detection of cell apoptosis
Cell apoptosis was measured by flow cytometry using the Annexin V-FITC/PI Apoptosis Kit (MultiSciences Biotech Co., Ltd). Briefly, PC-9 and H1975 cells were exposed to 160 μM scutellarin for 48 hours. After treatment, cells were harvested and washed with ice-cold PBS, then resuspended in 500 μL binding buffer. Subsequently, cells were stained using 5 μL Annexin V-FITC and 10 μL PI solution, followed by incubation for 5 mins at 37 ℃ in a dark atmosphere. Finally, the number of apoptotic cells was examined by FACS (Becton-Dickinson, Bedford, MA, USA).
2.5 Western blot analysis
Cells were lysed in a standard RIPA buffer supplemented with 1% protease and phosphatase inhibitors. Protein concentration was measured by the BCA assay. Equal amounts of proteins were separated by 8%-12% SDS-PAGE, then transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% skimmed milk for one hour, the membranes were overnight incubated with primary antibodies, followed by incubation with anti-rabbit secondary antibody for another one hour. Immunoreactivity was visualized utilizing an enhanced chemiluminescence (ECL) system, and protein bands were quantified by the Image-Pro Plus 6.0.
2.6 In vivo xenograft and treatment experiments
Female BALB/c nude mice were purchased from Guangdong Medical Laboratory Animal Center (Fushan, Guangdong, China). The animal procedures were approved by the Animal Care and Use Committee of Guangdong Provincial Hospital of Chinese Medicine (the Ethics Approval Number 2016023) and the Declaration of the National Institutes of Health Guide for Care and Use of Laboratory Animals. To establish xenograft tumor model, mice were subcutaneously injected with 4×106 H1975-Luciferase cells (stably expressing fluorescence) suspended in 200 μL PBS. When tumor reached approximately 100 mm3 (Volume = Length ×width2 × 0.5), mice were randomly divided into three groups (n = 8): the vehicle; the low dose scutellarin (30 mg/kg); the high dose scutellarin (60 mg/kg). At this point, scutellarin was dissolved in PBS (PH 7.4), and was orally administered to each mouse once a day for three weeks. The tumor dimensions were measured using digital calliper every 3 days. Before treatment or after the last administration, the tumor size was monitored by in vivo bioluminescence imaging (IVIS Lumina LT Series III Pre-Clinical In Vivo Imaging System). After 3 weeks, all mice were humanely sacrificed and the tumors were resected for protein quantitation analysis.
2.7 Immunohistochemistry
Twenty surgically excised lung adenocarcinoma specimens and adjacent normal lung tissues were fixed in 4% paraformaldehyde at 4℃, then embedded in paraffin, and 4-μm paraffin sections were obtained. The sections were deparaffinized and serially rehydrated with xylene. The anti-gen retrieval was performed before the sections were incubated in 10% serum blocking solution. Then the slides were incubated with primary antibodies (p-AKT and p-ERK) in blocking solution overnight at 4 ℃. After washing and incubation with secondary antibody at room temperature for 30 m, sections were visualized with diaminobenzidine and couterstained with hematoxylin. Finally, these immune-stained slides were evaluated and scored by two independent pathologists.
2.8 Statistical Analysis
All data were presented as the mean ± SEM. The differences between the groups were analyzed using unpaired Student's t test or one-way ANOVA test in SPSS software (version 17.0). p < 0.05 was considered to be statistically significant.
3. Results
3.1 Effects of scutellarin on the proliferation and apoptosis on NSCLC cell lines
To determine the anti-tumor effect of scutellarin on NSCLC cells, the MTT assay was firstly employed. PC-9 and H1975 cells were treated with various concentrations of scutellarin (0, 5, 10, 20, 40, 80, 160 μM) for 24 or 48 hours. As shown in Fig. 1B, treatment of scutellarin clearly inhibited cell growth in a dose and time-dependent manner. In addition, the anti-proliferation effects of scutellarin on cervical cancer Hela cells and hepatocellular carcinoma HepG2 cells were confirmed by MTT assay. We found that scutellarin inhibited the cell viability of HepG2 and Hela cells (Fig. 1C), however, NSCLC cells were more sensitive to scutellarin than hepatocellular carcinoma and cervical cancer cells. Of note, human normal lung epithelial cell line Beas-2B was involved to determine the toxicity of scutellarin by MTT assay, and results showed that scutellarin exhibited no significant cytotoxic activity on Beas-2B cells (Fig. 1D). Additionally, we detected the cell apoptosis by flow cytometry using the Annexin V-FITC/PI Apoptosis Kit. Results showed that 160 μM scutellarin treatment significantly induced apoptosis, when compared with the control cells (Fig. 1E). Thus, scutellarin displayed a marked anti-tumor response to NSCLC cells.
3.2 Scutellarin induced autophagy in NSCLC cells
Considering that autophagy plays an essential role in cancers, here, we therefore examined whether scutellarin was able to alter the expression of autophagy-related proteins. Microtubule-associated protein light chain 3 (LC3), an excellent marker of autophagy, is widely used for monitoring autophagy [26]. During autophagy induction, the transition of the non-lipidated form of LC3 (LC3-I) to the lipidated form of LC3 (LC3-II) is indispensable [27]. Thus, the increase of LC3-II level or LC3-II/LC3-I ratio specifically signifies the induction of autophagy. As expected, results showed that 160 μM scutellarin increased LC3-II conversion in PC-9 and H1975 cells (Fig. 2A). Thus, these results implied that scutellarin induced autophagy in NSCLC cells.
Scutellarin induced autophagy in NSCLC cells. (A) PC-9 and H1975 cells were treated with 0, 40, 80, 160 μM scutellarin for 48 hours, the expression of autophagy marker LC3 was evaluated by western blotting with the indicated antibody. β-actin was used as a control. (B) PC-9 and H1975 cells were challenged with 0, 5, 10, 20 μM HCQ, LC3 and p62 expressions were determined by western blots. (C) PC-9 and H1975 cells were treated with 0, 1.25, 2.5, 5, 10, 20, 40 μM HCQ for 48 hours, and cell viability was measured by MTT assay. (D) PC-9 and H1975 cells were treated with 160 μM scutellarin alone, or 10 μM HCQ, or in combination for 48 hours. The LC3 expression was measured by western blots. (E) PC-9 and H1975 cells were treated with various concentrations of scutellarin alone, or in combination with 10 μM HCQ for 48 hours, cell viability was determined by MTT assay. (F) PC-9 and H1975 cells were treated with 160 μM scutellarin, or in combination with 10 μM HCQ for 48 hours, cell apoptosis was measured by flow cytometry. Data are representative of three independent experiments (mean ± SEM). *p<0.05.
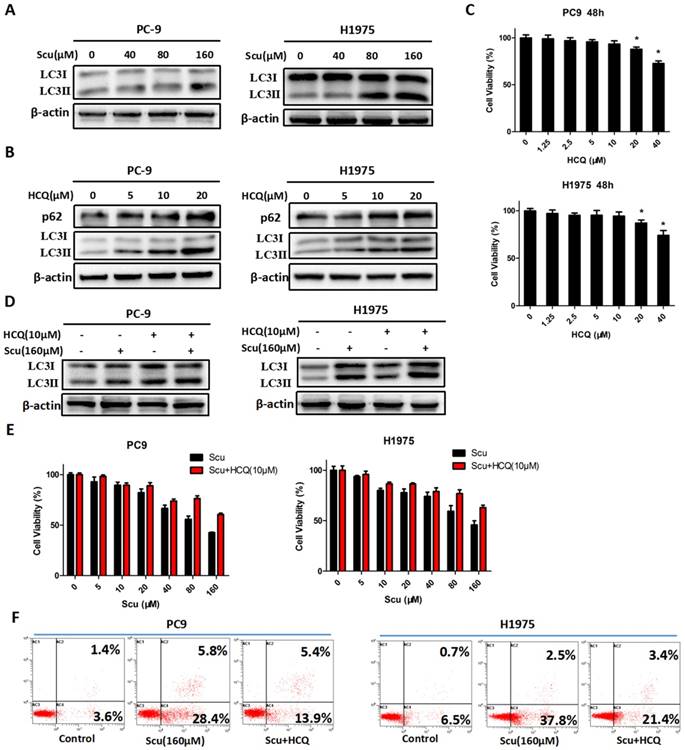
To further verify the role of autophagy in NSCLC cells, autophagy inhibitor HCQ was used. HCQ is generally utilized to inhibit autophagy by impairing the fusion of autophagosomes with lysosomes [28]. As shown in Fig. 2B, an increase in the level of LC3-II protein in PC-9 and H1975 cells treated with 5, 10, 20 μM HCQ was observed, while p62 expression was also up-regulated, suggesting that HCQ inhibited induction of autophagy. Consistently, 10 μM HCQ did not affect the cell viability of NSCLC cells (Fig. 2C). Thus, 10 μM HCQ was selected for further research. Here, we noted that 10 μM HCQ indeed enhanced the expression of LC3-II, and HCQ combined with scutellarin markedly increased the level of LC3-II, indicating that HCQ could block scutellarin-induced autophagy (Fig. 2D). We next examined whether HCQ treatment could affect the anti-proliferative function of scutellarin using MTT assay. As shown in Fig. 2E, 10 μM HCQ treatment had no significant effect on cell proliferation in PC-9 and H1975 cells. Interestingly, 10 μM HCQ slightly abolished the anti-proliferative ability of scutellarin, and the efficiency was better when the dose of scutellarin was higher. Collectively, our results indicated that scutellarin-induced autophagy is not cytoprotective, but antineoplastic.
It is noteworthy that autophagy and apoptosis might be connected with each other and occur simultaneously or sequentially under certain circumstances [29]. To investigate whether there exists interaction between autophagy and apoptosis, we examined the effect of autophagy inhibitor on scutellarin-induced apoptosis. Here, results showed that scutellarin plus HCQ treatment reduced scutellarin-induced apoptosis (Fig. 2F). Thus, scutellarin-induced autophagy promoted apoptosis, leading to cell death.
3.3 Activation of ERK1/2 signaling pathway is required for scutellarin-induced autophagy
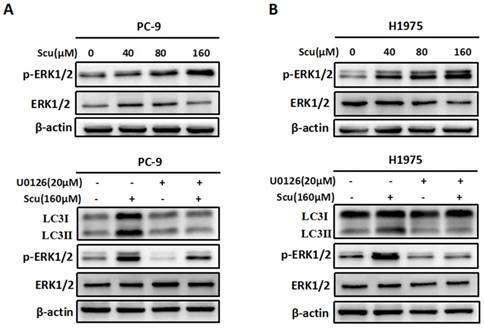
Accumulating evidences suggest that ERK1/2 signalling pathway has recently emerged as an important player in regulating autophagy [15]. To evaluate whether scutellarin-induced autophagy was associated with ERK1/2 signalling, we detected the level of p-ERK1/2 and ERK1/2 in PC-9 and H1975 cells treated with scutellarin. As shown in Fig. 3A, in accordance with LC3-II protein, scutellarin treatment dramatically increased the expression of p-ERK1/2, suggesting that elevated autophagy might be modulated by the ERK1/2 signalling. To exclude this possibility, we next determined the unclear relationship between autophagy and ERK1/2 using a specific ERK1/2 inhibitor, U0126. As exhibited in Fig. 3B, 20 μM U0126 attenuated p-ERK1/2 level, whereas total ERK1/2 showed no change, and scutellarin plus U0126 treatment decreased scutellarin-induced activation of ERK1/2. Moreover, scutellarin treatment caused autophagic responses in NSCLC cells, however, scutellarin in combination with U0126 remarkedly attenuated the expression of LC3-II. Overall, scutellarin triggered high autophagy partly by up-regulating ERK1/2 signaling pathway.
Activation of ERK1/2 signaling pathway is required for scutellarin-induced autophagy. (A) PC-9 and H1975 cells were exposed to 0, 40, 80, 160 μM scutellarin for 48 hours, and the appearance of p-ERK1/2, ERK1/2 were examined by western blots. (B) PC-9 and H1975 cells were treated with 160 μM scutellarin alone, or 20 μM U0126, or in combination for 48 hours, the expressions of LC3, ERK1/2, p-ERK1/2 were evaluated by western blots. Data are representative of three independent experiments.
3.4 Scutellarin inhibited AKT signaling pathway, and there existed crosstalk between ERK and AKT
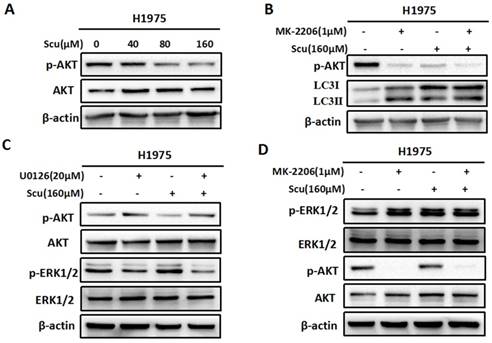
To further confirm whether scutellarin affects the AKT signaling, we treated H1975 cells with 0, 40, 80, 160 μM scutellarin. Results showed that scutellarin treatment increased AKT phosphorylation (p-AKT), whereas no difference was observed in the total AKT level (Fig. 4A). In addition, AKT inhibitor MK-2206 blocked p-AKT, and inhibition of AKT by MK-2206 induced autophagy (Fig. 4B), suggesting AKT pathway negatively regulated autophagy. Furthermore, we found that inhibition of ERK1/2 by U0126 increased the expression of p-AKT (Fig. 4C). Similarly, AKT inhibitor MK-2206 improved p-ERK1/2 level, indicating that there existed crosstalk between ERK1/2 and AKT pathways.
Scutellarin inhibited AKT signaling pathway, and there existed crosstalk between ERK and AKT. (A) H1975 cells were exposed to 0, 40, 80, 160 μM scutellarin for 48 hours, the AKT and p-AKT expression were analysed by western blots. (B) H1975 cells were treated with 160 μM scutellarin alone, or 1 μM MK-2206, or in combination for 48 hours, p-AKT and LC3 levels were determined by western blots. (C) H1975 cells were treated with 160 μM scutellarin in the presence or absence 20 μM U0126, the indicated proteins were detected by western blots. (D) Western bloting analysis of p-ERK1/2, ERK1/2, p-AKT and AKT levels in the indicated cells.
3.5 Scutellarin suppressed tumor growth in mouse xenograft model
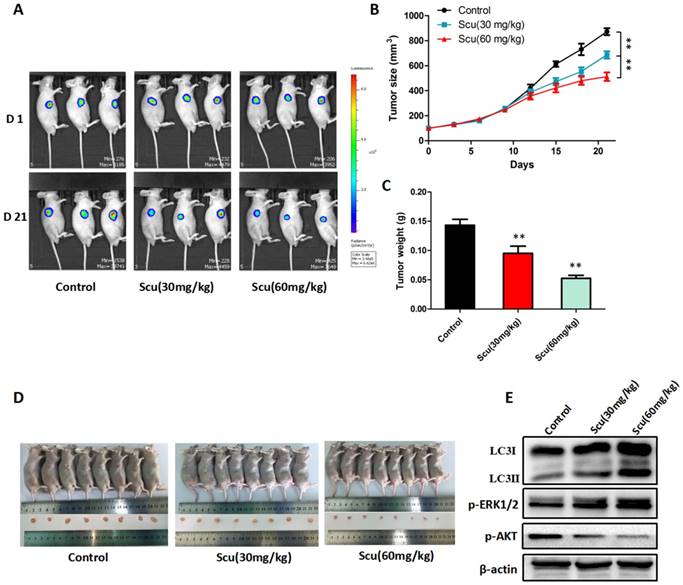
To further investigate the anti-tumor activity of scutellarin in vivo, H1975-Luciferase cells were subcutaneously injected into BALB/c nude mice. As a result, mice treated with 30, 60 mg/kg scutellarin showed significant suppression of the tumor growth, compared with vehicle controls (Fig. 5A, 5D). Notably, 60 mg/kg scutellarin reduced about 50% of the tumor size, with better efficacy than 30 mg/kg scutellarin (Fig. 5B, 5C). In addition, we measured the expression of LC3, p-ERK1/2, ERK1/2, and p-AKT in tumors, and found that 30, 60 mg/kg scutellarin treatment could up-regulate LC3-II and p-ERK1/2 level, and down-regulate p-AKT (Fig. 5E). Taken together, these observations demonstrated that scutellarin suppressed tumor growth in mouse xenograft model, in accordance with in vitro cell experiments.
Scutellarin suppressed tumor growth in mouse xenograft model. H1975-Luciferase cells expressing luciferase were subcutaneously implanted into BALB/c nude mice. When tumor reached approximately 100 mm3 (Volume = Length ×width2 × 0.5), mice were randomly divided into three groups (n = 8): the vehicle; the low dose scutellarin (30 mg/kg); the high dose scutellarin (60 mg/kg). After 21 days treatment, the tumors were collected. (A) The tumor sizes were monitored by IVIS, representative bioluminescence images of tumor in each group are shown. (B) Quantification of tumor volume was showed. (C) Tumor weight in nude mice. (D) Mice were humanely sacrificed, and representative images of tumors isolated from nude mice. (E) Western blot assay to confirm the expression of LC3, ERK1/2, p-ERK1/2 in the indicated group of tumor samples. Data are representative of three independent experiments (mean ± SEM). **p<0.01.
3.6 Clinical correlation of AKT and ERK in NSCLC
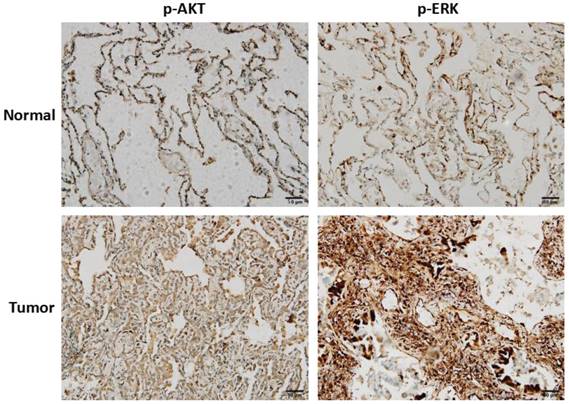
To clarify the clinical correlation of AKT and ERK in NSCLC, twenty surgically excised lung adenocarcinoma specimens and adjacent normal lung tissues were assessed using immunohistochemistry. As shown in Fig. 6, high-expressions of p-AKT and p-ERK were observed in lung adenocarcinoma specimens compared with normal lung tissues. Result of p-AKT expression was consistent with our in vitro cell experiment, indicating that AKT played as an oncogene. In most cases, activated ERK pathway is involved in directing cell proliferation, survival and apoptosis [12]. Here, we found that ERK was positively expressed in normal lung tissues, and significantly higher expressed in tumor tissues.
Clinical correlation of AKT and ERK in NSCLC. Representative images of immunohistochemistry staining of p-AKT and p-ERK expressions in lung adenocarcinoma tissues and adjacent normal lung tissues.
3. Discussion
This study, to our best knowledge, for the first time investigated the tumor-suppressive effect of scutellarin on NSCLC cell lines. The data showed that scutellarin was capable of inhibiting the proliferation of NSCLC cells, PC-9 and H1975, promoted cell apoptosis, and induced autophagy. Mechanistically, scutellarin-induced autophagy was tightly correlated with the activation of the ERK1/2 signaling pathway and suppression of AKT pathway. Interestingly, scutellarin treatment specifically killed NSCLC cells, however, the anti-proliferative activities of scutellarin on hepatocellular carcinoma cells (HepG2 and 97H cells) and cervical cancer cells (Hela cells) were not obvious.
Autophagy, also known as “self-eating”, acts as a janus-faced player due to its double-edged functions in cancer therapy [30]. In most contexts, autophagy facilitates tumorigenesis, where cancers induce autophagy to survive under microenvironmental stress and become more aggressive [31]. For example, in Kras-driven lung cancer, deletion of Atg7 that is an essential autophagy gene caused metabolic impairment, resulting in reduction of tumor cell proliferation and tumor burden [32]. This suggests taht autophagy maintains lung tumor metabolism that is required for lung tumorigenesis, and autophagy inhibition might be a therapeutic approach for cancer treatment. As such, novel components targeting inhibiting autophagy have thereby attracted great interest. However, growing evidence suggests that, under certain circumstances, excessive or persistent autophagy not only can promote autophagic cell death, but also potentiate the cytotoxicity of chemotherapeutic drugs [33]. Thus, the role of autophagy in cancer remains a foremost challenge for us to understand.
During autophagy activation, lots of double-membrane electron-dense autophagosomes that capture unnecessary contents fuse with lysosomes or vacuoles to form autolysosomes, where these dysfunctional organelles are degraded [34]. In particular, accumulation of LC3-II is regarded as the induction of autophagy [26]. Accordingly, our results demonstrated that scutellarin induced autophagy in PC-9 and H1975 cells, suggesting that scutellarin was a novel autophagy inducer. Intriguingly, treatment with autophagy inhibitor HCQ slightly disabled the tumor-suppressive effect of scutellarin, when compared with the scutellarin treatment alone. Thus, scutellarin-induced autophagy is not cytoprotective, but antineoplastic.
Apoptosis and autophagy are two different forms of programmed cell death that both regulate cell survival and cell death. Although a well-defined mechanism remains lacking, the relationships between autophagy and apoptosis have been: (i) autophagy precedes apoptosis [4]; (ii) autophagy plays a cytoprotective role to delay apoptosis, thus, inhibition of autophagy can enhance anti-cancer drugs-induced apoptotic cell death [35]; (iii) autophagy and apoptosis both promote cell death [10]. Here, we demonstrated that scutellarin induced apoptosis and autophagy, and inhibition of autophagy by HCQ attenuated scutellarin-induced apoptosis. Taken together, scutellarin-induced autophagy could regulate apoptosis, leading to cell death.ERK is an effector of frequently mutant oncogene Ras that is associated with proliferation in a broad range of human tumors [36]. Thus, ERK has been tightly proposed as a tumor-promoting player in human cancers. Recently, small-molecule inhibitors targeting Ras-ERK signalling has yielded substantial improvement in melanoma patients [37]. However, many reports did not always support the role of ERK in tumorigenesis. For example, resveratrol can induce apoptosis in thyroid cancer cells by activating ERK1/2 pathway, and activated ERK1/2 triggers tumor-suppressor p53, leading to apoptotic death [38]. Therefore, whether ERK1/2 signaling is friend or foe in cancer remains elusive. In this study, we found that scutellarin significantly increased the expression of p-ERK1/2 in NSCLC cells, suggesting that scutellarin activated ERK1/2 pathway.
Growing evidence shows that ERK has been implicated in triggering the induction of autophagy [15]. Also, scutellarin was an autophagy inducer that could promote autophagic death in PC-9 and H1975 cells. To this end, we proposed a hypothesis whether scutellarin-induced autophagy was associated with activation of ERK1/2. Consistent with this possibility, we observed that ERK1/2 inhibitor U0126 repressed scutellarin-induced autophagy. Thus, scutellarin induced autophagy in NSCLC cells through activating ERK1/2 signaling pathway. In addition to ERK1/2 pathway, scutellarin inhibited AKT signaling pathway, and AKT inhibitor MK-2206 could induced autophagy. Moreover, there also existed crosstalk between ERK and AKT pathways. Furthermore, in vivo xenograft mice experiment proved that scutellarin treatment significantly reduced tumor growth when compared with the controls. In addition, following scutellarin treatment, the expressions of LC3-II and p-ERK1/2 were elevated, whereas p-AKT was decreased. These data indicated that scutellarin suppressed tumor growth in vitro and in vivo by activating ERK1/2 signaling and inhibiting AKT signaling.
4. Conclusions
In conclusion, our results demonstrated that scutellarin induced apoptosis and autophagy in NSCLC cells through activating ERK1/2 and inhibiting AKT signaling pathways. Our study for the first time uncovered the tumor-suppressive function of scutellarin on NSCLC cells, and might provide a potential novel therapy for treatment of patients with NSCLC.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No81403142), the Special Fund of Guangdong Provincial Hospital of Chinese Medicine for Scientific and Technological Research of Traditional Chinese Medicine (Grant No YK2013B2N09), the Terry Fox Foundation Cancer Research Funding (Grant No YN2014TF04), and the Science and Technology Planning Project of Guangdong Province (Grant No 2016A020226048, No 2014A020212264, No 2014A020221080 and No 2013B021800248).
Author Contributions
Chao-Yue Sun and Cai-Yun Li designed the experiments and drafted the manuscript; Xiao-Feng Li, Ying Zhu, Zu-Qing Su performed the experiments; Xie-Qi Wang analyzed the results; Qing-Lian He obtained the funding; Guang-Juan Zheng, Bing Feng supervised the study and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7-30
2. Miller KD, Siegel RL, Lin CC. et al. Cancer treatment and survivorship statistics, 2016. CA: a cancer journal for clinicians. 2016;66(4):271-289
3. Chen CW, Wu MH, Chen YF. et al. A Potent Derivative of Indolizino[6,7-b]Indole for Treatment of Human Non-Small Cell Lung Cancer Cells. Neoplasia. 2016;18(4):199-212
4. Liu G, Pei F, Yang F. et al. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. International journal of molecular sciences. 2017:18 (2)
5. Wirawan E, Vanden Berghe T, Lippens S. et al. Autophagy: for better or for worse. Cell research. 2012;22(1):43-61
6. Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35(1):1-11
7. Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216-9
8. Zhan L, Zhang Y, Wang W. et al. Autophagy as an emerging therapy target for ovarian carcinoma. Oncotarget. 2016;7(50):83476-83487
9. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913-30
10. Li X, Lu Q, Xie W. et al. Anti-tumor effects of triptolide on angiogenesis and cell apoptosis in osteosarcoma cells by inducing autophagy via repressing Wnt/beta-Catenin signaling. Biochemical and biophysical research communications. 2018;496(2):443-449
11. Wang Z, Guo Q, Wang R. et al. The D Domain of LRRC4 anchors ERK1/2 in the cytoplasm and competitively inhibits MEK/ERK activation in glioma cells. Journal of hematology & oncology. 2016;9(1):130
12. Zeng Z, Xu J, Zheng W. Artemisinin protects PC12 cells against beta-amyloid-induced apoptosis through activation of the ERK1/2 signaling pathway. Redox biology. 2017;12:625-633
13. Deschenes-Simard X, Kottakis F, Meloche S. et al. ERKs in cancer: friends or foes? Cancer research. 2014;74(2):412-9
14. Wu Y, Chen H, Li R. et al. Cucurbitacin-I induces hypertrophy in H9c2 cardiomyoblasts through activation of autophagy via MEK/ERK1/2 signaling pathway. Toxicology letters. 2016;264:87-98
15. Bartholomeusz C, Rosen D, Wei C. et al. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer research. 2008;68(22):9302-10
16. Yeh PS, Wang W, Chang YA. et al. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer letters. 2016;370(1):66-77
17. Pan CW, Jin X, Zhao Y. et al. AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. The EMBO journal. 2017;36:995-1010
18. Butler DE, Marlein C, Walker HF. et al. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget. 2017;8:56698-56713
19. Dent P. Crosstalk between ERK, AKT, and cell survival. Cancer Biol Ther. 2014;15:245-6
20. Wang W, Ma X, Han J. et al. Neuroprotective Effect of Scutellarin on Ischemic Cerebral Injury by Down-Regulating the Expression of Angiotensin-Converting Enzyme and AT1 Receptor. PloS one. 2016;11(1):e0146197
21. Wang Z, Yu J, Wu J. et al. Scutellarin protects cardiomyocyte ischemia-reperfusion injury by reducing apoptosis and oxidative stress. Life sciences. 2016;157:200-207
22. Yuan Y, Zha H, Rangarajan P. et al. Anti-inflammatory effects of Edaravone and Scutellarin in activated microglia in experimentally induced ischemia injury in rats and in BV-2 microglia. BMC Neurosci. 2014;15:125
23. Ke Y, Bao T, Wu X. et al. Scutellarin suppresses migration and invasion of human hepatocellular carcinoma by inhibiting the STAT3/Girdin/Akt activity. Biochemical and biophysical research communications. 2017;483(1):509-515
24. Zhu PT, Mao M, Liu ZG. et al. Scutellarin suppresses human colorectal cancer metastasis and angiogenesis by targeting ephrinb2. Am J Transl Res. 2017;9(11):5094-5104
25. Li H, Huang D, Gao Z. et al. Scutellarin inhibits the growth and invasion of human tongue squamous carcinoma through the inhibition of matrix metalloproteinase-2 and -9 and αvβ6 integrin. International journal of oncology. 2013;42(5):1674-81
26. Gugnoni M, Sancisi V, Manzotti G. et al. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell death & disease. 2016;7(12):e2520
27. Eritja N, Chen BJ, Rodríguez-Barrueco R. et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy. 2017;13(3):608-624
28. Kondo Y, Kanzawa T, Sawaya R. et al. The role of autophagy in cancer development and response to therapy. Nature reviews Cancer. 2005;5(9):726-34
29. Maiuri MC, Zalckvar E, Kimchi A. et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature Reviews Molecular Cell Biology. 2007;8(9):741-752
30. Singh SS, Vats S, Chia AY. et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2017;37(9):1142-1158
31. White E. The role for autophagy in cancer. The Journal of clinical investigation. 2015;125(1):42-6
32. Guo JY, Teng X, Laddha SV. et al. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes & development. 2016;30(15):1704-17
33. Zhang P, Zheng Z, Ling L. et al. w09, a novel autophagy enhancer, induces autophagy-dependent cell apoptosis via activation of the EGFR-mediated RAS-RAF1-MAP2K-MAPK1/3 pathway. Autophagy. 2017;13(7):1093-1112
34. Li Y, Zhang Y, Wang L. et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy. 2017;13(7):1145-1160
35. Jing Z, Sui X, Yao J. et al. SKF-96365 activates cytoprotective autophagy to delay apoptosis in colorectal cancer cells through inhibition of the calcium/CaMKIIγ/AKT-mediated pathway. Cancer letters. 2016;372(2):226-238
36. Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011;30(32):3477-88
37. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nature reviews Drug discovery. 2014;13(12):928-42
38. Shih A, Davis FB, Lin HY. et al. Resveratrol Induces Apoptosis in Thyroid Cancer Cell Lines via a MAPK- and p53-Dependent Mechanism. J Clin Endocrinol Metab. 2002;87(3):1223-32
Author contact
![]() Corresponding authors: Guang-Juan Zheng, E-mail: zhengguangjuanedu.cn; Bing Feng, E-mail: bing19831com
Corresponding authors: Guang-Juan Zheng, E-mail: zhengguangjuanedu.cn; Bing Feng, E-mail: bing19831com