Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(18):3373-3381. doi:10.7150/jca.25083 This issue Cite
Short Research Communication
Is Ras a potential target in treatment against cutaneous squamous cell carcinoma?
Li Li#, Min Li#, Song Xu, Wenbo Bu, Mengli Zhang, Heng Gu ![]() , Xu Chen
, Xu Chen ![]()
Jiangsu Key Laboratory of Molecular Biology for Skin Diseases and STIs, Institute of Dermatology, Chinese Academy of Medical Science & Peking Union Medical College, Nanjing, 210042, China
# These authors contribute to this work equally.
Received 2018-1-22; Accepted 2018-7-2; Published 2018-9-8
Abstract
Hyperactive retrovirus-associated DNA sequence (Ras) genes have been found in human cancers and are involved in cancer pathogenesis. Salirasib, one anti-Ras compound, was reported to exhibit antitumoral effects, but its role remains unclear in cutaneous squamous cell carcinoma (cSCC). In our study, salirasib treatment led to deregulation of c-Raf, ERK and Akt signaling, blockage of MTOR signaling, interruption on Beclin 1-related autophagy regulation, activation of apoptosis and down-regulation of some cell cycle regulatory proteins in primary human epidermal keratinocyte (HEK)s, but did not exhibit similar effects in the human cSCC cell line COLO-16. MEK inhibitor U0126 can lead to dephosphorylation of MTOR and Rictor in COLO-16 cells; however, c-Raf was not yet down-regulated after salirasib treatment in the presence of U0126. Furthermore, we verified that the Ras activity could be suppressed by salirasib, and there was no loss-of-function mutation in c-Raf in HEKs and COLO-16 cells. In summary, salirasib does not exhibit antitumoral effects in the cSCC cells in assays in vitro. We speculated that the disability of signaling transmission from Ras to c-Raf in COLO-16 cells might contribute to the ineffective performance of salirasib.
Keywords: cutaneous squamous cell carcinoma, keratinocyte, salirasib, Ras, p53
Introduction
Hyperactive retrovirus-associated DNA sequence (Ras) genes have been found in human cancers and are involved in cancer pathogenesis [1]. The in vitro activity of salirasib (S-trans, transfarnesylthiosalicylic acid, FTS), an anti-Ras compound, was reported in types of human tumour cell lines. Recently, salirasib was evaluated for preclinical, phase I and II trials in the treatment of pancreatic, hepatologic and lung cancers, respectively [2-4]. Although Ras activation was reported to be involved in the development of human cutaneous squamous cell carcinoma (cSCC), the performance of salirasib has not been evaluated [5]. Herein, we reported that salirasib caused a series of events including deregulation of ERK and Akt signalling, blockage of mechanistic target of rapamycin (MTOR) signalling, interruption of Beclin 1-related autophagy regulation, activation of apoptosis and down-regulation of some cell cycle regulatory proteins in primary human epidermal keratinocyte (HEK)s, but did not exhibit similar effects in the human cSCC cell line COLO-16 because of the interruption of signaling transmission from Ras to c-Raf.
Materials and Methods
Cell culture
HEKs were cultured in Keratinocyte-SFM (Gibco, CA, USA). COLO-16 [6], A431 [7] and HaCaT [8] cells, three cell lines that were reported to be p53 mutated, were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (Gibco, CA, USA).
Human skin samples
cSCC samples were obtained from surgical excision. The samples were used to be assayed after pathology examination. The tissues were obtained under informed consent of patients. This study conformed to principles of ethics and protected the privacy of the patients' personal health information. This study was approved by Ethics Committee (2012-KY/LC-003).
Reagents and antibodies
Compounds included E64d, pepstatin, acridine orange (AO), dimethylsulphoxide (DMSO) (all from Sigma-Aldrich, St. Louis, MO, USA), salirasib, torin 1 (both from Tocris, Bristol, UK), pp242 (Abcam, Cambridge, MA, USA) and U0126 (Cell Signaling Technology, Danvers, MA, USA). Primary and secondary antibodies were from Cell Signaling Technology, MA USA.
Western blot
Western blot was performed as previously described [9].
AO staining assay
AO staining assay was performed as previously described [9].
β-galactosidase staining assay
Cell senescence was evaluated using the Senescence β-gal Staining Kit (Beyotime Biotechnology, Haimen, Jiangsu, China) according to the manufacturer's instructions. The senescence-associated β-galactosidase (SA-β-gal)-positive cells were observed and counted under a microscope at a magnification of × 400.
Ras activity assay
The Ras activity was assessed by the Ras Activation ELISA ASSAY Kit #17-497 (Merck, Darmstadt, Germany) according to the manufacturer's introductions. The spots were visualized by the ChemiDoc™ XRS+ system (Bio-Rad Laboratories, Hercules, CA, USA). The area intensities were quantified using Quantity One software.
RNA interference
HEKs were transiently transfected with p53 siRNA (siP53) using Lipofectamine 2000 (Invitrogen, CA, USA) according to the manufacturer's instructions. Scrambled siRNA was used as negative control (NC). After transfection for 48 hours (h), cells were treated with or without salirasib for 6 h. siP53: 5'-GUA CCA CCA UCC ACU ACA ATT-3' (sense), 5'-UUG UAG UGG AUG GUG GUA CTT-3' (antisense). NC: 5'-UUC UCC GAA CGU GUC ACG UTT-3' (sense), 5'-ACG UGA CAC GUU CGG AGA ATT-3' (antisense).
Co-Immunoprecipitation
Anti-Beclin 1 antibody was added to the cell lysate (1: 50) and rotated gently overnight at 4°C. The mixture was then incubated with SureBeads Protein G (Bio-Rad Laboratories) at 4°C for 4 h. The SureBeads were washed with PBST three times and resuspended in boiling 1 × SDS sample buffer. After centrifugation, the supernatant was analysed by western blot.
Cell proliferation assay
The Cell Counting Kit-8 (CCK-8) (Beyotime) was used according to the manufacturer's instructions. Cells were seeded in 96-well plates at a density of 5×103 cells per well. Prior to detection, the CCK-8 reagent was added and incubated for 2 h at 37°C. The optical density was measured at 450 nm.
Exome sequencing
Refer to the supporting information.
Statistical analysis
Results were obtained from 3-6 independent experiments for statistical analysis. T tests or ANOVA were performed for data analysis. The analysis results were considered significant if p-values < 0.05.
Results
Salirasib impeded autophagy flux and inhibited senescence in HEKs
Salirasib treatment increased the conversion of microtubule-associated protein 1 light chain 3 (LC3)-I to LC3-II (LC3-II /LC3-I) in HEKs and COLO-16 cells (Figure 1A). Next, LC3-II turnover was assayed to monitor the autophagy flux. Salirasib treatment inhibited LC3-II turnover (LC3-II/β-actin, comparison in the presence and absence of E64d and pepstatin) in HEKs, whereas no change was observed in COLO-16 cells (Figure 1B). AO was used to stain autophagosomes in situ. Similar to the results of the LC3-II accumulation assay, AO positive vacuoles were decreased after salirasib treatment only in HEKs (Figure 1C). Next, cellular senescence was detected. We did not observe obvious change in the quantity of SA-β-gal-positive COLO-16 cells after salirasib treatment. However, we observed that salirasib treatment led to significant shrinkage of the cell appearance and decreased SA-β-gal-positive staining in HEKs (Figure 1D).
Effects of salirasib on autophagy and senescence in HEKs and COLO-16 cells. (A) HEKs and COLO-16 cells were treated with 0, 25, 50 and 75 μM salirasib for 6 h. LC3-II(A/B)/LC3-I(A/B) ratios were calculated. (B) HEKs and COLO-16 cells were treated with 0, 25, 50 and 75 μM salirasib in the presence and absence of 10 μg /mL E64d and 10 μg /mL pepstatin for 6 h. LC3-II(A/B)/β-actin ratios were calculated to represent LC3-II turnover (autophagy flux). (C) HEKs and COLO-16 cells were treated with 0 and 50 μM salirasib in the presence and absence of 10 μg /mL E64d and 10 μg /mL pepstatin for 6 h. The mean of the red/green fluorescence ratios of cells was calculated. Bars = 5 μm. Autophagosomes were marked with red fluorescence, and nuclei and cytoplasm were visualized in green fluorescence. (D) HEKs and COLO-16 cells were treated with 0 and 50 μM salirasib for 6 h. SA-β-Gal-positive cells were dyed blue. * p < 0.05. Values represent the mean ± SD of 3-6 experiments.
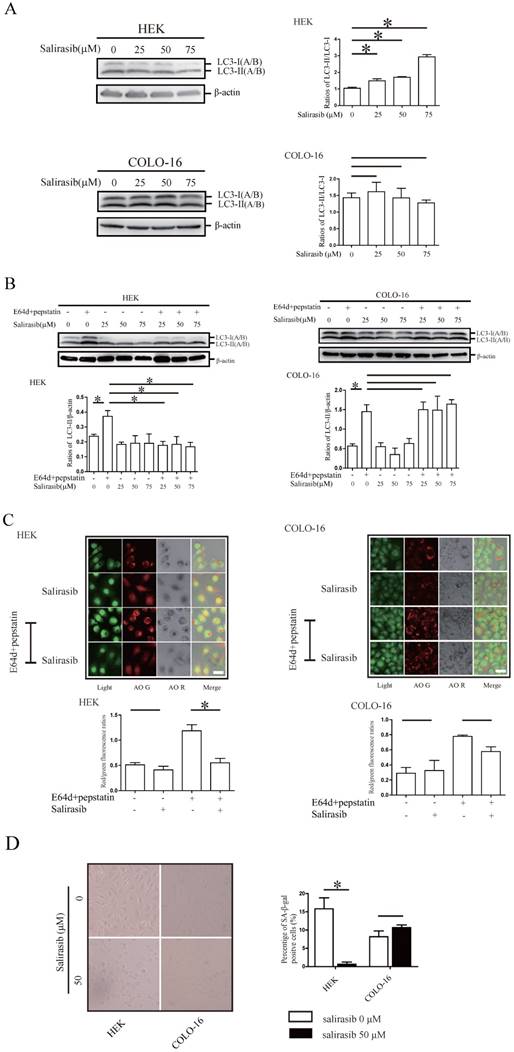
Pathway signaling and cell viability regulated by salirasib in HEKs were not sensitive to salirasib treatment in COLO-16 cells
MTOR and Rictor with their downstream target proteins showed a decrease of phosphorylation in HEKs but not in COLO-16 cells (Figure 2A). We validated that MTOR complex (MTORC) in COLO-16 cells were sensitive to the MTOR inhibitors torin 1 or pp242, and the inhibitory effect was not affected by salirasib treatment (Figure 2B-C). In HEKs, salirasib treatment led to the inhibition of Raf/MEK/ERK and Akt pathways, which behaved as the down-regulation of c-Raf expression and decreased the phosphorylation of MEK, ERK and Akt. However, the two pathways were not inhibited by salirasib treatment in COLO-16 cells (Figure 2D). Cleavage of apoptotic markers caspase 3 and PARP was observed in HEKs but not in COLO-16 cells (Figure 2E). Then down-regulation of some cell cycle regulatory proteins Cyclin D1, Cyclin B1 and Myt1, and phosphorylation of Histone H3 were found in HEKs, suggesting aberrant regulation of the cell cycle. The inconsistent effect was found in COLO-16 cells (Figure 2E). Consistent with the results of apoptosis assay, salirasib significantly inhibited cell viability of HEKs in 25, 50 and 75 μM doses, while only 75 μM salirasib led to a low-level impairment on the viability of COLO-16 cells (Figure 2F).
Effects of salirasib on MTORC signaling, related signalling pathways and cell viability in HEKs and COLO-16 cells. (A, D, E) HEKs and COLO-16 cells were treated with 0, 25, 50 and 75 μM salirasib for 6 h. Phosphorylation and protein levels of MTOR, Rictor, p70 S6 Kinase, S6 Ribosomal Protein, 4E-BP1 and PKCα were examined for the detection of MTORC activity, and the ratios of phospho-MTOR/MTOR and phospho-Rictor/Rictor represent protein activities (A). Phosphorylation or protein levels of Ras, c-Raf, MEK, ERK, Akt (D), PRAP, Caspase-3, p53, Cyclin D1, Cyclin B1, Myt1 and Histone H3 (E) were examined. (B) COLO-16 cells were treated with 0, 0.5, 1.0 and 2.0 μM torin1 or pp242 for 6 h. Phosphorylation or protein levels of MTOR, Rictor, p70 S6 Kinase, S6 Ribosomal Protein, 4E-BP1, PKCα and LC3 were examined. (C) COLO-16 cells were treated with 0 and 50 μM salirasib with or without 1.0 μM torin 1 or pp242 for 6 h. Phosphorylation or protein levels of MTOR, Rictor and LC3 were examined. (F) HEKs and COLO-16 cells were treated with 0, 25, 50 and 75 μM salirasib for 6 h. Cell proliferation was determined by CCK-8 assay. The protein expression of the indicated signalling pathways was detected through western blot. Protein levels were normalized to those for β-actin. * p < 0.05. Values represent the mean ± SD of 3-6 experiments.

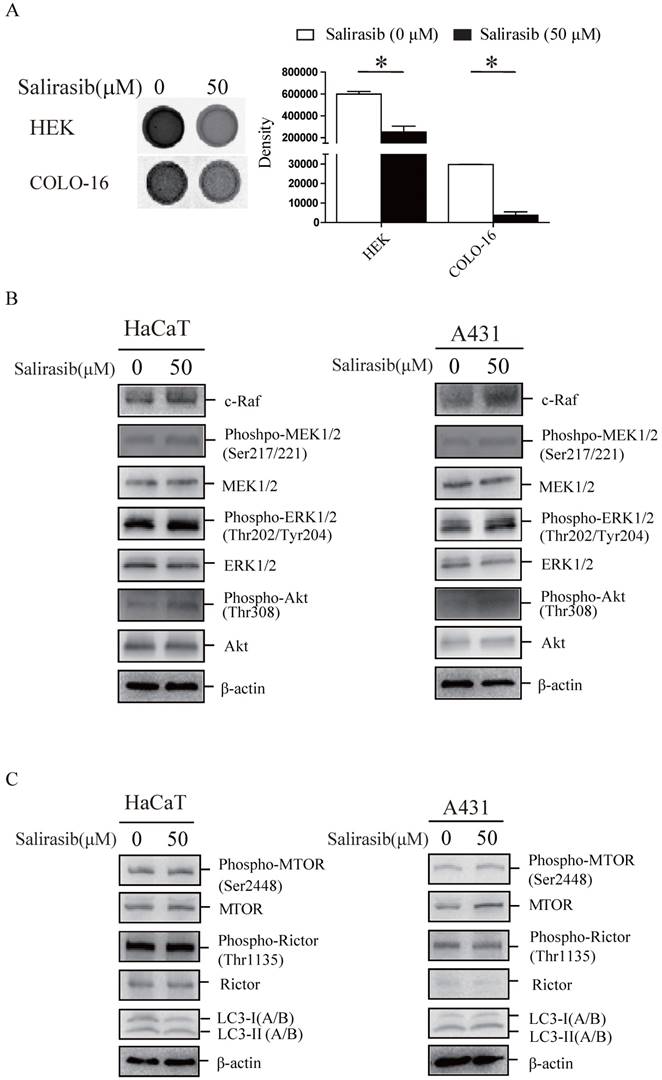
Disability of signalling transmission from Ras to c-Raf in COLO-16 cells might exist
First, we verified that the Ras activity in HEKs and COLO-16 cells could be suppressed by salirasib (Figure 3A). Second, salirasib exited similar effects on Raf/MEK/ERK, Akt and MTORC signalling in HaCaT and A431 cells (Figure 3B-C). Furthermore, we identified p53 exonic mutations in 9 cSCC tissue samples from 10 cases of patients by exome sequencing (Table 1), but pathogenic mutations of Ras and c-Raf were not been found in any samples, suggesting that Ras and c-Raf mutations might be rare in cSCC.
Effects of salirasib on Ras activities in HEKs and COLO-16 cells and analysis of signalling pathways in HaCaT and A431 cells. (A) COLO-16 cells were incubated with 0 and 50 μM salirasib for 6 h and Ras activities were measured. The area intensities were quantified to represent Ras activities. (B-C) HaCaT and A431 cells were incubated with 0 and 50 μM salirasib for 6 h. Phosphorylation or protein levels of c-Raf, MEK, ERK, Akt (B), MTOR, Rictor and LC3 (C) were examined.
p53 exonic mutations identified in 9 cSCC tissue samples from 10 cases of patients. F: female M: male
| Sex | Age (years) | Allele 1 | Allele 2 | Allele 3 | |||
|---|---|---|---|---|---|---|---|
| Nucleotide change | Amino-acid change | Nucleotide change | Amino-acid change | Nucleotide change | Amino-acid change | ||
| F | 75 | c.C98>G | p.P33R | ||||
| M | 73 | c.C98>G | p.P33R | ||||
| M | 93 | c.T89>A | p.I30N | c.C98>G | p.P33R | ||
| M | 89 | c.C98>G | p.P33R | ||||
| M | 92 | c.T89>A | p.I30N | c.C55>T | p.P19S | c.C98>G | p.P33R |
| F | 78 | c.C98>G | p.P33R | ||||
| F | 87 | c.C98>G | p.P33R | ||||
| M | 91 | c.C98>G | p.P33R | ||||
| F | 82 | c.C98>G | p.P33R | ||||
| F | 70 | ||||||
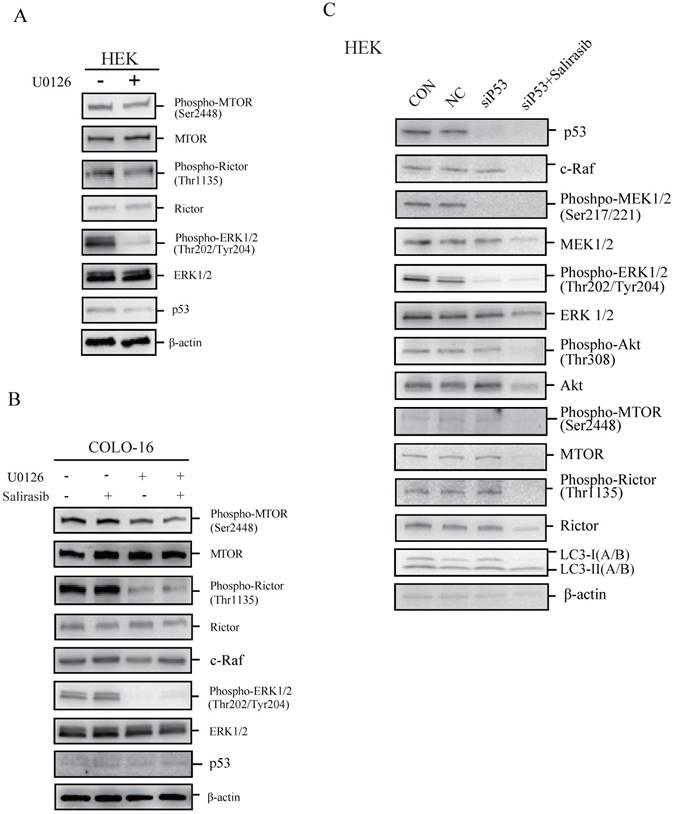
p53 loss-of-function might not contribute to the insensitiveness of COLO-16 cells to salirasib treatment
To determine if p53 plays a key role in regulating cellular responses to salirasib treatment, we performed the following investigations. We found that the MEK inhibitor U0126 led to dephosphorylation of MTOR and Rictor, indicating that the ERK pathway normally regulates MTOR signalling in HEKs and COLO-16 cells (Figure 4A-B). In COLO-16 cells with p53 loss-of-function, c-Raf was not yet down-regulated after salirasib treatment in the presence of U0126, suggesting that the target of salirasib is upstream of ERK (Figure 4B). However, in HEKs with p53 silenced, salirasib still led to the down-regulation of c-Raf, MEK, ERK, Akt as well as MTOR and Rictor (Figure 4C). In addition, in HEKs, U0126 decreased p53 expression, and p53 knock-down caused dephosphorylation of MEK and ERK, suggesting a positive feedback loop between p53 and ERK (Figure 4A, C).
The performance of salirasib on ERK-inhibited or p53-silenced HEKs or COLO-16 cells. (A-B) HEKs or COLO-16 cells were treated with 0 and 5 μM U0126 (A) with or without 50 μM salirasib (B) for 6 h. ERK activities were inhibited by U0126, and phosphorylation and protein levels of MTOR and Rictor were examined for the detection of MTORC activity. Protein levels of p53 (A-B) and c-Raf (B) were also examined. (C) Phosphorylation or protein levels of p53, c-Raf, MEK, ERK, Akt, MTOR, Rictor and LC3 were examined in normal mock (CON), NC, and siP53-transfected (with or without salirasib treatment) HEKs to determine the effect of salirasib under p53 silenced conditions.
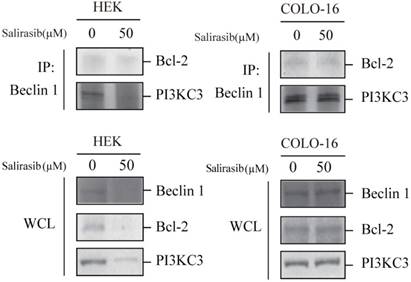
Salirasib decreased combination between Beclin 1 and PI3KC3 in HEKs
In addition to MTOR signalling, the Beclin 1 complex mainly consisted of Beclin 1 and PI3 kinase class III (PI3KC3), which facilitates the autophagy process, and the function is inactivated by Bcl-2 (an apoptotic inhibitor). Salirasib down-regulated the expression of Beclin 1, PI3KC3 and Bcl-2 and decreased the combination between Beclin 1 and PI3KC3 in HEKs but not in COLO-16 cells (Figure 5).
Effects of salirasib on combination between Beclin 1 and PI3KC3 in HEKs. The interplay of Beclin 1, PI3KC3 and Bcl-2 complex was analysed by Co-IP. The immunoprecipitated proteins were detected with Bcl-2 and PI3KC3 antibodies. Proteins in whole cell lysate (WCL) were detected with Beclin 1, Bcl-2 and PI3K antibodies.
Discussion
Salirasib was reported to induce autophagy in HeLa, HCT-116 and DLD-1 cell lines [10]. Therefore, we first determined the autophagy level because of the crucial role of autophagy in initiation and development of tumours. The findings demonstrated that salirasib promoted LC3-I to LC3-II conversion and impeded autophagy flux in HEKs but did not affect the autophagy of COLO-16 cells. Autophagy was reported to be involved in cellular senescence [11]. Cell senescence can be reflected through the change of cells stained with SA-β-gal [12]. The result of SA-β-gal assay indicated that salirasib might lead to a more intense biological effect in normal keratinocytes compared with cSCC cells.
MTOR was reported to be involved in autophagy activation in response to salirasib treatment [13]. Therefore, the activities of MTORC related signalling were evaluated in salirasib-treated cells. The performance of MTOR signalling in salirasib-treated HEKs was consistent with previous reports; however, the MTOR signalling of COLO-16 cells exhibited a novel nonresponse to salirasib. Salirasib is an S-farnesylcysteine analogue and could interfere with Ras membrane association by competing directly with farnesylated Ras [14]. Salirasib suppresses Ras-activated signalling including the Raf/MEK/ERK and Akt pathways, and modulates the cell cycle process, apoptosis and autophagy [15]. While in this study, we found that salirasib did not exhibit similar effects on the two pathways in COLO-16 cells. The effects of salirasib on apoptosis and cell cycle in HEKs also differed from COLO-16 cells. Whether salirasib inhibited the expression of cell cycle regulatory proteins or promoted apoptosis in HEKs via suppressing Raf/MEK/ERK cascades and Akt signalling is unclear and needs further investigation.
Since induced effects of salirasib on autophagy and cell death have been revealed in many tumour cells [10], we speculated that dysfunction of some key proteins led to the insensitivity of COLO-16 cells. We observed that salirasib did not work in reported p53 dysfunction cells including COLO-16, A431 (another epidermal carcinoma cell line) and HaCaT cells. p53 protein was at an extremely low basal level in COLO-16 cells (Figure 2E). Loss-of-function of p53 was reported to induce cell proliferation via activation of Raf/MEK/ERK signalling [16]. The findings indicate that the nonresponse of c-Raf to its upstream signalling component Ras, but not p53 loss-of-function, contributes to the abnormal response to salirasib in COLO-16 cells. We confirmed that there no loss-of-function mutant in c-Raf in four cells lines; therefore, the c-Raf nonresponse is caused by disability of signalling transmission between them.
The findings of Co-IP might explain the results that MTOR inhibition did not induce autophagy, and apoptosis was triggered in salirasib-treated HEKs. Our observations demonstrated that the novel regulation of autophagy by salirasib is caused by its multiple targets on autophagy regulation.
In summary, salirasib does not exhibit antitumoral effects to cSCC cells in assays in vitro. We speculated that the interruption of signaling transmission from Ras to c-Raf in COLO-16 cells might contribute to the ineffective performance of salirasib (Figure 6). Considering the potential value of the Ras/Raf pathway in treatment against cSCC, its mechanism is worth further investigation. The Ras function in cSCC needs to be further clarified before it is considered as a target in pharmaceutical approaches.
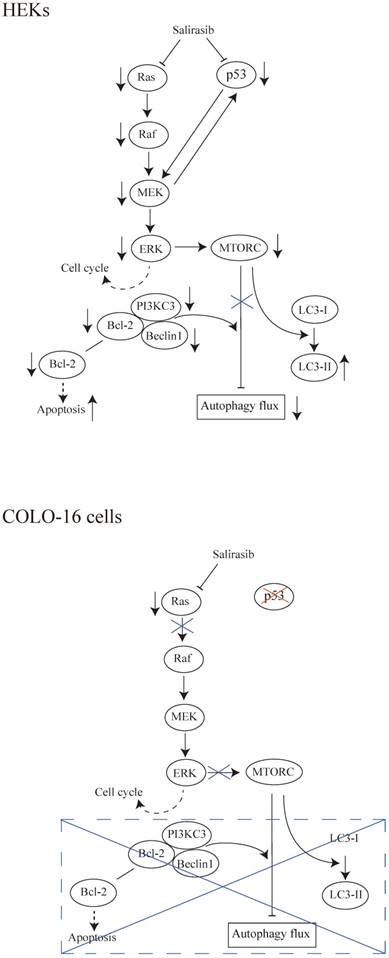
Schematic representations of the results in this study. (HEKs) Salirasib inhibited Ras/Raf/MEK/ERK and p53 signalling. Inhibited ERK might contribute to the down-regulation of cell cycle regulatory proteins. The linkage between p53 and MEK was observed. The inhibition of MTOR signalling in response to its upstream pathway promoted the conversion from LC3-I to LC3-II. However, the autophagy flux was inhibited by salirasib treatment. Decreased expression of the Beclin 1 and PI3KC3 and formation of the Beclin 1-PI3KC3 complex impeded autophagy flux, in agreement with previous findings. Decreased Bcl-2 expression might facilitate apoptosis. (COLO-16 cells) In cells with a p53-mutation, salirasib treatment decreased the Ras activity, but did not lead to the inhibition of Raf/MEK/ERK signalling. Therefore, the inconsistent effects on cell cycle regulatory proteins were observed. MTORC is silent in response to the upstream signalling. MTORC- and Beclin 1-dependent autophagy processes and Bcl-2-related apoptosis were not disturbed under this condition.
Supplementary Material
Supplementary methods.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (No. 81773342, 81673083, 81703153), CAMS Innovation Fund for Medical Sciences (CIFMS-2017-I2M-1-017 and 2016-I2M-1-005), PhD Programs Foundation of Ministry of Education of China (No. 20131106120046), the Jiangsu Provincial Special Program of Medical Science (BL2012003), the Open Foundation of State Key Laboratory of Pollution Control and Resource Reuse (PCRRF17032). Wenbo Bu, Xu Chen and Song Xu are supported by the PUMC Youth Fund and Fundamental Research Funds for the Central Universities (3332015116, 3332014008, 3332015026, 2016RC320005, 2016ZX320014).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295-308
2. Badar T, Cortes JE, Ravandi F, O'Brien S, Verstovsek S, Garcia-Manero G. et al. Phase I study of S-trans, trans-farnesylthiosalicylic acid (salirasib), a novel oral RAS inhibitor in patients with refractory hematologic malignancies. Clin Lymphoma Myeloma Leuk. 2015;15:433-8 e2
3. Laheru D, Shah P, Rajeshkumar NV, McAllister F, Taylor G, Goldsweig H. et al. Integrated preclinical and clinical development of S-trans, trans-Farnesylthiosalicylic Acid (FTS, Salirasib) in pancreatic cancer. Invest New Drugs. 2012;30:2391-9
4. Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG. et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6:1435-7
5. Jia J, Li C, Luo S, Liu-Smith F, Yang J, Wang X. et al. Yes-Associated Protein Contributes to the Development of Human Cutaneous Squamous Cell Carcinoma via Activation of RAS. J Invest Dermatol. 2016;136:1267-77
6. Pierceall WE, Mukhopadhyay T, Goldberg LH, Ananthaswamy HN. Mutations in the p53 tumor suppressor gene in human cutaneous squamous cell carcinomas. Mol Carcinog. 1991;4:445-9
7. Thongrakard V, Titone R, Follo C, Morani F, Suksamrarn A, Tencomnao T. et al. Turmeric toxicity in A431 epidermoid cancer cells associates with autophagy degradation of anti-apoptotic and anti-autophagic p53 mutant. Phytother Res. 2014;28:1761-9
8. Cordani N, Pozzi S, Martynova E, Fanoni D, Borrelli S, Alotto D. et al. Mutant p53 subverts p63 control over KLF4 expression in keratinocytes. Oncogene. 2011;30:922-32
9. Chen X, Li M, Li L, Xu S, Huang D, Ju M. et al. Trehalose, sucrose and raffinose are novel activators of autophagy in human keratinocytes through an mTOR-independent pathway. Sci Rep. 2016;6:28423
10. Schmukler E, Grinboim E, Schokoroy S, Amir A, Wolfson E, Kloog Y. et al. Ras inhibition enhances autophagy, which partially protects cells from death. Oncotarget. 2013;4:145-55
11. Kwon Y, Kim JW, Jeoung JA, Kim MS, Kang C. Autophagy Is Pro-Senescence When Seen in Close-Up, but Anti-Senescence in Long-Shot. Mol Cells. 2017;40:607-12
12. Zhang F, Zhang M, Wang A, Xu M, Wang C, Xu G. et al. TWEAK increases SIRT1 expression and promotes p53 deacetylation affecting human hepaticstellate cell senescence. Cell Biol Int. 2017;41:147-54
13. Makovski V, Haklai R, Kloog Y. Farnesylthiosalicylic acid (salirasib) inhibits Rheb in TSC2-null ELT3 cells: a potential treatment for lymphangioleiomyomatosis. Int J Cancer. 2012;130:1420-9
14. Quah SY, Tan MS, Teh YH, Stanslas J. Pharmacological modulation of oncogenic Ras by natural products and their derivatives: Renewed hope in the discovery of novel anti-Ras drugs. Pharmacol Ther. 2016;162:35-57
15. Blum R, Elkon R, Yaari S, Zundelevich A, Jacob-Hirsch J, Rechavi G. et al. Gene expression signature of human cancer cell lines treated with the ras inhibitor salirasib (S-farnesylthiosalicylic acid). Cancer Res. 2007;67:3320-8
16. Drosten M, Sum EY, Lechuga CG, Simon-Carrasco L, Jacob HK, Garcia-Medina R. et al. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc Natl Acad Sci U S A. 2014;111:15155-60
Author contact
![]() Corresponding authors: Xu Chen: chenxcams.cn, Heng Gu: guhengcams.cn
Corresponding authors: Xu Chen: chenxcams.cn, Heng Gu: guhengcams.cn