Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(18):3394-3399. doi:10.7150/jca.26068 This issue Cite
Research Paper
Antitumor activity of sorafenib plus CDK4/6 inhibitor in pancreatic patient derived cell with KRAS mutation
Taehyang Lee, Kyung Kim, Jeeyun Lee, Se Hoon Park, Young Suk Park, Ho Yeong Lim, Won Ki Kang, Joon Oh Park ![]() , Seung Tae Kim
, Seung Tae Kim ![]()
Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
Received 2018-3-15; Accepted 2018-7-22; Published 2018-9-8
Abstract
KRAS mutation has been known as crucial marker for growth and maintenance of pancreatic cancer (PC) and targeting the KRAS is inevitable component for realizing precision medicine to PC. We established patient-derived tumor cells (PDCs) from patient with KRAS G12R mutant PC. Through the PDC, we investigated the therapeutic impact of sorafenib alone, LEE001 alone and the combination of sorafenib and LEE001 in KRAS mutant PC. For the validation, we also tested a cell viability assay for sorafenib, LEE001, and sorafenib plus LEE001 in KRAS G12R transfected HEK293T cells. Based on MTT proliferation assays using PDCs, values of IC50 were 6.07 uM to sorafenib and > 10.00 uM to LEE001, respectively. The value of IC50 of the combination (sorafenib plus LEE001) was 3.19 uM. Cell proliferation of PDC was significantly inhibited by sorafenib plus LEE001, as compared to sorafenib monotherapy and LEE001 monotherapy. In the validation through KRAS G12R transfected HEK293T cells, consistent to findings in PDCs, combinations of sorafenib plus LEE001 had most effective inhibitory effect in KRAS G12R transfected HEK293T cells. Furthermore, on analyzing the regulation of targeted downstream pathways upon exposure to sorafenib, LEE001, and sorafenib plus LEE001 by immunoblot assay using KRAS G12R transfected HEK293T cells, AKT phosphorylation was distinctively decreased in KRAS G12R transfected HEL293 cells after only sorafenib plus LEE001. This study suggests that the combination of RAF and CDK4/6 inhibitors might be a novel treatment strategy for KRAS G12R mutant pancreatic cancer. The antitumor effect of RAF plus CDK4/6 inhibitors also needs to be evaluated in other subtypes of KRAS mutation in pancreatic cancer.
Keywords: KRAS mutation, CDK4/6 inhibitor, sorafenib, pancreatic cancer
Introduction
Pancreatic ductal adenocarcinoma (PDA) is challenge to manage, with 5 year rates of survival lower than 10% for patients with cancers of all stages.[1] Recent sequencing studies had shown detailed genetic information of PDA with mutational activation of the KRAS oncogene found in ~95% of patients.[2-5] KRAS mutation has been known as crucial marker for PDC growth and maintenance and targeting the KRAS is inevitable component for realizing precision medicine to PDA.[6-9] KRAS mutation constitutively activates down-stream pathways including RAS/RAF/MEK/ERK signaling.[10] The unregulated signaling in these pathways lead to increased proliferation, decreased apoptosis, disrupted cellular metabolism, and increased angiogenesis which in turn leads to tumor cell proliferation.[11, 12] However, despite vigorous efforts, an effective anti-RAS treatment strategy has yet to be introduced to the clinic.[13, 14]
Cyclin D expression is the rate-limiting step in cell cycle progression from G1 into S phase.[15] Cyclin D complexes with and activates cyclin dependent kinase (CDK) 4 and 6, which phosphorylate and inactivate the tumor suppressor retinoblastoma protein (Rb).[16] CDK4 and CDK6 play key roles in mammalian cell proliferation, where they help to drive the progression of cells into the DNA synthetic (S) phase of the cell division cycle. Some cancer specific mutations such as those affecting receptor tyrosine kinase (RTK), RAS, RAF, and PI3K can enhance cyclin D-dependent CDK4/6 activity. Thus, cell type specific RTK, RAS/RAF/MEK/ERK, and PI3K/AKT inhibitor can increase the threshold for CDK4/6 activation and synergize with CDK4/6 inhibitors to induce G1 phase cell cycle arrest.[17] CDK4/6 inhibitors may prove useful in the treatment of a variety of cancer subtypes based on overcoming the tumor suppressive activity of pRb in cancer cells.
Sorafenib, a multi-kinase inhibitor, has been shown to inhibit tumor growth and tumor angiogenesis by targeting RAF kinase, vascular endothelial growth factor receptor, c-kit and platelet derived growth factor receptor.[18, 19] Also, sorafenib revealed the impressive benefit to non-small cell lung cancer (NSCLC) patients with KRAS mutation in previous clinical trial.[20] LEE-001 (Ribociclib) is orally bioavailable and highly selective CDK4/6 targeting agent exhibit IC50 value in the low nanomolar range. Herein, through PDC, we investigated the therapeutic impact of sorafenib alone, LEE001 alone and the combination of sorafenib and LEE001 in KRAS mutant pancreatic cancer.
Patients and Methods
Cell line and patient-derived cell (PDC) culture. HEK293T cells were purchased from American Type Culture Collection (ATCC) and HEK293T cells were maintained in RPMI1640 containing 10% FBS and 1% antibiotic-anti-mycotic solution (Gibco BRL, Paisley, UK).
A 67 years old man presented initially with stage IV, KRAS mutant pancreatic cancer. He had multiple pulmonary metastatic lesions and pleural effusion at the diagnosis. As this time, pleural tapping for pleural effusion was performed and we generated PDCs from the patient after pathologic tumor confirmation. The tumor sample has been analyzed by target sequencing and then tumor was conformed to harbor the KRAS G12R mutation. We also confirmed the KRAS G12R mutation in the patient's matched PDCs by ddPCR (Fig. S1). Pleural effusions (1L) were collected from metastatic pancreatic cancer patient and there were divided into 50-mL tubes, centrifuged at 1500 rpm for 10 min for gathering the cells. The cells were washed twice with phosphate-buffered saline. Cell pellets were resuspended in RPMI1640 containing 10% FBS, 1% antibiotic-anti-mycotic solution (Gibco BRL, Paisley, UK), 0.5 g/ml of hydrocortisone (Sigma Aldrich St. Louis, MO, USA), 5 g/ml of insulin (PeproTech, Rocky Hill, NJ, USA) and 5 ng of EGF (PeproTech Rocky Hill, NJ, USA) and plated into 75-cm2 culture flasks. The medium was changed every 3 days, and all cell lines were maintained at 37˚C in a 5% CO2-humidifed atmosphere. PDCs were passaged using TrypLE Express (Gibco BRL) to detach cells when they reached 80-90% confluence.
Targeted gene sequencing. We conducted genomic analysis for tumor sample biopsied from the metastatic pancreatic patient originating PDC. Formalin-fixed paraffin embedded (FFPE) samples containing >40% tumor cellularity were dissected under microscopy from 4-μm-thick unstained sections (10 to 20 slides) or from fresh frozen tissues by comparison with a hematoxylin and eosin-stained slide. Briefly, DNA was extracted using standard procedures (Qiagen, Hilden, Germany) and extracted genomic DNA was sheared to 150-200 bp using Covaris S220 (Covaris, Woburn, MA, USA), and targeted genes were captured using a custom panel capture library (Agilent Technologies, Santa Clara, CA, USA) covering 2.5 Mb of exonic regions for the Illumina Paired-End Sequencing Library kit. We performed DNA sequencing of 100 or 101-bp paired-end reads using the Illumina HiSeq 2,500 sequencer (Illumina, San Diego, CA, USA). We aligned the sequencing reads to the human reference genome (GRCh37/hg19), excluded duplicated reads, and extracted uniquely mapped and properly paired reads with an insert size. Somatic alterations were detected by CancerSCAN and actionable variants included in this panel were selected based on publicly available databases such as My Cancer Genome® (http://www.mycancergenome.org/)
Digital PCR. We used the Bio-Rad QX200 ddPCR system (Bio-Rad, Hercules, CA, USA). The ddPCR probe mastermix and primers targeting KRAS G12R mutation with KRAS wild type were all purchased from Thermo Fisher Scientific (Waltham, MA, USA). The primer sequences are proprietary to the company. Data was processed using QuantaSoft v.1.6 (Bio-Rad, Hercules, CA, USA). To determine the false-positive rate, 2 repeats of 50 ng of the wild type cell line DNA and Milli-Q water (EMD Millipore, Billerica, MA, USA) as no-template control were used.
Plasmid construct and Transfection. KRAS G12R mutant construct was purchased from Origene (RC400106, Rockville, MD, USA). For transfection, 5*105 cells were seeded on 6 well plate. 1 day after, medium changed to RPMI1640 without antibiotic-anti-mycotic solution, and then 4 ug plasmid were transfected with lipofectamine3000 (Thermo Fisher scientific, Waltham, MA, USA) according to manufactural's guide. Transfection efficiency was confirmed by immunoblotting.
Cell treatment and viability assay. After pathologic confirmation, cells were seeded at a density of 1-2 × 106 cells/10-mm dish or 5,000 cells/well in 96-well plates for immunoblot analysis and cell proliferation assays and treated for 3 days with various doses of sorafenib and LEE001. Inhibition of cell proliferation was determined using Cell Titer Glo (Promega, Madison, WI, USA) according to the manufacturer's protocol. Interactions between drugs were presented as the combination index (CI), calculated by dividing the expected growth inhibition rate by the observed growth inhibition rate: CI <1.0 indicates antagonistic cytotoxicity; CI=1.0 is additive cytotoxicity; and CI >1.0 is synergistic cytotoxicity.
Immunoblot analysis. Total proteins were isolated using RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) containing a protease inhibitor cocktail (Roche, Basel, Switzerland) and phosphatase inhibitor cocktail (Roche, Basel, Switzerland), and protein concentrations were determined using a BCA Protein Assay (Pierce, Appleton, WI, USA). Aliquots containing 30 μg of protein were subjected to 4-12% SDS-polyacrylamide gel electrophoresis and electrotransferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.1% v/v Tween 20, and probed overnight at 4°C with specific antibodies; RAS (#3965) p-RAF (#2696) RAF (#14814), P-AKT (#4060), AKT (#9272) from Cell Signaling Technology (Beverly, MA, USA), and -actin (#sc-47778) from Santa Cruz (Dallas, Texas, USA). Horseradish peroxidase-conjugated anti-rabbit (#170-6515, Bio-rad, Hercules, CA, USA) or mouse IgG (#170-6516, Bio-rad, Hercules, CA, USA) was used as a secondary antibody, and signals were detected by chemiluminescence using ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA) and visualized using LAS-4000 (GE healthcare, Hammersmith,UK).
Results
Cell viability assay using patient derived cells. To investigate therapeutic impacts of sorafenib monotherapy, LEE001 monotherapy and the combination of sorafenib and LEE001 in KRAS mutant pancreatic cancer, we performed a cell viability assay using the KRAS G12R mutant pancreatic PDC line (Figure 1). At first, we tested whether PDCs were sensitive to RAF inhibitor (sorafenib) and CDK4/6 inhibitor (LEE001) respectively. Based on MTT proliferation assays, values of IC50 were 6.07 uM to sorafenib and > 10.00 uM to LEE001, respectively. As monotherapy, LEE001 did not suppress cell viability of PDC.
Anti-cancer effect of sorafenib monotherapy, LEE001 monotherapy and the combination of sorafenib and LEE001 in KRAS G12R mutant pancreatic cancer PDC.
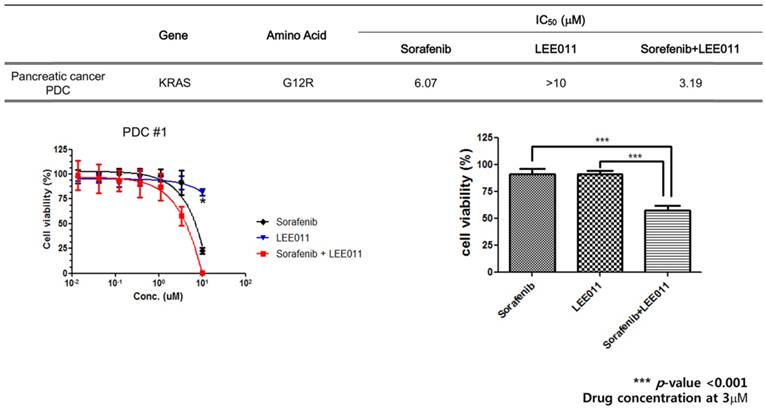
Next, we conducted a cell viability assay for combination (sorafenib plus LEE001) in the same PDCs. The value of IC50 of the combination was 3.19 uM. Cell proliferation of PDC was significantly inhibited by sorafenib plus LEE001, as compared to sorafenib monotherapy and LEE001 monotherapy. The calculated combination index (CI) for sorafenib and LEE001 showed synergistic effects of the two agents on PDCs with the KRAS G12R mutation (Table 1).
Combination effect of sorafenib and LEE001
| RAF inhibitor (Sorafenib) | CDK4/6 inhibitor (LEE001) | Combination (1;1) | index | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration (uM) | MGI* | p-value | Concentration (uM) | MGI | p-value | Expected | Observed | p-value | |
| 0.01 | 0.976 | 0.453 | 0.01 | 0.988 | 0.762 | 0.964 | 0.987 | 0.724 | 0.98 |
| 0.03 | 0.972 | 0.465 | 0.03 | 0.955 | 0.192 | 0.928 | 0.956 | 0.030 | 0.97 |
| 0.1 | 0.999 | 0.982 | 0.1 | 0.902 | 0.015 | 0.901 | 0.934 | 0.006 | 0.96 |
| 0.3 | 0.993 | 0.888 | 0.3 | 0.955 | 0.151 | 0.949 | 0.906 | 0.002 | 1.05 |
| 1 | 0.988 | 0.777 | 1 | 0.926 | 0.025 | 0.915 | 0.870 | 0.001 | 1.05 |
| 3 | 0.912 | 0.202 | 3 | 0.915 | 0.067 | 0.835 | 0.577 | <0.005 | 1.45 |
| 10 | 0.227 | <0.005 | 10 | 0.817 | <0.005 | 0.185 | 0.003 | <0.005 | 70.41 |
*MGI, mean growth inhibition rate = growth rate of treated group/growth rate of untreated group
MGI index : <1: antagonistic effect 1-1.2: additional effect, >1.2: synergistic effect
P value was calculated by paired t test compared with no treatment, GraphPad Prism 5.0.
Expected: Growth inhibition rate of treatment A x growth inhibition rate of treatment B.
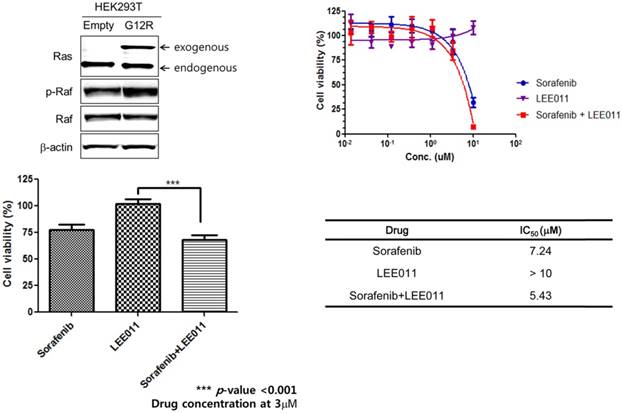
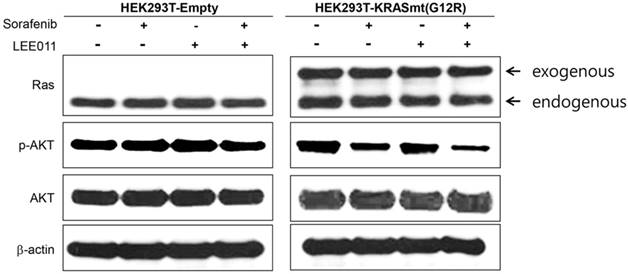
MTT assay and immunoblot assay using KRAS G12R transfected HEK293T. To verify the combination effect of sorafenib and LEE001 in PDC with KRAS G12R mutation, we tested a cell viability assay for sorafenib, LEE001, and sorafenib plus LEE001 in KRAS G12R transfected HEK293T cells (Figure 2). Consistent to findings in PDCs, combinations of sorafenib plus LEE001 had most effective inhibitory effect in KRAS G12R transfected HEK293T cells. Furthermore, we analyzed the regulation of targeted downstream pathways upon exposure to sorafenib, LEE001, and sorafenib plus LEE001 by immunoblot assay using KRAS G12R transfected HEK293T cells (Figure 3). After only sorafenib plus LEE001, AKT phosphorylation was distinctively decreased in KRAS G12R transfected HEL293 cells.
Verification of sorafenib plus LEE011 effect on KRAS G12R mutant transfected HEK293T cell.
Downregulation of p-AKT upon sorafenib plus LEE011 treatment in KRAS G12R mutant overexpressed HEK293T cells.
Discussion
KRAS mutation has been known as being present in 70~95% of pancreatic cancers.[2-5] Although about 90% of pancreatic cancers harbor activated driver oncogenic KRAS, effective overcoming treatment strategy against KRAS mutation has not been developed until now. This present study showed that RAF inhibitor (sorafenib) and CDK4/6 inhibitor (LEE001) might have the anti-tumor activity in PDC with KRAS G12R mutation. This finding was consistent to KRAS G12R transfected HEK293T cells. After sorafenib plus LEE001, AKT phosphorylation was also distinctively decreased in KRAS G12R transfected HEL293 cells. These findings suggest that RAF inhibitor (sorafenib) and CDK4/6 inhibitor (LEE001) might be a promising treatment strategy in metastatic pancreatic cancer patients with KRAS mutation.
KRAS mutations constitutively activate the RAS/RAF/ERK signal pathway. The activation of this signaling modulates the activity of target transcription factors such as cyclin D1 and cyclin D1/CDK complex.[21, 22] CDK4/6 activation is linked to promoting tumor progression. Loss of the cyclin dependent kinase inhibitor 2 (CDKN2A) tumor suppressor gene function by mutation or promoter methylation is found in 95% of pancreatic tumors.[23] CDKN2A is associated with the inhibition of CDK4/6.[24, 25] In other word, 95% of pancreatic tumor needs to inhibit the CDK4/6. However, previous study of CDK4/6 inhibitor in KRAS mutant pancreatic cell lines showed concern that though CDK4/6 inhibitor monotherapy supressed cell proliferation, it appeared to also increase epithelial mesenchymal transition (EMT) in cell lines.[26] Considering these findings, we tried to test the combination of RAF inhibitor (sorafenib) and CDK4/6 inhibitor (LEE001) in KRAS mutant pancreatic cancer. The anti-tumor effect of CDK4/6 inhibitors has been being actively explored in various tumor types such as melanoma, neuroblastoma, liposarcoma and mantle cell lymphoma.[27, 28] CDK4/6 inhibitors is regarded as having more potent antitumor activity when in combination with other agents that either strengthen the cytostatic effect of CDK4/6 inhibitors or convert reversible cytostasis into irreversible growth arrest or cell death. In the present study, the calculated combination index (CI) showed synergistic effects of the combination of RAF inhibitor and CDK4/6 inhibitor in PDCs with the KRAS G12R mutation (Table 1). These findings suggested that combination treatment with RAF and CDK4/6 inhibitors might be a novel treatment strategy for patients with KRAS G12R-mutant pancreatic cancer. Through PDC with KRAS G12R mutation and KRAS G12R transfected HEK293T cells, we confirmed the more potent antitumor effect of sorafenib plus LEE001. Furthermore, in immunoblot assay using KRAS G12R transfected HEK293T cells, only sorafenib plus LEE001, decreased AKT phosphorylation.
The activating point mutation of the KRAS oncogene on codon 12 remains the major events. The single nucleotide mutation on codon 12 of exon 2 induces replacement of the GGT sequence (encoding for glycine) by the GAT sequence (aspartic acid-G12D-c35 G>A), GTT (valine-G12V-c35 G>T), CGT (arginine G12R-c34 G>C), or GCT (alanine-G12A-c35 G>C). A point mutation can also occur, but less frequently, on codon-13 (G13D) or -61 (Q61L or Q61H).[29-31] KRAS mutation subtypes might induce different tumor biology in the same tumor type.[32] The RAS protein is differentially coupled to downstream signaling pathways depending on the types of mutation. Herein, we dealt with only one subtype of KRAS mutation. Thus, the finding of the present study is not applied to other KRAS mutant subtypes of pancreatic cancer. Furthermore, the KRAS G12R mutant subtype is not common KRAS mutant type in pancreatic cancer.
Generalization of our results is limited because they are based on a single case and a specific subtype of KRAS mutation. Also, the finding in the present study is not validated in other cell lines with KRAS G12R mutation. Nevertheless, this study suggests that the combination of RAF and CDK4/6 inhibitors might be a novel treatment strategy for KRAS G12R mutant pancreatic cancer. The antitumor effect of RAF plus CDK4/6 inhibitors needs to be evaluated and validated in other subtypes of KRAS mutation in pancreatic cancer.
Supplementary Material
Supplementary figure.
Acknowledgements
Funding
Support was provided by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C3418, HI16C1990). Support was also provided by grants (GF01140111) of Samsung Medical Center.
Support was also provided by a grant from Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1A6A3A11934805, 2016R1A6A3A11932444 and 2016R1D1A1A09918622)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vincent A, Herman J, Schulick R. et al. Pancreatic cancer. Lancet. 2011;378:607-620
2. Biankin AV, Waddell N, Kassahn KS. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399-405
3. Jones S, Zhang X, Parsons DW. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-1806
4. Waddell N, Pajic M, Patch AM. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495-501
5. Forrester K, Almoguera C, Han K. et al. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327:298-303
6. Ying H, Kimmelman AC, Lyssiotis CA. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-670
7. Collins MA, Bednar F, Zhang Y. et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639-653
8. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897-909
9. Aguirre AJ, Bardeesy N, Sinha M. et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112-3126
10. van Krieken JH, Jung A, Kirchner T. et al. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2008;453:417-431
11. Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2-27
12. Vasan N, Boyer JL, Herbst RS. A RAS renaissance: emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res. 2014;20:3921-3930
13. Stephen AG, Esposito D, Bagni RK. et al. Dragging ras back in the ring. Cancer Cell. 2014;25:272-281
14. Cox AD, Fesik SW, Kimmelman AC. et al. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828-851
15. Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463-3469
16. Kato J, Matsushime H, Hiebert SW. et al. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331-342
17. Sherr CJ. Cancer cell cycles. Science. 1996;274:1672-1677
18. Wilhelm SM, Carter C, Tang L. et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-7109
19. Wilhelm SM, Adnane L, Newell P. et al. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129-3140
20. Kim ES, Herbst RS, Wistuba II. et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44-53
21. Yamamoto T, Ebisuya M, Ashida F. et al. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006;16:1171-1182
22. Cheng M, Sexl V, Sherr CJ. et al. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci U S A. 1998;95:1091-1096
23. Schutte M, Hruban RH, Geradts J. et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126-3130
24. Asghar U, Witkiewicz AK, Turner NC. et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130-146
25. Witkiewicz AK, Knudsen KE, Dicker AP. et al. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10:2497-2503
26. Liu F, Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012;11:2138-2148
27. Leonard JP, LaCasce AS, Smith MR. et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119:4597-4607
28. Dickson MA, Tap WD, Keohan ML. et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J Clin Oncol. 2013;31:2024-2028
29. Forbes SA, Bindal N, Bamford S. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945-950
30. Delpu Y, Hanoun N, Lulka H. et al. Genetic and epigenetic alterations in pancreatic carcinogenesis. Curr Genomics. 2011;12:15-24
31. di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220-1229
32. Ihle NT, Byers LA, Kim ES. et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228-239
Author contact
![]() Corresponding authors: Seung Tae Kim, M.D., Ph.D., Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel.: +82-2-3410-1779; Fax: +82-2-3410-1754; Email: shty1edu and Joon Oh Park, M.D., Ph.D., Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel: +82-2-3410-1779; Fax: +82-2-3410-1754; E-mail: oncoparkedu
Corresponding authors: Seung Tae Kim, M.D., Ph.D., Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel.: +82-2-3410-1779; Fax: +82-2-3410-1754; Email: shty1edu and Joon Oh Park, M.D., Ph.D., Division of Hematology/Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea. Tel: +82-2-3410-1779; Fax: +82-2-3410-1754; E-mail: oncoparkedu