Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(21):3874-3885. doi:10.7150/jca.25581 This issue Cite
Research Paper
Ethanol Enhances Estrogen Mediated Angiogenesis in Breast Cancer
Rachana Maniyar1, Sanjukta Chakraborty1, Robert Suriano1,2 ![]()
1. Department of Microbiology and Immunology, New York Medical College, Valhalla, New York, United States of America
2. Division of Natural Sciences, College of Mount Saint Vincent, Bronx. New York, United States of America
Received 2018-2-16; Accepted 2018-8-20; Published 2018-10-6
Abstract
Angiogenesis, a highly regulated process, is exploited by tumors like breast cancer to ensure a constant supply of oxygen and nutrients and is key for tumor survival and progression. Estrogen and alcohol independently have been observed to contribute to angiogenesis in breast cancer but their combinatorial effects have never been evaluated. The exact mechanism by which estrogen and alcohol contribute to breast cancer angiogenesis remains to be elucidated. In this study, we defined the in vitro effects of the combination of estrogen and alcohol in breast cancer angiogenesis using the tubulogenesis and scratch wound assays. Conditioned media, generated by culturing the murine mammary cancer cell line, TG1-1, in estrogen and ethanol, enhanced tubule formation and migration as well as modulated the MAP Kinase pathway in the murine endothelial cell line, SVEC4-10. Additionally, estrogen and ethanol in combination enhanced the expression of the pro-angiogenic factors VEGF, MMP-9, and eNOS, and modulated Akt activation. These observations suggest that TG1-1 cells secrete pro-angiogenic molecules in response to the combination of estrogen and ethanol that modulate the morphological and migratory properties of endothelial cells. The data presented in this study, is the first in attempting to link the cooperative activity between estrogen and ethanol in breast cancer progression, underscoring correlations first made by epidemiological observations linking the two.
Keywords: breast cancer, angiogenesis, estrogen, alcohol, ethanol
Introduction
Breast cancer remains one of the most commonly occurring cancers in women. The estimated breast cancer cases and deaths in 2018 stand at 268,670 and 41,400 respectively; making it second only to lung cancer in the number of cancer related deaths in the United States of America1. Factors known to influence the development of breast cancer include genetics, evidenced by the positive association between breast cancer and mutated BRCA1 and BRCA2 genes2-4, and the female sex hormone, estrogen5-7. In addition to its essential physiological roles, estrogen is known to sustain breast cancer development and progression by enhancing cell viability and motility8. The actions of estrogen are primarily initiated by its binding to estrogen receptor alpha (ERα), resulting in the activation of key cellular signaling pathways including the MAPK, NFκB and AP-1 pathways9,10. Other long-known risk factors for breast cancer include early menarche, late menopause, age at first birth, intervals between births as well as number of births, all of which implicate estrogen exposure as a key factor for breast cancer development11-15. This led to the development of anti-estrogens like tamoxifen as a mainstay treatment strategy for breast cancer. Anti-estrogens like tamoxifen, in addition to inhibiting the various cellular effects of estrogen, have been demonstrated to inhibit angiogenesis, promoting a better prognosis in treated patients, and enhanced cell death in animal models16-19.
Interestingly, in addition to the aforementioned risk factors, epidemiological observations positively correlate breast cancer incidence with alcohol consumption. A meta-analysis of 53 epidemiological studies, including 58,515 women with invasive breast cancer and 95,067 healthy controls demonstrated a consistently higher relative risk of breast cancer in the alcohol consuming group. Moreover, the relative risk increased with increasing levels of alcohol intake20. Another meta-analysis spanning 33 years revealed a similar result and estimated that an average of one drink per day leads to a monotonic 10% rise in breast cancer risk21. On the molecular level, experimental studies have suggested that by-products of ethanol metabolism, namely acetaldehyde, play a role in increasing oxidative stress, epigenetic changes, and DNA damage, thus promoting tumor development22,23. A study by Williams et al evaluating risk of invasive breast cancer in women who consume alcohol demonstrated an increased risk in invasive breast cancer in certain populations that drank more than 7 alcoholic beverages per week24 Although epidemiological studies link alcohol and increased breast cancer incidence, a concrete mechanism is yet to be established.
A significant observation was made wherein increased risk and incidence of breast cancer was positively correlated in women with ER positive breast cancer who reported increased alcohol consumption25-27. This observation was further corroborated in a study, that included 38,454 women, and observed a positive correlation between breast cancer risk and moderate alcohol consumption in postmenopausal women on hormone replacement therapy25,28. Together, these studies provide anecdotal evidence suggesting a relationship between alcohol and estrogen that promotes breast cancer; however, the mechanisms regulating this risk have not been fully elucidated.
One biological mechanism that is targeted by both alcohol and estrogen in tumors is angiogenesis; a process indispensable for tumor growth and progression. Ethanol consumption has been demonstrated to promote mammary tumor growth, leading to increased cellular proliferation and enhanced expression of VEGF, an essential protein involved in angiogenesis29,30. Estrogen, over and above increasing viability and proliferation of breast cancer cells via the ER-alpha pathway, is also known to promote endothelial cell activity; consequently neo vascularization and angiogenesis 8,31. We reported earlier that estrogen mobilizes endothelial progenitor cells (EPCs) from the bone marrow to the tumor site thus increasing breast tumor tissue neo-vascularization32.
Based on our current knowledge we speculate that a cooperative mechanism exists between estrogen and alcohol which acts to increase angiogenesis. To date, studies addressing this interaction between estrogen and alcohol on neo-vascularization in breast cancer have not been undertaken. This formed the basis for this study; an effort to explain a mechanism for this potential relationship between alcohol and estrogen in enhancing angiogenesis. We hypothesized that both alcohol and estrogen play complementary roles to enhance angiogenesis in breast cancer, thereby promoting it. To that end, we used in vitro model systems to explore the effects of ethanol, estrogen and a combination of the two on both a murine mammary cancer cell line, TG1-1, and a murine endothelial cell line, SVEC4-10. Treated TG1-1 cells were assayed for the effect of either estrogen, ethanol, or their combination on expression of various pro-angiogenic factors, as well as the effects on the PI3K/Akt cell-signaling pathway. In addition, conditioned media from treated TG1-1 cells was harvested and used to determine the effects of TG1-1 secretory factors on SVEC4-10 cells. To that end, SVEC4-10 cells were cultured in TG1-1 conditioned media after which tubulogenesis, migration, proliferation and activation of relevant signaling pathways was assayed for. This study, the first of its kind, demonstrates an enhanced expression of angiogenic markers when cells are treated with a combination of alcohol and estrogen.
Materials and Methods
Cell Culture and Treatments
The TG1-1 murine mammary cancer cell line33, a kind gift from Dr. Rakesh Jain (Harvard University), was developed from spontaneous tumors in transgenic FVB mice expressing the c-neu oncogene under the control of the mouse mammary tumor virus promoter. SVEC4-10, a murine endothelial cell line, was purchased from ATCC (CRL-2181). Both cell lines were maintained in complete 1X DMEM (Corning #10-013-CV) supplemented with 10% (v/v) Fetal Bovine Serum (FBS) (BioTC #FBS-02), 2mM L-glutamine (Corning #25-005-CI), 1X penicillin/streptomycin (P/S) (Corning #30-002-CI) and 25μg/ml Plasmocin prophylactic (Invivogen #ant-mpp). For ethanol treatments, TG1-1 cells were incubated with 200 proof absolute ethanol (Fisher Scientific #BP2818-100) at a concentration of 0.2% and 0.5%, since physiologically relevant doses are set to be at 0.3%34. For estrogen treatment, TG1-1 cells were incubated with β-estradiol (Sigma #E8875-1G) at a concentration of 10-8 M. Treatments were carried out for 24 hours either in combination or as stand-alone treatments. SVEC4-10 cells were treated for 24 hours with the conditioned media generated from alcohol and/or estrogen treated TG1-1 cells.
Conditioned Media Experiments
Conditioned media was generated by treating TG1-1 cells with 0.2% or 0.5% ethanol and/or 10-8M estrogen in starvation media; phenol red free RPMI (Corning #17-105-CV) containing 5% charcoal stripped FBS (Sigma #F6765) and 1X P/S. The media was collected at the end of 24 hours, centrifuged to remove cell debris, and stored at -80oC until further use. SVEC4-10 cells were plated in complete DMEM. Once 70% confluence was reached, the SVEC4-10 cells were incubated in starvation media overnight. The following day, starvation media was removed and TG1-1 conditioned media added to the SVEC4-10 cells and incubated for 24 hours. At the end of 24 hours, cells were lysed and whole cell lysates analyzed for protein expression by western blots.
Western Blot
Whole cell lysates were made from TG1-1 and SVEC4-10 cells using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.2% sodium deoxycholate, 0.1% SDS, 0.5% NP-40) supplemented with HALT protease/phosphatase inhibitor cocktail (Thermofisher #78440). Samples were placed on ice for 45 minutes with intermittent vortexing. The lysates were then centrifuged for 20 minutes at 14000 rpm at 4oC. Protein concentrations were determined using the Bio-rad Protein Assay Dye Reagent Concentrate (Bio-Rad #5000006). Fifteen micrograms of total protein were loaded per sample and resolved on 10% SDS gels under reducing conditions and transferred onto Immobilon-P membranes (Millipore #IPVH00010) at 220mA for 2 hours at 4oC. Membranes were blocked using 5% dried milk in 1X TBST (200mM Tris-HCl, 150mM NaCl, pH 7.4, and 0.1% Tween-20 added fresh/liter of 1XTBS (TBS-T)) for 2 hours on a shaker at room temperature. They were incubated with primary antibodies (VEGF, eNOS, MAPK, AKT, MEK, FAK) overnight at a 1:1000 dilution (unless indicated otherwise) in 2% bovine serum albumin made in 1X TBST at 4oC. The next day, membranes were washed three times with 1X TBST, followed by incubation with horse radish peroxidase conjugated secondary antibodies, made at a 1:10,000 dilution in 2% dried milk in 1X TBST, for 2 hours, on a shaker, at room temperature. After 4 washes in 1XTBST, the membrane was developed using Pierce ECL western blotting substrate (Thermofisher #32106), and detected on CL-Xposure Film (Thermofisher #34091). Films were scanned and band intensity analyzed using the ImageJ Software.
Tubulogenesis Assay
Twenty four well plates were coated with 100μl/well of growth factor reduced matrigel (Corning #356231) and incubated for 30 minutes at 37oC to enable polymerization. Overnight starved SVEC4-10 cells were re-suspended in treated or untreated TG1-1 conditioned media as well as unconditioned starvation media and complete DMEM as controls, and plated at 50,000 cells in 500μl/well. The plates were incubated overnight. The following day, excess media was carefully removed and tubulogenesis observed under the microscope in bright field at 500X magnification. Four fields of view were counted per well (2 wells/treatment). Using Image J's Angiogenesis Analyzer toolset, the number of master segments and total segment length were determined for each field of view and averaged.
XTT Assay
SVEC4-10 cells were plated at 10,000 cells/well in a 96 well plate and allowed to adhere overnight. The next day, starvation media was added and the cells were starved for 14-18 hours after which the starvation media was replaced by TG1-1 conditioned media as well as unconditioned starvation media and complete DMEM serving as controls. Twenty four hours post treatment, conditioned media was removed and 100µl serum free clear DMEM containing 25µl of the XTT (Thermo Fisher #X6493) reagent (1mg/ml) with 25mM Phenazine Methosulfate (PMS) (Fisher Scientific #AC130160010) was added to each well. The plate was incubated at 37oC for 4 hours and OD was measured at 450nm with a reference wavelength of 630nm.
Scratch Wound (Migration) Assay
SVEC4-10 cells were plated at 500,000 cells/well in a 6 well plate and incubated until a 60% confluent monolayer was observed. Subsequently, the cells were starved for 14-18 hours in starvation media followed by treatment with TG1-1 conditioned media for 24 hours. Prior to start of the treatment, two vertical wounds were created using a 20µl sterile pipette tip. Horizontal lines were made, using a sharpie, on the underside of the plate to allow for visualization at the same spots at 0 hours and 24 hours post treatment. Pictures were taken in the bright field phase contrast at 100X magnification, and migration distance calculated using the scale on the microscope.
Statistical Analysis
Data was normalized to estrogen treatment groups. The ethanol and estrogen combination treatments were compared to the estrogen stand-alone treatment using a Student's T Test where possible; P<0.05 was considered to be statistically significant. Error bars on all figures represent standard error of mean.
Results
SVEC4-10 cells demonstrate enhanced tubulogenesis when cultured in conditioned media obtained from estrogen and alcohol treated TG1-1 cells
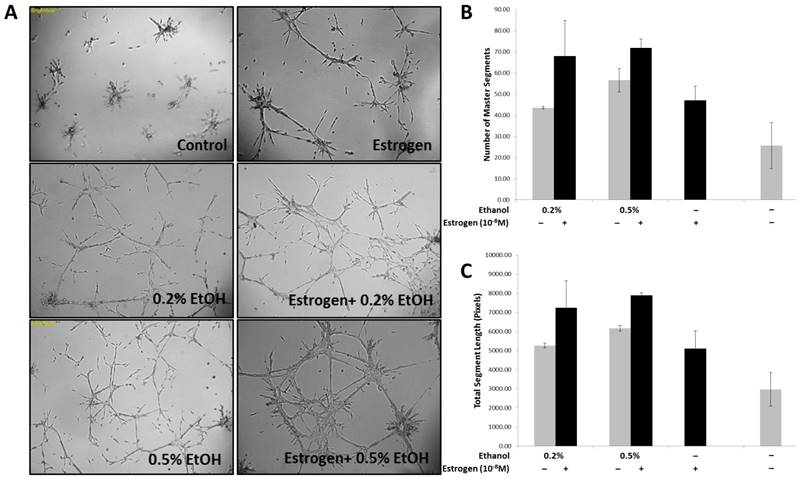
Previous observations by our laboratory demonstrated that estrogen stimulates bone marrow derived endothelial progenitor cell (BM-EPCs) migration from the bone marrow thus resulting in enhanced neo-vascularization in breast cancer32. Based on our observations, and other groups29,35-38 stating that ethanol can influence angiogenesis, we sought to determine if estrogen and ethanol in combination act to enhance angiogenesis in breast cancer, by using an in vitro tubulogenesis assay. In addition to recruitment from the bone marrow, angiogenesis is a complex process determined by the ability of endothelial cells to differentiate and form stable tubules in response to pro-angiogenic factors. In an effort to mimic the tumor microenvironment, conditioned media from TG1-1 cells treated with ethanol, in the presence and absence of estrogen, as well as estrogen alone, was added onto SVEC4-10 cells for 24 hours. Enhanced tubule formation was observed in SVEC4-10 cells when treated with conditioned media generated (Fig 1A) in presence of a combination of ethanol at a concentration of 0.2% or 0.5% with estrogen as compared to estrogen alone. This was further quantified; number of master segments formed (Fig 1B) and the total segment length (Fig 1C) determined. There was an increase in the number of master segments as well as total segment length, commonly used to represent the extent of tubulogenesis39,40. Our results provide evidence that TG1-1 cells, treated with estrogen and alcohol in combination presumably secrete pro-angiogenic factors which result in increased SVEC4-10 endothelial cell differentiation and formation of stable tubules on a basement membrane matrix.
Enhanced Tubulogenesis in SVEC4-10 cells in presence of alcohol and estrogen A) Representative brightfield images (50X) of SVEC4-10 cells cultured in alcohol (Ethanol/EtOH) and/or estrogen (10-8M) treated TG1-1 conditioned media; B) Number of master segments, and C) Total Segment Length in pixels, analyzed using angiogenesis analyzer by Image J. (n=2) Error bars represent ±SEM.
Ethanol enhances estrogen mediated expression of pro-angiogenic markers in TG1-1 breast cancer cells
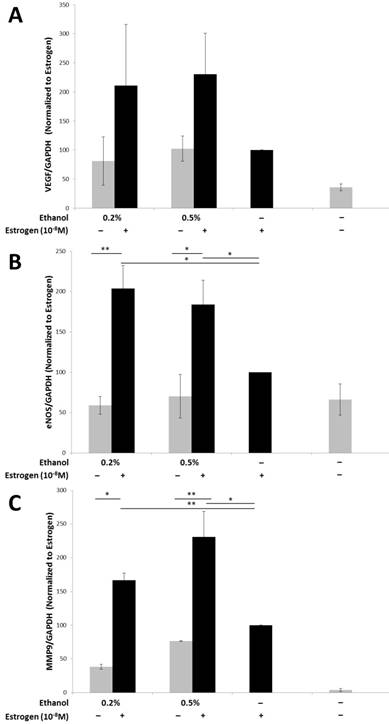
Angiogenesis is a dynamic process that requires the interplay between various pro-angiogenic factors, which are required both in vivo and in vitro, such as that observed during tubule formation41. Vascular endothelial growth factor (VEGF), is considered to be the most important pro-angiogenic factor, playing a critical role in angiogenesis30,42-44. In order to determine the levels of VEGF protein expression that could have contributed to the enhanced tubulogenesis observed, TG1-1 whole cell lysates were used and levels of VEGF determined by a western blot analysis (Fig 2A). We observed an increase in the expression levels of VEGF protein when TG1-1 cells were treated with a combination of ethanol at 0.2% and 0.5% and estrogen, in a dose dependent manner, compared with estrogen treatment alone. Another molecule that plays an important role in angiogenesis is endothelial nitric oxide synthase (eNOS)45-49. eNOS on converting L-arginine to L-citrulline produces nitric oxide (NO) which plays a crucial role in vascular remodeling and maintaining a vasodilatory tone50-52. We assessed the expression of eNOS in treated Tg1-1 cells, which was similar to that observed in VEGF. Enhanced eNOS expression was observed when TG1-1 cells were treated with a combination of ethanol at 0.2% and 0.5% and estrogen compared to estrogen alone (Fig 2B). In addition to VEGF and eNOS, we assessed the expression of MMP-9, which belongs to a family of proteins collectively known as matrix metalloproteinases. Matrix metalloproteinases play a role in the proteolytic degradation of the extracellular matrix leading to tumor invasion, metastasis, as well as tissue remodeling that occurs during angiogenesis as observed in many cancers53-56. Expression of MMP-9 in TG1-1 cells (Fig 2C) was greatly enhanced upon treating the cells with a combination of ethanol, at 0.2%, 0.5% as well as 1.0%, and estrogen, in a dose dependent manner, when compared to estrogen alone. Taken together these results demonstrate that ethanol and estrogen enhance the expression of pro-angiogenic and ECM remodeling factors in tumor cells, which are in part responsible for the stimulated proliferation and differentiation of endothelial cells and thus, angiogenesis.
Enhanced pro-angiogenic factor expression in TG1-1 breast cancer cells in presence of alcohol and estrogen Western blot analysis of whole cell lysates from TG1-1 cells treated with Ethanol and Estrogen. Whole cell lysates were assayed for expression of A) VEGF; B) eNOS and C) MMP-9. Expression was determined by normalizing to TG1-1 protein lysates treated with estrogen alone (n=3) Error bars represent ±SEM a Student T test was performed. *p<0.05, **p<0.01
Ethanol and estrogen induced TG1-1 secretory factors enhance activation of MAP kinase cell survival pathway in SVEC4-10 cells
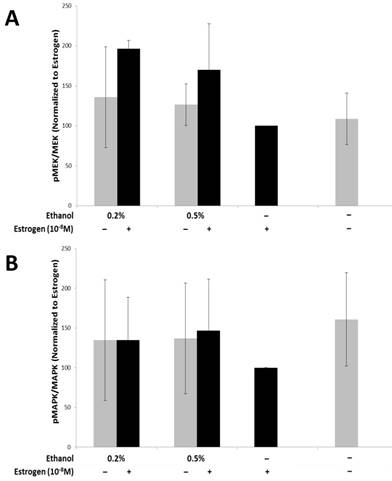
The MAPK pathway has a wide array of downstream effector molecules that affect cellular functions, such as cell proliferation, differentiation, and cell survival, which has been observed to play a role in angiogenesis57-59. In order to evaluate if secretory factors from treated TG1-1 cells would lead to activation of the MAPK pathway in SVEC4-10 cells, we treated the cells with conditioned media harvested from TG1-1 cell cultures followed by western blot analyses. The MAPK pathway targets assayed for were Mek/pMek and MAPK/pMAPK (Fig 3A and B). There was an upregulation of activation of MEK1/2, as denoted by the p-MEK/MEK ratio (Fig 3A), in SVEC4-10 cells cultured in conditioned media generated from TG1-1 cells treated with a combination of 0.2%, 0.5% and 1.0% ethanol and estrogen as compared to estrogen alone. Surprisingly, this increase in MEK1/2 activation did not carry on downstream to its effector, MAPK, as no major differences were noted in expression of MAPK/pMAPK (Fig 3B). One plausible explanation is that there may be a regulating factor playing a role, which may be a result of an inhibitory phosphorylation of MEK1 by cdk560.
Enhanced MEK Activation in SVEC4-10 cells in the presence of alcohol and estrogen Western blot analysis from whole cell lysates of SVEC4-10 cells cultured in conditioned media from ethanol and estrogen treated TG1-1 cells. Whole cell lysates were assayed for expression of A) MEK Activation; and B) MAPK Activation. Expression was determined by normalizing to SVEC4-10 protein lysates treated with TG1-1 conditioned media cultured in the presence of estrogen alone (n=2) Error bars represent ±SEM.
Ethanol and estrogen induced TG1-1 secretory factors enhance expression of the cellular migration mediator FAK in SVEC4-10 Cells
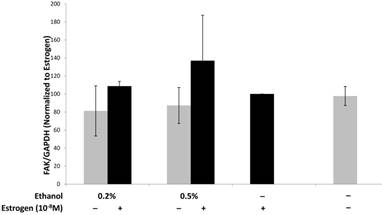
Endothelial focal adhesion kinase (FAK), induced by VEGF, plays a critical role during tumor angiogenesis, as it is involved in cell signaling via the PI3 Kinase/Akt signaling pathway and maintaining endothelial cell adhesion during angiogenesis61-64. The importance of FAK in angiogenesis was also observed in experiments using FAK knockout mice in which angiogenesis was inhibited63,65. We evaluated the expression of FAK in SVEC4-10 cells upon treatment with TG1-1 conditioned media. FAK expression in SVEC4-10 cells was enhanced in a dose dependent manner upon culturing the cells with CM derived from TG1-1 cells treated with a combination of ethanol and estrogen as compared to estrogen alone (Fig 4). These data indicate that enhanced FAK expression could play a role in endothelial cell migration by modulating cytoskeletal proteins, such as actin and microtubules, which are both directly involved in cell motility.66,67
Alcohol and estrogen enhance expression of FAK in SVEC4-10 cells. Western blot analysis from whole cell lysates of SVEC4-10 cells cultured in conditioned media from ethanol and estrogen treated TG1-1 cells. Whole cell lysates were assayed for expression of FAK, a marker for migration. Expression was determined by normalizing to SVEC4-10 protein lysates treated with TG1-1 conditioned media cultured in the presence of estrogen alone (n=2) Error bars represent ±SEM.
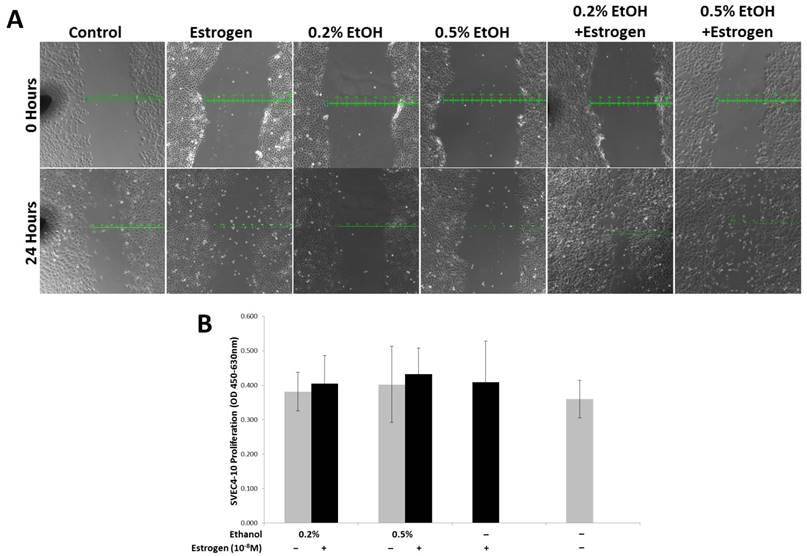
Ethanol and estrogen induced TG1-1 secretory factors enhance migration but not proliferation in SVEC4-10 cells
In order to evaluate the migratory potential of SVEC4-10 cells in presence of ethanol and estrogen treated TG1-1 conditioned media, we performed a scratch wound assay. Treating SVEC4-10 cells with secretory factors of ethanol and estrogen treated TG1-1 cells demonstrated an enhanced migration compared to ethanol or estrogen alone, as observed by the scratch wound results. In presence of conditioned media generated with 0.2% and 0.5% ethanol in combination with estrogen, the wound closed by 300µm and 400μm respectively. This migration was higher as compared to conditioned media generated by treating TG1-1 cells with ethanol alone, where the wound closed by 100μm for both 0.2% and 0.5% and estrogen alone, where the wound closed by 200μm (Fig 5A). Migration and proliferation form the two foundation pillars for successful angiogenesis and frequently occur simultaneously during angiogenesis68-70. To evaluate the effects of TG1-1 secretory factors on proliferation of SVEC4-10 cells, we performed an XTT Proliferation assay on SVEC4-10 cells cultured in the presence of Tg1-1 conditioned media. Surprisingly, no striking differences in cell proliferation were observed when comparing the different treatment conditions (Fig 5B). These data indicate that TG1-1 secretory factors enhance migration of endothelial cells in presence of ethanol and estrogen in combination.
SVEC4-10 cell migration is enhanced in the presence of alcohol and estrogen. A) Representative bright field (100X) image of a scratch wound assay using SVEC4-10 cells in presence of conditioned media from ethanol and estrogen treated TG1-1 cells for 24 hours, demonstrating enhanced migration in presence of ethanol and estrogen. (n=2) B) XTT assay using SVEC4-10 cells cultured in presence of conditioned media from ethanol and estrogen treated TG1-1 cells for 24 hours (n=3) Error bars represent ±SEM.
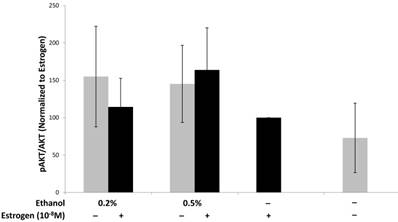
Ethanol and estrogen enhance activation of the pro-survival PI3K/AKT signaling pathway in TG1-1 breast cancer cells
We have provided evidence that treating TG1-1 breast cancer cells with a combination of ethanol and estrogen leads to an increase in the expression of pro-angiogenic factors. Interestingly, the pro-angiogenic factors (VEGF, MMP-9, and eNOS) assayed for in this study require the PI3K/Akt signaling pathway, amongst other signaling pathways, to control their expression71-75. Additionally, AKT phosphorylation leads to the activation of pro-survival pathways leading to enhanced cancer progression8,76,77. We thus wanted to evaluate the effects of ethanol and estrogen on activation of Akt in TG1-1 cells when treated with a combination of ethanol and estrogen. Following treatment of TG1-1 cells, whole cell lysates were prepared followed by western blot analyses in order to determine the expression of AKT and phosphorylated Akt (Fig 6). Treatment of TG1-1 cells with a combination of 0.5% and 1.0% ethanol and estrogen led to enhanced activation of AKT compared to estrogen alone.
Enhanced pro-survival pathway activation in TG1-1 breast cancer cells in presence of ethanol and estrogen Western blot analysis using whole cell lysates from TG1-1 cells treated with Ethanol and Estrogen. Expression of activated Akt was determined by normalizing to protein lysates from TG1-1 cells treated with estrogen alone (n=3) Error bars represent ±SEM.
Discussion
Angiogenesis is a highly regulated process exploited by tumors in order to sustain their growth and development, ultimately providing the necessary vasculature needed for metastasis30,78,79. Various factors, such as estrogen and ethanol have been shown to modulate angiogenesis. Our laboratory has demonstrated that estrogen mobilizes bone marrow derived endothelial progenitor cells (BM-EPCs) thus resulting in increased neo-vascularization of mammary tumor tissue in mice32. Studies have demonstrated that ethanol as well is directly linked to the process of angiogenesis by means of increasing the expression of VEGF, monocyte chemoattractant protein 1 (MCP-1), and hypoxia inducible factor 1 alpha (HIF-1α)29,38. Interestingly, despite the available literature, no studies have been undertaken to address the possible correlation between estrogen and ethanol, in combination, on the process of angiogenesis. This comes as a surprise being that literature exists in which epidemiological observations have been made linking estrogen and alcohol in increasing breast cancer initiation and progression7,20,21,80,81.
In this study we demonstrate, for the first time, that ethanol enhances estrogen-mediated angiogenesis in breast cancer using the estrogen responsive murine mammary cancer cell line, TG1-1, and the murine endothelial cell line, SVEC4-10. Expression of pro-angiogenic molecules is regulated by multiple cell signaling pathways, including the PI3K/Akt pathway, which is known to play a prominent role in angiogenesis72,77,82. We observed an enhanced activation of the PI3K/Akt pathway, as determined by the phosphorylation status of Akt in TG1-1 when treated with estrogen and ethanol. The enhanced expression of Akt/pAkt observed in response to both ethanol and estrogen correlates with the enhanced expression of pro-angiogenic factors observed in figure 2. Activation of the PI3K/Akt pathway in tumor cells is known in literature to modulate expression of various pro-angiogenic factors72,73,77. Treatment of TG1-1 cells in the combination of estrogen and ethanol resulted in an enhanced expression of VEGF, eNOS, and MMP-9 compared to treatment with either estrogen or ethanol alone.
Vascular endothelial growth factor (VEGF) plays a pivotal role in tumor angiogenesis43,44,46,83. VEGF is also known to function in an autocrine signaling loop in hormone responsive cancers84-87 and activate endothelial nitric oxide synthase (eNOS).83 Expression of eNOS is classically restricted to the vascular endothelium but has been shown to be expressed in cancers and is positively correlated with cancer aggressiveness88,89. The enhanced expression of eNOS that we observed in response to both estrogen and ethanol could be caused, in part, by the autocrine function of VEGF. Lastly, we observed a substantial increase in the expression of MMP-9 in TG1-1 cells upon treatment with the combination of estrogen and ethanol, compared to either estrogen or ethanol alone. Tumor derived matrix metalloproteinase 9 (MMP-9) has been known to play a crucial role in tumor progression, tissue remodeling, and angiogenesis90-92. The Williams study demonstrated an increased invasive breast cancer risk in women who drink more than 7 alcoholic drinks per week24. MMP-9 has been shown to lead to proteolytic activation of TGFβ, consequently leading to increased invasion as well as angiogenesis90,93, and release of VEGF from ovarian cancer cells94. Based on these observations, we conclude that ethanol and estrogen, in combination, result in activation of the PI3K/Akt pathway, which results in enhanced expression of pro-angiogenic molecules. We speculate that increased MMP9 as a result of the combination of estrogen and ethanol points to increased invasiveness; however this needs to be evaluated further.
Having assayed for the expression of various pro-angiogenic factors, we focused our attention on characterization of SVEC4-10 endothelial cells in response to conditioned media generated by culturing TG1-1 in estrogen and ethanol. The two parameters that we assayed for were tubulogenesis, a direct measure of angiogenesis in vitro, and SVEC4-10 migration and proliferation. We observed increased tubule formation in SVEC4-10 endothelial cells treated with conditioned media generated from TG1-1 cells cultured in a combination of ethanol and estrogen, when compared to TG1-1 cells cultured in either estrogen or ethanol alone. We evaluated migration using the classical scratch wound assay. As with tubulogenesis, enhanced migration of SVEC4-10 cells was observed when cultured in conditioned media obtained from TG1-1 cultured in both ethanol and estrogen together but, no differences in SVEC4-10 proliferation were observed under the same experimental conditions.
The observations made with respect to enhanced tubulogenesis and migration of SVEC4-10 cells prompted us to explore the MAP-Kinase pathway, as is it known to have numerous downstream effectors that target cellular processes including proliferation, migration and survival.57 Surprisingly, our data indicate that the media generated from TG1-1 cells treated with a combination of ethanol and estrogen, while demonstrating enhanced MEK activation, failed to translate into activation of its downstream effector MAPK. We believe that this is a phenomenon observed due to the role of cdk5 in angiogenesis. Cdk5 is known to mediate endothelial cell migration95. Phosphorylation of MEK1 by Cdk5, has been demonstrated to lead to its inactivation and thus a downregulation of MAPK activation, pushing cells towards a more migratory phenotype60. Keeping this in mind, it was not surprising when we observed an enhanced migratory capacity in the SVEC4-10 cell plated in ethanol and estrogen treated TG1-1 conditioned media, but no proliferation differences. Lastly, to validate enhanced migratory capacity of SVEC4-10 cells, we assayed for the expression of focal adhesion kinase (FAK) in response to TG1-1 conditioned media. Enhanced expression of FAK was also observed in cells cultured in conditioned media generated from both ethanol and estrogen treated TG1-1 cells. This is significant as FAK associates with paxillin and cellular integrins leading to enhanced endothelial cell migration63,96,97.
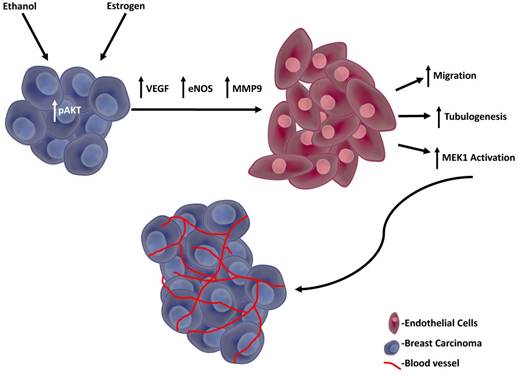
The possible mechanism we propose in outlined in Figure 7, in which the enhanced angiogenic events are initiated by activated AKT in response to both estrogen and ethanol, thus leading to the secretion of pro-angiogenic factors from TG1-1 murine breast cancer cells. In turn, these pro-angiogenic factors act upon SVEC-4-10 murine endothelial cells, resulting in enhanced activation of the MAP kinase pro-survival pathway and FAK expression, consequently enhancing cellular migration and tubulogenesis.
Proposed model TG1-1 cells, in presence of ethanol and estrogen, activate its pro-survival AKT pathway, leading to enhanced secretion of pro-angiogenic factors, such as VEGF, eNOS and MMP9. These proangiogenic factors in turn act on SVEC4-10 endothelial cells resulting in enhanced activation of MEK thus leading to increased endothelial cell migration and tubulogenesis. Overall, the events described consequently lead to enhanced breast cancer angiogenesis and progression.
Thus, the experimental observations made in this study support various epidemiological observations in that increased alcohol consumption in post-menopausal women on estrogen replacement therapy have an increased risk of developing breast cancer. We demonstrate that alcohol acts as a promoter of estrogen driven biological processes observed in breast cancer. Specifically, we propose a mechanism by which estrogen and alcohol, in combination, can lead to enhanced angiogenesis in breast cancer. The mechanism reported in this study may indirectly be responsible for the increased risk of breast cancer associated with these women, supporting the later stages that involve a sustained blood supply via angiogenesis. This study does not limit itself to post-menopausal women; in premenopausal women, increased alcohol intake could also enhance various estrogen mediated processes as well as enhance the risk of developing estrogen responsive cancers.
A potentially different pro angiogenic mechanism may be via the NMDA glutamate receptor. The NMDA receptor, vital for cancer survival and growth is known to be expressed on various cancer cells including the estrogen responsive MCF-7 breast cancer cells98-101. Estrogen is known to upregulate the expression and activity of this receptor in the central nervous tissue102,103. One can speculate that it may have a similar effect on estrogen responsive breast cancer cells. Ethanol has also been demonstrated to upregulate NMDA receptor expression104. This study opens up multiple avenues for further exploration. Alcohol metabolism begins with the conversion of alcohol to acetaldehyde by the enzymatic action of alcohol dehydrogenase105. Alcohol dehydrogenase activity is known to be lower in women than in men, leading to a higher blood alcohol level in women106. This prolonged exposure could potentiate the pro-tumorigenic effect seen in breast cancer patients. Additionally, the effects of acetaldehyde in combination with estrogen remain to be studied. This study does not include the kinetics and effects of alcohol metabolites in combination with estrogen, an important future direction to pursue. Our lab has established an in vivo model linking estrogen to enhanced angiogenesis using the estrogen responsive TG1-1 breast cancer cell line32. Based on our previous study, we are now planning on performing animal studies aimed at addressing the systemic effects of estrogen and ethanol on breast cancer angiogenesis; the concentration of ethanol in serum and tumor tissue and the effect of both on angiogenesis in breast cancer. Our study opens up a new area of exploration into other components of the tumor microenvironment, such as macrophages. Anti-inflammatory tumor associated M2 macrophages play a role in angiogenesis and tumor progression107-109.and, can be studied in presence of both ethanol and estrogen. This will provide novel intervention strategies that target alcohol metabolism pathways in an effort to dampen angiogenesis and improve patient prognosis.
Acknowledgements
We gratefully acknowledge the unstinted philanthropic support from Dr. Raj K. Tiwari.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30 doi:10.3322/caac.21442
2. Wooster R, Bignell G, Lancaster J. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789-792 doi:10.1038/378789a0
3. Tavtigian SV, Simard J, Rommens J. et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet. 1996;12(3):333-337 doi:10.1038/ng0396-333
4. Foulkes WD, Stefansson IM, Chappuis PO. et al. Germline BRCA1 Mutations and a Basal Epithelial Phenotype in Breast Cancer. CancerSpectrum Knowl Environ. 2003;95(19):1482-1485 doi:10.1093/jnci/djg050
5. Cauley JA, Lucas FL, Kuller LH, Stone K, Browner W, Cummings SR. Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Study of Osteoporotic Fractures Research Group. Ann Intern Med. 1999;130(4):270-277
6. Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol. 2006;102(1-5):89-96 doi:10.1016/j.jsbmb.2006.09.004
7. Toniolo PG, Levitz M, Zeleniuch-Jacquotte A. et al. A prospective study of endogenous estrogens and breast cancer in postmenopausal women. J Natl Cancer Inst. 1995;87(3):190-197
8. Ho J-Y, Chang F-W, Huang FS. et al. Estrogen Enhances the Cell Viability and Motility of Breast Cancer Cells through the ERα-ΔNp63-Integrin β4 Signaling Pathway. PLoS One. 2016;11(2):e0148301. doi:10.1371/journal.pone.0148301
9. Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 2006;238(1):1-14 doi:10.1016/j.canlet.2005.06.018
10. Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7(8):497-508
11. Hsieh C-C, Trichopoulos D, Katsouyanni K, Yuasa S. Age at menarche, age at menopause, height and obesity as risk factors for breast cancer: Associations and interactions in an international case-control study. Int J Cancer. 1990;46(5):796-800 doi:10.1002/ijc.2910460508
12. Apter D, Reinilä M, Vihko R. Some endocrine characteristics of early menarche, a risk factor for breast cancer, are preserved into adulthood. Int J Cancer. 1989;44(5):783-787 doi:10.1002/ijc.2910440506
13. Apter D, Vihko R. Early Menarche, a Risk Factor for Breast Cancer, Indicates Early Onset of Ovulatory Cycles. J Clin Endocrinol Metab. 1983;57(1):82-86 doi:10.1210/jcem-57-1-82
14. MacMahon B, Cole P, Lin TM. et al. Age at first birth and breast cancer risk. Bull World Health Organ. 1970;43(2):209-221
15. Albrektsen G, Heuch I, Hansen S, Kvåle G. Breast cancer risk by age at birth, time since birth and time intervals between births: exploring interaction effects. Br J Cancer. 2005;92(1):167-175 doi:10.1038/sj.bjc.6602302
16. Speirs V. Tamoxifen and angiogenesis. Breast Cancer Res. 2001;3(1):68462. doi:10.1186/bcr-2001-68462
17. Haran EF, Maretzek AF, Goldberg I, Horowitz A, Degani H. Tamoxifen Enhances Cell Death in Implanted MCF7 Breast Cancer by Inhibiting Endothelium Growth. Cancer Res. 1994:54 (21)
18. Gagliardi A, Collins DC. Inhibition of Angiogenesis by Antiestrogens. Cancer Res. 1993:53 (3)
19. Lindner DJ, Borden EC. Effects of tamoxifen and interferon-β or the combination on tumor-induced angiogenesis. Int J Cancer. 1997;71(3):456-461 doi:10.1002/(SICI)1097-0215(19970502)71:3<456::AID-IJC25>3.0.CO;2-C
20. Collaborative Group on Hormonal Factors in Breast Cancer. Alcohol, tobacco and breast cancer - collaborative reanalysis of individual data from 53 epidemiological studies, including 58 515 women with breast cancer and 95 067 women without the disease. Br J Cancer. 2002;87(11):1234-1245 doi:10.1038/sj.bjc.6600596
21. Ellison RC, Zhang Y, Mclennan CE, Rothman KJ. Exploring the Relation of Alcohol Consumption to Risk of Breast Cancer. Am J Epidemiol. 154(8).
22. Seitz HK, Pelucchi C, Bagnardi V, Vecchia C La. REVIEW Epidemiology and Pathophysiology of Alcohol and Breast Cancer: Update 2012. doi:10.1093/alcalc/ags011.
23. Esteller M. Epigenetics in Cancer. N Engl J Med. 2008;358(11):1148-1159 doi:10.1056/NEJMra072067
24. Williams LA, Olshan AF, Tse CK, Bell ME, Troester MA. Alcohol intake and invasive breast cancer risk by molecular subtype and race in the Carolina Breast Cancer Study. Cancer Causes Control. 2016;27(2):259-269 doi:10.1007/s10552-015-0703-4
25. Chen WY, Colditz GA, Rosner B. et al. Use of postmenopausal hormones, alcohol, and risk for invasive breast cancer. Ann Intern Med. 2002;137(10):798-804
26. Hong J, Holcomb VB, Dang F, Porampornpilas K, Núñez NP. Alcohol consumption, obesity, estrogen treatment and breast cancer. Anticancer Res. 2010;30(1):1-8
27. Zhang SM, Lee I-M, Manson JE, Cook NR, Willett WC, Buring JE. Alcohol consumption and breast cancer risk in the Women's Health Study. Am J Epidemiol. 2007;165(6):667-676 doi:10.1093/aje/kwk054
28. Strumylaite L, Sharp SJ, Kregzdyte R, Poskiene L, Bogusevicius A, Pranys D. The Association of Low-To-Moderate Alcohol Consumption with Breast Cancer Subtypes Defined by Hormone Receptor Status. PLoS One. 2015;10(12):e0144680. doi:10.1371/journal.pone.0144680
29. Lu Y, Ni F, Xu M. et al. Alcohol promotes mammary tumor growth through activation of VEGF-dependent tumor angiogenesis. Oncol Lett. 2014;8(2):673-678 doi:10.3892/ol.2014.2146
30. Prager GW, Poettler M. Angiogenesis in cancer. Hamostaseologie. 2011;32(2):105-114 doi:10.5482/ha-1163
31. Morales DE, McGowan KA, Grant DS. et al. Estrogen Promotes Angiogenic Activity in Human Umbilical Vein Endothelial Cells In Vitro and in a Murine Model. Circulation. 1995:91 (3)
32. Suriano R, Chaudhuri D, Johnson RS. et al. 17 -Estradiol Mobilizes Bone Marrow-Derived Endothelial Progenitor Cells to Tumors. Cancer Res. 2008;68(15):6038-6042 doi:10.1158/0008-5472.CAN-08-1009
33. Duda DG, Fukumura D, Munn LL. et al. Differential Transplantability of Tumor-Associated Stromal Cells. Cancer Res. 2004;64(17):5920-5924 doi:10.1158/0008-5472.CAN-04-1268
34. Izevbigie EB, Ekunwe SI, Jordan J, Howard CB. Ethanol Modulates the Growth of Human Breast Cancer Cells In Vitro. Exp Biol Med. 2002;227(4):260-265 doi:10.1177/153537020222700406
35. Bora PS, Kaliappan S, Xu Q. et al. Alcohol linked to enhanced angiogenesis in rat model of choroidal neovascularization. FEBS J. 2006;273(7):1403-1414 doi:10.1111/j.1742-4658.2006.05163.x
36. Tan W, Bailey AP, Shparago M. et al. Chronic alcohol consumption stimulates VEGF expression, tumor angiogenesis and progression of melanoma in mice. Cancer Biol Ther. 2007;6(8):1211-1217
37. Wang S, Xu M, Li F. et al. Ethanol promotes mammary tumor growth and angiogenesis: the involvement of chemoattractant factor MCP-1. Breast Cancer Res Treat. 2012;133(3):1037-1048 doi:10.1007/s10549-011-1902-7
38. Castro GD, de Castro CR, Maciel ME. et al. Ethanol-induced oxidative stress and acetaldehyde formation in rat mammary tissue: Potential factors involved in alcohol drinking promotion of breast cancer. Toxicology. 2006;219(1-3):208-219 doi:10.1016/j.tox.2005.11.019
39. DeCicco-Skinner KL, Henry GH, Cataisson C. et al. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J Vis Exp. 2014(91):e51312. doi:10.3791/51312
40. Khoo CP, Micklem K, Watt SM. A comparison of methods for quantifying angiogenesis in the Matrigel assay in vitro. Tissue Eng Part C Methods. 2011;17(9):895-906 doi:10.1089/ten.TEC.2011.0150
41. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628-635 doi:10.1038/nprot.2010.6
42. Wei L-H, Kuo M-L, Chen C-A. et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003;22:1517-1527 doi:10.1038/sj.onc
43. Nö JE, Christensen J, Mooney DJ, Polverini PJ. Vascular Endothelial Growth Factor (VEGF)-Mediated Angiogenesis Is Associated with Enhanced Endothelial Cell Survival and Induction of Bcl-2 Expression. 1999. doi:10.1016/S0002-9440(10)65284-4.
44. Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular Endothelial Growth Factor and Angiogenesis. Pharmacol Rev. 2004;56(4):549-580 doi:10.1124/pr.56.4.3
45. Smith RS, Lin K-F, Agata J, Chao L, Chao J. Human Endothelial Nitric Oxide Synthase Gene Delivery Promotes Angiogenesis in a Rat Model of Hindlimb Ischemia. Arterioscler Thromb Vasc Biol. 2002:22 (8)
46. Xing Y, Lai J, Liu X. et al. Netrin-1 restores cell injury and impaired angiogenesis in vascular endothelial cells upon high glucose by PI3K/AKT-eNOS. J Mol Endocrinol. 2017;58(4):167-177 doi:10.1530/JME-16-0239
47. Lu A, Wang L, Qian L. The role of eNOS in the migration and proliferation of bone-marrow derived endothelial progenitor cells and in vitro angiogenesis. Cell Biol Int. 2015;39(4):484-490 doi:10.1002/cbin.10405
48. Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6(7):521-534 doi:10.1038/nrc1910
49. Fukumura D, Gohongi T, Kadambi A. et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98(5):2604-2609 doi:10.1073/pnas.041359198
50. MacLauchlan S, Yu J, Parrish M. et al. Endothelial nitric oxide synthase controls the expression of the angiogenesis inhibitor thrombospondin 2. doi:10.1073/pnas.1104357108.
51. Bir SC, Xiong Y, Kevil CG, Luo J. Emerging role of PKA/eNOS pathway in therapeutic angiogenesis for ischaemic tissue diseases. Cardiovasc Res. 2012;95(1):7-18 doi:10.1093/cvr/cvs143
52. Lee PC, Salyapongse AN, Bragdon GA. et al. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am J Physiol - Hear Circ Physiol. 1999:277 (4)
53. Hawinkels LJAC, Zuidwijk K, Verspaget HW. et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur J Cancer. 2008;44(13):1904-1913 doi:10.1016/j.ejca.2008.06.031
54. Rao JS, Gondi C, Chetty C, Chittivelu S, Joseph PA, Lakka SS. Inhibition of invasion, angiogenesis, tumor growth, and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol Cancer Ther. 2005:4 (9)
55. Bergers G, Brekken R, McMahon G. et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737-744 doi:10.1038/35036374
56. Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114(5):623-633 doi:10.1172/JCI22087
57. Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim Biophys Acta - Mol Cell Res. 2007;1773(8):1213-1226 doi:10.1016/j.bbamcr.2006.10.005
58. Yang Y-H, Wang Y, Lam KSL. et al. Suppression of the Raf/MEK/ERK Signaling Cascade and Inhibition of Angiogenesis by the Carboxyl Terminus of Angiopoietin-Like Protein 4. doi:10.1161/ATVBAHA.107.157776.
59. Genersch E, Hayess K, Neuenfeld Y, Haller H. Sustained ERK phosphorylation is necessary but not sufficient for MMP-9 regulation in endothelial cells: involvement of Ras-dependent and -independent pathways. J Cell Sci. 2000:113 Pt 23:4319-4330
60. Sharma P, Veeranna, Sharma M. et al. Phosphorylation of MEK1 by cdk5/p35 down-regulates the mitogen-activated protein kinase pathway. J Biol Chem. 2002;277(1):528-534 doi:10.1074/jbc.M109324200
61. Alexopoulou AN, Ho-Yen CM, Papalazarou V, Elia G, Jones JL, Hodivala-Dilke K. Tumour-associated endothelial-FAK correlated with molecular sub-type and prognostic factors in invasive breast cancer. BMC Cancer. 2014;14:237. doi:10.1186/1471-2407-14-237
62. Tavora B, Batista S, Reynolds LE. et al. Endothelial FAK is required for tumour angiogenesis - Supplementary Data. EMBO Mol Med. 2010;2(12):516-528 doi:10.1002/emmm.201000106
63. Chen XL, Nam J-O, Jean C. et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell. 2012;22(1):146-157 doi:10.1016/j.devcel.2011.11.002
64. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598-610 doi:10.1038/nrc3792
65. Tavora B, Batista S, Reynolds LE. et al. Endothelial FAK is required for tumour angiogenesis. EMBO Mol Med. 2010;2(12):516-528 doi:10.1002/emmm.201000106
66. Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J Biol Chem. 2012;287(52):43359-43369 doi:10.1074/jbc.M112.423905
67. Etienne-Manneville S. Actin and Microtubules in Cell Motility: Which One is in Control? Traffic. 2004;5(7):470-477 doi:10.1111/j.1600-0854.2004.00196.x
68. Shikatani EA, Trifonova A, Mandel ER. et al. Inhibition of Proliferation, Migration and Proteolysis Contribute to Corticosterone-Mediated Inhibition of Angiogenesis. Addison CL, ed. PLoS One. 2012;7(10):e46625. doi:10.1371/journal.pone.0046625
69. Qutub AA, Popel AS. Elongation, proliferation & migration differentiate endothelial cell phenotypes and determine capillary sprouting. BMC Syst Biol. 2009;3:13. doi:10.1186/1752-0509-3-13
70. Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvasc Res. 1977;14(1):53-65 doi:10.1016/0026-2862(77)90141-8
71. Michell BJ, Griffiths JE, Mitchelhill KI. et al. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr Biol. 1999;9(15):845-S1 doi:10.1016/S0960-9822(99)80371-6
72. Zeiher AM, Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601-605 doi:10.1038/21224
73. Cheng C-Y, Hsieh H-L, Hsiao L-D, Yang C-M. PI3-K/Akt/JNK/NF-κB is essential for MMP-9 expression and outgrowth in human limbal epithelial cells on intact amniotic membrane. Stem Cell Res. 2012;9(1):9-23 doi:10.1016/j.scr.2012.02.005
74. Dellinger MT, Brekken RA, Downes A, Thorpe P, Detmar M. Phosphorylation of Akt and ERK1/2 Is Required for VEGF-A/VEGFR2-Induced Proliferation and Migration of Lymphatic Endothelium. Kume T, ed. PLoS One. 2011;6(12):e28947. doi:10.1371/journal.pone.0028947
75. Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci. 2011;4:51. doi:10.3389/fnmol.2011.00051
76. Jiang B-H, Liu L-Z. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008;8(1):19-26
77. Kitamura T, Asai N, Enomoto A. et al. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol. 2008;10(3):329-337 doi:10.1038/ncb1695
78. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213-219
79. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674 doi:10.1016/j.cell.2011.02.013
80. Seitz HK, Pelucchi C, Bagnardi V, Vecchia C La. Epidemiology and Pathophysiology of Alcohol and Breast Cancer: Update 2012. Alcohol Alcohol. 2012:47 (3)
81. Purohit V. Moderate alcohol consumption and estrogen levels in postmenopausal women: a review. Alcohol Clin Exp Res. 1998;22(5):994-997
82. Hisamoto K, Ohmichi M, Kurachi H. et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2001;276(5):3459-3467 doi:10.1074/jbc.M005036200
83. Song Z-J, Gong P, Wu Y-E. Relationship between the expression of iNOS,VEGF, tumor angiogenesis and gastric cancer. World J Gastroenterol World J Gastroenterol. 2002;8(4):591-595
84. Weigand M, Hantel P, Kreienberg R, Waltenberger J. Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis. 2005;8(3):197-204 doi:10.1007/s10456-005-9010-0
85. Bachelder RE, Wendt MA, Mercurio AM. Vascular Endothelial Growth Factor Promotes Breast Carcinoma Invasion in an Autocrine Manner by Regulating the Chemokine Receptor CXCR4. Cancer Res. 2002:62 (24)
86. Steiner H, Berger AP, Godoy-Tundidor S. et al. An autocrine loop for vascular endothelial growth factor is established in prostate cancer cells generated after prolonged treatment with interleukin 6. Eur J Cancer. 2004;40(7):1066-1072 doi:10.1016/j.ejca.2003.11.033
87. Matsuura M, Onimaru M, Yonemitsu Y. et al. Autocrine loop between vascular endothelial growth factor (VEGF)-C and VEGF receptor-3 positively regulates tumor-associated lymphangiogenesis in oral squamoid cancer cells. Am J Pathol. 2009;175(4):1709-1721 doi:10.2353/ajpath.2009.081139
88. Charles N, Ozawa T, Squatrito M. et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6(2):141-152 doi:10.1016/j.stem.2010.01.001
89. Orucevic A, Bechberger J, Green AM, Shapiro RA, Billiar TR, Lala PK. Nitric-oxide production by murine mammary adenocarcinoma cells promotes tumor-cell invasiveness. Int J cancer. 1999;81(6):889-896
90. Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget. 2014;5(9):2736-2749 doi:10.18632/oncotarget.1932
91. Bergers G, Brekken R, McMahon G. et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737-744 doi:10.1038/35036374
92. Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9(2):267-285 doi:10.1111/J.1582-4934.2005.TB00355.X
93. Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163-176
94. Belotti D, Paganoni P, Manenti L. et al. Matrix Metalloproteinases (MMP9 and MMP2) Induce the Release of Vascular Endothelial Growth Factor (VEGF) by Ovarian Carcinoma Cells. Cancer Res. 2003:63 (17)
95. Liebl J, Weitensteiner SB, Vereb G. et al. Cyclin-dependent kinase 5 regulates endothelial cell migration and angiogenesis. J Biol Chem. 2010;285(46):35932-35943 doi:10.1074/jbc.M110.126177
96. Abu-Ghazaleh R, Kabir J, Jia H, Lobo M, Zachary I. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem J. 2001;360(Pt 1):255-264
97. Le Boeuf F, Houle F, Sussman M, Huot J. Phosphorylation of focal adhesion kinase (FAK) on Ser732 is induced by rho-dependent kinase and is essential for proline-rich tyrosine kinase-2-mediated phosphorylation of FAK on Tyr407 in response to vascular endothelial growth factor. Mol Biol Cell. 2006;17(8):3508-3520 doi:10.1091/mbc.E05-12-1158
98. North WG, Gao G, Memoli VA, Pang RH, Lynch L. Breast cancer expresses functional NMDA receptors. Breast Cancer Res Treat. 2010;122(2):307-314 doi:10.1007/s10549-009-0556-1
99. Deutsch SI, Tang AH, Burket JA, Benson AD. NMDA receptors on the surface of cancer cells: Target for chemotherapy? Biomed Pharmacother. 2014;68(4):493-496 doi:10.1016/j.biopha.2014.03.012
100. Cao Y, Lin S-H, Wang Y, Chin YE, Kang L, Mi J. Glutamic Pyruvate Transaminase GPT2 Promotes Tumorigenesis of Breast Cancer Cells by Activating Sonic Hedgehog Signaling. Theranostics. 2017;7(12):3021-3033 doi:10.7150/thno.18992
101. Huang W, Choi W, Chen Y. et al. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013;23(5):724-727 doi:10.1038/cr.2013.15
102. Tang B, Ji Y, Traub RJ. Estrogen alters spinal NMDA receptor activity via a PKA signaling pathway in a visceral pain model in the rat. Pain. 2008;137(3):540-549 doi:10.1016/j.pain.2007.10.017
103. El-Bakri NK, Islam A, Zhu S. et al. Effects of estrogen and progesterone treatment on rat hippocampal NMDA receptors: relationship to Morris water maze performance. J Cell Mol Med. 8(4):537-544.
104. Chandrasekar R. Alcohol and NMDA receptor: current research and future direction. Front Mol Neurosci. 2013;6:14. doi:10.3389/fnmol.2013.00014
105. Bagnardi V, Blangiardo M, La Vecchia C, Corrao G. Alcohol consumption and the risk of cancer: a meta-analysis. Alcohol Res Health. 2001;25(4):263-270
106. Chrostek L, Jelski W, Szmitkowski M, Puchalski Z. Gender-related differences in hepatic activity of alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase in humans. J Clin Lab Anal. 2003;17(3):93-96 doi:10.1002/jcla.10076
107. Kuribayashi K, Motoyama S, Sasaki T. et al. C-reactive protein reduces the relative number of tumor-associated M2 macrophages and intratumoral angiogenesis in mice. Tohoku J Exp Med. 2014;233(4):249-255
108. Chen P-C, Cheng H-C, Wang J. et al. Prostate cancer-derived CCN3 induces M2 macrophage infiltration and contributes to angiogenesis in prostate cancer microenvironment. Oncotarget. 2014;5(6):1595-1608 doi:10.18632/oncotarget.1570
109. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MPJ, Donners MMPC. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis. 2014;17(1):109-118 doi:10.1007/s10456-013-9381-6
Author contact
![]() Corresponding author: Robert Suriano, Email: robert.surianoedu
Corresponding author: Robert Suriano, Email: robert.surianoedu