Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(7):1734-1744. doi:10.7150/jca.26380 This issue Cite
Research Paper
Metformin Induces Apoptosis and Inhibits Proliferation through the AMP-Activated Protein Kinase and Insulin-like Growth Factor 1 Receptor Pathways in the Bile Duct Cancer Cells
Jin Lee1 ![]() , Eun Mi Hong1, Jung Han Kim2, Jang Han Jung1, Se Woo Park1, Dong Hee Koh1, Min Ho Choi1, Hyun Joo Jang1, Sea Hyub Kae1
, Eun Mi Hong1, Jung Han Kim2, Jang Han Jung1, Se Woo Park1, Dong Hee Koh1, Min Ho Choi1, Hyun Joo Jang1, Sea Hyub Kae1
1. Division of Gastroenterology, Department of Internal Medicine, Hallym University College of Medicine, Gyeonggi-Do, Korea
2. Division of Gastroenterology, Department of Internal Medicine, Hallym University College of Medicine, Seoul, Korea
Received 2018-3-30; Accepted 2019-1-19; Published 2019-4-5
Abstract
Background/Aims: Metformin has been found to have antineoplastic activity in some cancer cells. This study was performed to determine whether metformin inhibits the proliferation of bile duct cancer cells by inducing apoptosis and its effects on the expression of gene-related proteins involved in cancer growth.
Methods: Human extrahepatic bile duct cancer cells (SNU-245 and SNU-1196) were cultured. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays were performed to determine the effect of metformin on the cell proliferation. Apoptosis was measured by a cell death detection enzyme-linked immunosorbent assay and a caspase-3 activity assay. Expression levels of various proteins, with or without specific small interfering ribonucleic acid-induced gene disruption, were measured by Western blot analysis. The migratory activity of the cancer cells was evaluated by wound healing assay.
Results: Metformin suppressed cell proliferation in bile duct cancer cells by inducing apoptosis. Metformin inhibited mammalian target of rapamycin (mTOR) by activation of tuberous sclerosis complex 2 (TSC-2) through phosphorylation of adenosine monophosphate-activated protein kinase at threonine-172 (AMPKThr172). Hyperglycemia impaired metformin-induced AMPKThr172 activation and enhanced phosphorylation of AMPK at serine-485 (AMPKSer485). Metformin blocked the inhibitory effect of insulin-like growth factor 1 receptor (IGF-1R)/insulin receptor substrate 1 (IRS-1) pathway on TSC-2, and hyperglycemia impaired metformin-induced inhibition of IGF-1R/IRS-1 pathway and modulated the invasiveness of bile duct cancer cells; however, this effect was impaired by hyperglycemia.
Conclusions: Metformin has antineoplastic effects in bile duct cancer, and hyperglycemic environment interrupts the effect of metformin. In addition, AMPK and IGF-1R play a key role in the proliferation of bile duct cancer cells.
Keywords: bile duct cancer, metformin, apoptosis, AMP-activated protein kinase, insulin-like growth factor 1 receptor
Introduction
Cholangiocarcinoma is the most common malignant tumor of the extrahepatic biliary system, and the second most common intrahepatic malignant tumor following hepatocellular carcinoma, The 5-year survival rate is less than 20% because the late development of symptoms results in delayed diagnosis. Non-surgical treatments such as chemotherapy and radiotherapy can be considered when operation is difficult. However, these treatments are largely ineffective [1, 2].
Although various bile duct diseases (primary sclerosing cholangitis, choledochal cysts, cholelithiasis, clonorchiasis infection) and chronic liver diseases (chronic hepatitis B and C, liver cirrhosis) are documented as the risk factors for intrahepatic or extrahepatic cholangiocarcinoma, it is unclear whether diabetes and obesity are associated with cholangiocarcinoma [3-8]. However, recent some studies have provided the strong evidence that metabolic syndrome is associated with development of cholangiocarcinoma. An epidemiologic study based on the large-scale data revealed that the incidence of intrahepatic cholangiocarcinoma (ICC) was increased in elderly people (over 65 years of age) with metabolic syndrome [9]. The use of metformin in patients with type 2 diabetes mellitus has been shown to reduce the incidence of various cancers, including liver cancer [10-15]. Statin, another metabolic syndrome treatment agent, has been known to reduce the risk of liver cancer [16, 17], and previous our study also found that statin inhibits the growth in bile duct cancer cells and induces apoptosis [18]. Another recent epidemiological study demonstrated that metformin therapy can reduce the risk of liver metastasis of ICC in diabetic patients by 60%, which suggest the possibility that metformin could be used in the treatment of biliary cancer [19].
Molecular biologic mechanisms of the antitumor activity of metformin, which have been suggested in pancreatic cancer, involve phosphorylation of adenosine monophosphate-activated protein kinase at threonine-172 (AMPKThr172) through liver kinase B1 (LKB1) signal pathway, which induces activation of tuberous sclerosis complex 2 (TSC-2) and subsequently inhibits mammalian target of rapamycin (mTOR) activity [15, 20]. In addition, metformin is known to inhibit insulin-like growth factor 1 receptor (IGF-1R) activation, thereby lowering the activity of insulin receptor substrate 1 (IRS-1), and blocking signal transmission through the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/ protein kinase B (Akt) pathway. In hyperglycemic conditions, the activity of IGF-1R pathway is activated, inducing phosphorylation of Akt and AMP-activated protein kinase at serine-485 (AMPKSer485), inhibiting the activation of AMPKThr172, and blocking the growth inhibition effect of metformin [20].
In bile duct cancer, there is no research on how metformin actually inhibits cancer cell growth, induces apoptosis, or affects expression of gene-related proteins involved in cancer growth. Metformin has been widely used as a treatment for type 2 diabetes and its safety has been demonstrated, with the side effects of hypoglycemia being very mild. We performed this study to determine whether metformin inhibits the proliferation of bile duct cancer cells by inducing apoptosis, and its effects on the expression of gene-related proteins involved in cancer growth, and to identify how metformin affect molecular mechanisms involved in the inhibition of cancer cell growth.
Materials and Methods
Materials
Roswell Park Memorial Institute (RPMI) 1640 medium (2.0 g/L, 10 mM glucose), Low fasting glucose (FG) (1.0 g/L, 5 mM) or high glucose (HG) (4.5 g/L, 25 mM) Dulbeccos's modified eagle medium (DMEM), fetal bovine serum (FBS), trypsin/ethylenediaminetetraacetic acid (EDTA), and penicillin/ streptomycin were obtained from Gibco (Grand Island, NY, USA). Metformin was obtained from Cayman (Ann Arbor, MI, USA), and used following dissolution in phosphate buffered solution (PBS). Dimethyl sulfoxide (DMSO) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma Chemicals (St. Louis, MO, USA). Bax, Bcl-2, TSC-2, mTOR, phospho-AMPKThr172, phospho-AMPKSer485, AMPK, phospho-Akt, Akt, and β-actin antibodies were obtained from Cell Signaling (Danvers, MA, USA). The Pro-IGF-1R and IRS-1 antibodies and goat anti-rabbit IgG-horseradish peroxidase (HRP) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The Luminate Forte Western HRP western Blotting Detection kit was obtained from Millipore (Danvers, MA, USA).
Cell culture
Bile duct cancer cells (SNU-245, harvested from well-differentiated common bile duct cancer without major gene mutations; SNU-1196, harvested from hilar cancer, moderate-differentiated, with gene mutations in p15, p16, and p53) were purchased from the Korean Cell Line Bank (KCLB). KCLB authenticated the absence of mycoplasma or bacteria contamination and the cell line short tandem repeat (STR) profile [21]. Cells were cultured in RPMI 1640 media supplemented with 10% FBS, 2 mM glutamine, 100 μg/mL streptomycin, and 100 IU/mL penicillin and for regular culture. FG (5 mM) or HG (25 mM) DMEM was switched for the indicated experiments. FG corresponds to normal fasting blood sugar level (100 mg/dL), and HG corresponds to 450 mg/dL which is approximately 2 or 3 times higher than normal human post-prandial blood glucose level. The cell viability test examined the effect of metformin on cell proliferation under HG or FG condition in SNU-245 and SNU-1196 cells. The medium was changed twice per week, and the cells were maintained in 37°C incubator with 5% CO2. The cells were subcultured when confluent (every 5-7 days) using trypsin (2.5 g/L) and EDTA (1 g/L).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays
Cell proliferation was measured using the MTT assay. Cells were seeded at a density of 5 × 104 cells/ mL in a 96-well plate. After incubation for 24 h, cells were treated with various concentrations of metformin in serum-free medium for 24 or 48 h. MTT (0.5 mg/mL) was added to each well and incubated for an additional 4 h at 37°C. After the medium was removed, 100 μL DMSO was added to each well. The plate was gently shaken for 10 min, and the color intensity (proportional to the number of live cells) was evaluated with a DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA) at 570 nm wavelength.
Cell apoptosis assay
Cell apoptosis was assessed by the detection of mono-oligonucleosomes (histone-associated deoxyribonucleic acid (DNA) fragments) using the Cell Death Detection enzyme-linked immunosorbent assay (ELISA) Plus kit (Roche Applied Science, Mannheim, Germany), according to the manufacturer's instructions. Cells were seeded in a 96-well plate at a density of 1 × 104 cells/well and incubated for 24 h. Cells were treated with various concentrations of metformin for 24 or 48 h. After the medium was removed, cells were treated with 100 μL lysis buffer, which was included in the kit, for 30 min and centrifuged at 200 × g at 4°C for 10 min. The supernatant (cell lysate solutions) was placed in the wells of a streptavidin-coated plate supplied by the manufacturer. A mixture of anti- histone-biotin and anti-DNA-peroxidase antibodies was added to the cell lysate and incubated for 2 h. After washing, 100 μL 2,2′-azinobis-3-ethyl-benzothiazoline-6-sulfonic acid (ABST) substrate was added to each well for 20 min. Absorbance at 405 nm was measured with a DTX 880 Multimode Detector.
Caspase-3 activity assay
A Caspase-3 Activity Assay kit (BioVision, Mountain View, CA, USA) was used to measure caspase-3 activity according to the manufacturer's instructions. Cells were plated on 60 mm dishes at a density of 2 × 106 cells/mL and treated with various concentrations of metformin for 24 or 48 h. Cells were washed with PBS and harvested with lysis buffer, which was included in the kit. Cells were incubated on ice for 10 min. Cell lysates were then centrifuged at 4°C and 12,000 × g, and the supernatant was then transferred to a new tube and stored on ice. Protein content was analyzed using the Bradford assay (Sigma, St Louis, MO, USA). Assays were performed in 96-well plates containing 90 μg of protein in 50 μL lysis buffer, followed by the addition of 5 µL 4 mM N-Acetyl-Asp-Glu-Val-Asp p-nitroanilide (DEVD-pNA). The samples were incubated at 37°C for 2 h. Absorbance was measured at 405 nm using a DTX 880 Multimode Detector.
Western blotting
Cells were washed with PBS and harvested with lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1 μM phenylmethylsulfonyl fluoride [PMSF], 5 g/mL aprotinin, 5 g/mL leupeptin). Protein content was analyzed by Bradford assay. SDS-polyacrylamide gel electrophoresis (PAGE) was performed with a 4% stacking gel and a 10% resolving gel, followed by transfer to PVDF membranes (GE Healthcare, Herrcules, CA, USA). The membranes were blocked for 1 h at room temperature in blocking solution (5% skim milk in Tris-buffer with Tween-20 [TBS-T]: 200 mM Tris, 500 mM NaCl, pH 7.5, 0.05% v/v Tween-20) and incubated overnight at 4°C in 5% bovine serum albumin (BSA) in TBS-T with antibodies targeting Bax, Bcl-2, TSC-2, mTOR, phospho-AMPKThr172, phospho-AMPKSer485, AMPK, phospho-Akt, AKt or β-actin, and 5% skim milk in TBS-T with antibodies targeting Pro-IGF-1R and IRS-1. The membranes were washed with TBS-T and incubated with goat anti-rabbit IgG-HRP for 1 h at room temperature. The membranes were then washed again, incubated with Luminate Forte Western HRP western Blotting Detection kit (Millipore) for 5 min, and autoradiography was performed. The signal intensities for specific bands were quantified using Image J density analysis software (Version 1.51k, National Institutes of Health, Bethesda, MD, USA).
SiRNA transfection assay
SNU-245 and SNU-1196 cells were plated in 6-well plates at a density of 1 × 105 cells/well and allowed to attach overnight in a 37°C 5% CO2 incubator. Cells were grown to 60-80% confluency then treated with 60 pmols of either IGR-1R (sc-29358, Santa Cruz Biotechnology), AMPKα1 (sc-29673, Santa Cruz Biotechnology), AMPKα1/2 (sc-45312, Santa Cruz Biotechnology) or scrambled (sc-Santa cruz Biotechnology) siRNA in the supplied transfection media and incubated for 4 hours at 37°C in a 5% CO2 incubator. Afterwards, cells were incubated in a serum free media (5 mM or 25 mM glucose) containing 1% FBS and then treated with either metformin (5 or 10mM) for 24 hour. Cellular fractions were stored at -20°C until assayed.
Wound healing assay
SNU-245 cell were seeded into 6-well plate (Costar, Corning, Inc., Acton, MA) and grown until 80%~90% confluency. After 24hr of serum deprivation, a wound was produced at the center of the monolayer by gently scraping cells with a sterile plastic 200 μL pipette tip. The incubation medium was then removed and cells were washed with PBS, and then incubated with metformin. The wounded cell cultures were then incubated at 37℃ in a humidified and equilibrated (5% v/v CO2). Cells that migrated into the wounded area or protruded from the wound were visualized and photographed with an inverted microscope at the indicated times.
Statistical analyses
The results of each experiment are expressed as the mean ± standard deviation (SD) of duplicate cultures, and all results are representative of at least three separate experiments. One-way analysis of variance (ANOVA) for three or more unpaired groups or Student's t-test for two unpaired groups was used, and a P- value less than 0.05 was considered statistically significant.
Results
Metformin suppresses proliferation of bile duct cancer cells by inducing apoptosis
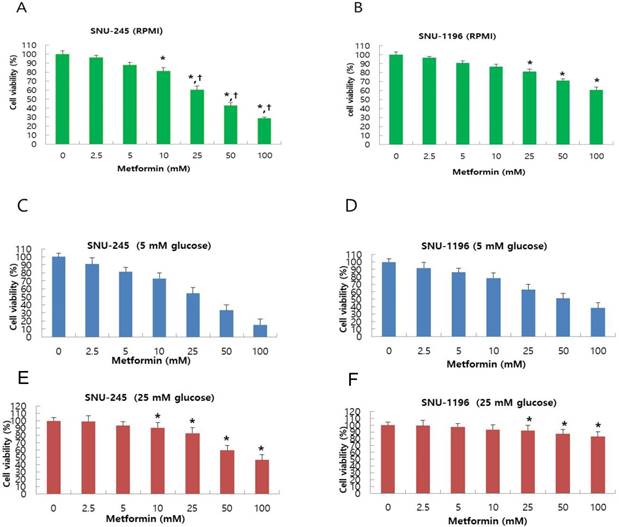
Metformin-induced inhibition of proliferation of bile duct cancer cell was measured using MTT assay after 24 h incubation. Metformin treatment dose-dependently suppressed bile duct cancer cell proliferation in both SNU-245 and SNU-1196 cell lines (Figure 1A-B). Bile duct cancer cells that did not harbor major mutations (SNU-245) were found to be more effectively suppressed by metformin treatment compared with cells containing several mutations in cancer-associated genes (SNU-1196) as well (Figure 1A-B). Both SNU-245 and SNU-1196 cells treated with metformin in HG condition showed higher cell viability compared to the cells in normal FG condition (Figure 1C-F).
Metformin suppresses proliferation in a dose-dependent manner, and HG condition reduces metformin-induecd growth suppression in SNU-245 and SNU-1196 cells. Cells were treated with indicated concentrations of metformin in RPMI media (10 mM glucose) (A, B) or in DMEM (5 mM or 25 mM glucose) (C, D, E, F) with 10% FBS for 24 h. The cell survival was evaluated by MTT assay. The data are expressed as the means ± SD. A, B) *P < 0.001, compared to untreated control cells and the cells treated with the lower concentrations of metformin; †P < 0.001, compared to SNU-1196 cells treated with the same concentrations of metformin. C, D, E, F) *P < 0.001, compared to the cells treated with the same concentrations of metformin in FG condition (5 mM).
The effect of metformin on apoptosis was evaluated by Cell Death Detection ELISA and caspase-3 activity assays. Metformin significantly and dose-dependently induced apoptosis of bile duct cancer cells at doses greater than 10 mM (Figure 2A-B). Caspase-3 activity, which plays a central role in the execution-phase of cell apoptosis, was also significantly and dose-dependently increased by metformin (Figure 2C-D). Additionally, the metformin-induced apoptotic effect was significantly prominent in SNU-245 cells compared with SNU-1196 cells (Figure 2A-D).
Metformin induces apoptosis in a dose-dependent manner, and up-regulates Bax expression and down-regulates Bcl-2 expression in SNU-245 and SNU-1196 cells. Cells were treated with indicated concentrations of metformin in RPMI media for 48 h. The data are expressed as the means ± SD. (A, B) Apoptosis (%) means relative percentage compared to untreated control (100%). (C, D) Caspase-3 activity (%) means relative activity compared to untreated control (100%). *P < 0.001, compared to untreated control cells and the cells treated with the lower concentrations of metformin; †P < 0.001, compared to SNU-1196 cells treated with the same concentrations of metformin. (E) Expression levels were analyzed by Western blotting. All results are representative of at least three separate experiments. *P < 0.01, compared to untreated control cells; **P < 0.001, compared to untreated control cells and the cells treated with the lower concentrations of metformin.
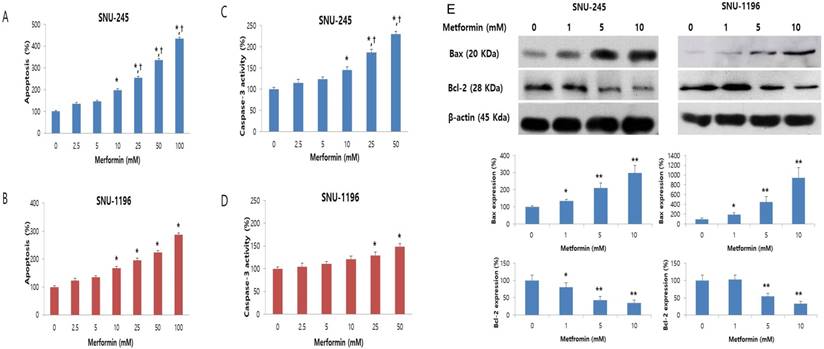
We also analyzed expression of the Bcl-2-like protein 4 (Bax, proapoptotic protein) and the B-cell lymphoma 2 (Bcl-2, antiapoptotic protein) by Western blot. Metformin significantly enhanced the expression of Bax and down-regulated the expression of Bcl-2 (Figure 2E), as compared to control cells, in both cell lines. These results indicate that metformin induces apoptosis and modulates proteins directly involved in bile duct cancer cell apoptosis.
Metformin inhibits mTOR by activation of AMPKThr172/TSC-2 pathway, and hyperglycemia impairs metformin-induced AMPKThr172 activation and enhances phosphorylation of AMPKSer485 in bile duct cancer cells
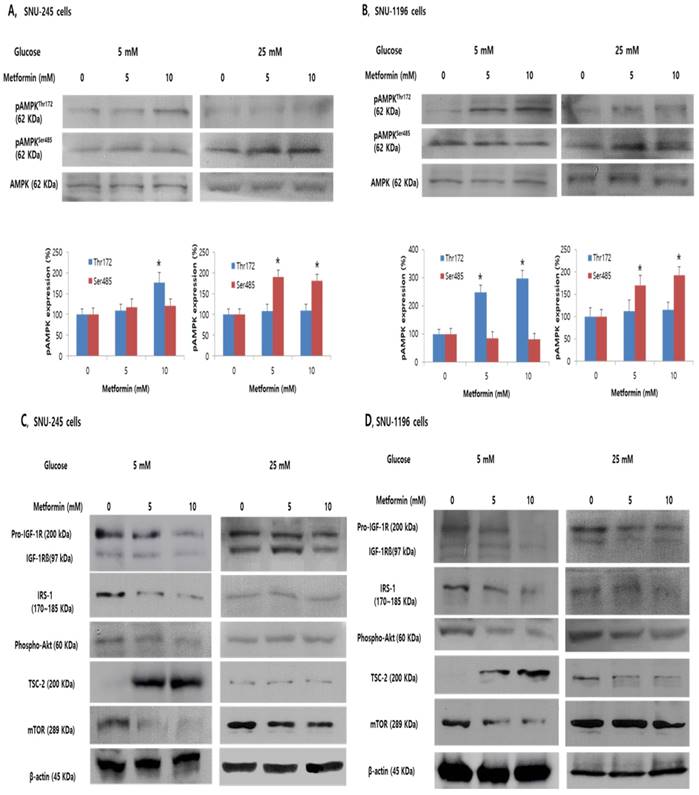
To evaluate whether metformin affects phosphorylation of AMPKThr172 and AMPKSer485 which are known to be directly related to TSC-2 activation or suppression, and to determine whether the reduced efficacy of metformin in hyperglycemic condition is related to altered AMPK activation, metformin-stimulated AMPK phosphorylation was evaluated in SNU-245 and SNU-1196 cells in either FG or HG conditions.
In FG condition, metformin induced AMPKThr172 phosphorylation in a dose-dependent manner and did not significantly affect AMPKSer485 phosphorylation (Figure 3A-B). Additionally, metformin induced TSC-2 activation and inhibited mTOR activity in both bile duct cancer cell lines in this condition (Figure 3C-D). However, exposure to hyperglycemic conditions inhibited the metformin-stimulated increase of phosphorylated AMPKThr172 and TSC-2, whereas AMPKSer485 phosphorylation was significantly increased, impairing metformin-induced mTOR inhibition in both SNU-245 and SNU-1196 cell lines (Figure 3A -D).
Metformin induces phosphorylation of AMPKThr172 and suppresses the inhibitory effects of IGF-1R/IRS-1/Akt on TSC-2. However, hyperglycemia impairs these effects in SNU-245 and SNU-1196 cells. Cells were treated with metformin in DMEM (5 mM or 25 mM glucose) with 1% FBS for 24 h. Expression levels were analyzed by Western blotting. All results are representative of at least three separate experiments. *P < 0.01, compared to untreated control cells; pAMPK, phosphoAMPK.
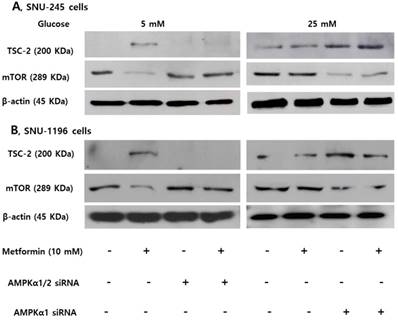
Expression of TSC-2 and mTOR were evaluated following small interfering ribonucleic acid (siRNA)- transfection against AMPKα (involved in phosphorylation at threonine-172) or AMPKα1 (involved in phosphorylation at serine-485) to clarify the role of AMPKThr172 and AMPKSer485 depending on the glucose level on the activities of TSC-2 and mTOR. The AMPKα silencer inhibited the metformin-induced TSC-2 activation and mTOR inactivation in FG condition, and the AMPKα1 silencer maintained metformin-induced TSC-2 activation and mTOR inactivation in HG condition in both SNU-245 and SNU-1196 cell lines (Figure 4).
AMPKThr172 and AMPKSer485 have a crucial role in modulating TSC-2 and mTOR expression in SNU-245 and SNU-1196 cells. Cells were treated with AMPKα1/2, AMPKα1 or scrambled siRNA in the supplied transfection media and incubated for 4 h. Afterwards, Cells were treated with metformin in either FG or HG media with 1% FBS for 24 h. Expression levels were analyzed by Western blotting. All results are representative of at least three separate experiments. The AMPKα siRNA inhibited the metformin-induced TSC-2 activation and mTOR inactivation in FG condition, and the AMPKα1 siRNA maintained metformin-induced TSC-2 activation and mTOR inactivation in HG condition in both SNU-245 and SNU-1196 cell lines.
Metformin blocks inhibitory effects of IGF-1R/IRS-1/Akt pathway on TSC2, and hyperglycemia impairs metformin-induced inhibition of the pathway in bile duct cancer cells
We evaluated the effects of metformin on IGF-1R, IRS-1, and Akt which is another pathway regulating TSC-2 and mTOR associated with tumor cell growth in either FG or HG condition. Metformin potently inhibited the activation of the IGF-1R (the IGF-1R is composed of two alpha subunits and two beta subunits), subsequently suppressing IRS-1 and Akt signalling, consequently, activating TSC-2 and suppressing mTOR in both SNU-245 and SNU-1196 cell lines at FG levels. However, in HG condition, metformin-induced inhibitory effects on IGF-1R and IRS-1 were abrogated. As a result, no inhibition of mTOR activity by metformin was observed (Figure 3C-D).
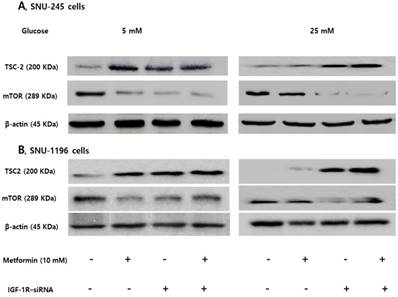
Expression of TSC-2 and mTOR were evaluated following siRNA transfection of IGF-1R to clarify the role of the IGF-1R/IRS-1 pathway on modulation of TSC-2 and mTOR. IGF-1R silencing induced TSC-2 activation and mTOR inactivation without metformin treatment in FG levels. However, IGF-1R silencing maintained metformin-induced TSC-2 activation and mTOR inactivation in HG conditions, which suggests that IGF-1R is another crucial pathway in modulating bile duct cancer cell proliferations in both low and hyperglycemic conditions (Figure 5).
IGF-1R silencing maintains metformin-induced TSC2 activation and mTOR inactivation in hyperglycemic conditions in SNU-245 and SNU-1196 cell. Cells were treated with either siRNA against IGR-1R or scrambled siRNA and incubated for 4 h. Afterwards, cells were treated with metformin in either 5 mM or 25 mM glucose-containing DMEM with 1% FBS for 24 h. Expression levels were analyzed by Western blotting. All results are representative of at least three separate experiments. IGF-1R silencing induced TSC-2 activation and mTOR inactivation without metformin treatment in FG conditions. However, IGF-1R silencing maintained metformin-induced TSC-2 activation and mTOR inactivation in HG conditions.
Metformin suppresses invasiveness of bile duct cancer cells, and the effect is impaired by hyperglycemia
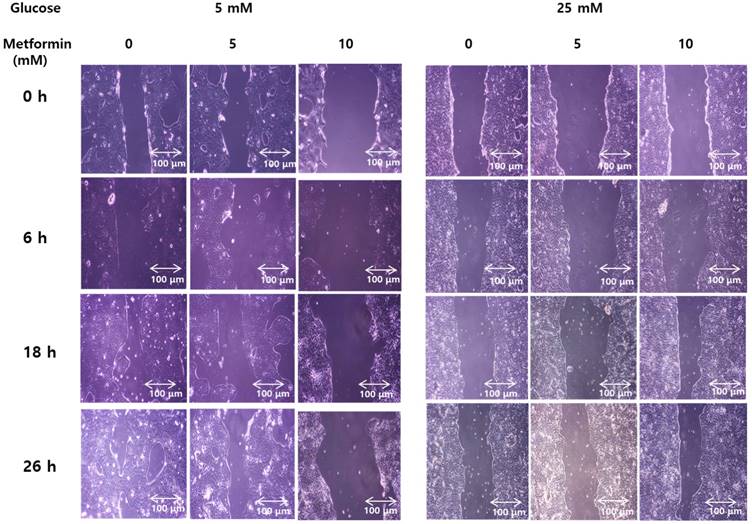
Wound healing assays weres performed to evaluate functional effect of metformin on invasiveness and migration of bile duct cancer cells in either normal or hypergylcemic conditions. In normal glucose levels, metformin inhibited growth of the cancer cells in a dose-dependent manner. However, at high glucose levels, this effect was markedly attenuated (Figure 6). This suggests that metformin inhibits growth and migration of bile duct cancer cells, although, this effects were impaired under hyperglycemic conditions.
Metformin weakens invasiveness of bile duct cancer cells, and the effect is impaired by hyperglycemia. SNU 245 cell were grown until 80%~90% confluency. A wound was produced at the center of the monolayer by gently scraping cells with a sterile pipette tip. The cells were incubated with metformin in either 5 mM or 25 mM glucose-containing DMEM with 1% FBS. The wound were visualized and photographed under the inverted microscope (x100 magnification) at indicated time. All results are representative of at least three separate experiments. In FG levels, metformin inhibited growth of the cancer cells in a dose-dependent manner. However, at HG levels, this effect was markedly attenuated.
Discussion
The incidence of metabolic disorders, such as obesity and diabetes mellitus, has been increasing as a result of the spread of the westernized lifestyle and changes in diet. These conditions are known to be associated with an increased risk of developing cancer and poor prognosis [22-24]. Altered levels of circulating factors including insulin, IGF-1, leptin, adiponectin, and interleukin-6 (IL-6) have been suggested to be strongly implicated [25].
Metformin is the drug of choice for the treatment of type 2 diabetes mellitus to improve hyperglycemia and insulin resistance, whose primary action is to inhibit hepatic glucose production through an LKB1/AMPK-mediated pathway [26]. Some studies using renal cells or podocytes have shown that hyperglycemia causes renal cell proliferation due to enhanced mTOR activity and inactivation of AMPK, and treatment of metformin stimulates AMPK phosphorylation and subsequently reduces the expression of mTOR. The authors concluded that metformin increases autophagy and prevents the renal cells from undergoing fibrosis, hypertrophy, and apoptosis [15, 27, 28]. Moreover, metformin is easily accessible with a good safety profile in clinical practice, and is well tolerated in patients with normal glycemic levels [29]. Long-term use of metformin has been reported to reduce various cancer risks in many epidemiologic studies in diabetic patients [10-14]. A meta-analysis study demonstrated that a 31% reduction in overall relative risk was noted in patients taking metformin compared with other antidiabetic drugs, and an inverse association was found to be significant for pancreatic and hepatocellular cancer [30]. Although the connections between metabolic syndromes and cholangiocarcinoma have remained inconclusive for years [4-6], a recent study using combined surveillance, epidemiology and Medicare data concluded that metabolic syndrome is associated with an increased risk of ICC in a population over 65 years of age [9]. Another case-control study proposed that diabetes and smoking are independent risk factors for ICC, and treatment with metformin is significantly associated with a 60% reduction in ICC risk in diabetic patients [19].
In pancreatic cancer, metformin has been shown to inhibit the growth of human cancer cells in a xenograft mouse model through a mechanism of disrupted cross-talk between the insulin receptor and a G-protein-coupled receptor [31, 32]. The known mechanism of antineoplastic activity of metformin is mainly mediated via the activation of the LKB1-AMPK signaling pathway, which, in turn, suppresses the Akt/mTOR pathway [33]. Additionally, metformin exerts its antitumor activity by regulating lipid metabolism, endothelial function, and immune functions [34-36]. Another suggested mechanism of the antitumor effect of metformin involves AMPKThr172 activation and inhibition of the IGF-1 signaling pathways. Hyperglycemic conditions potentiate IGF-1 responses, resulting in activation of an altered AMPK profile and impairment of metformin-induced inhibition of the growth promoting signals in pancreatic cancer cells [20].
In the case of cholangiocarcinoma, there has been only one in vitro study demonstrating that metformin exhibited a dose- and time-dependent antiproliferation effect on ICC cell lines, by mechanisms including apoptosis induction and cell cycle arrest, and mainly targeted the AMPK/mTOR pathway [37]. Our current study also showed similar results, providing additional evidence that metformin inhibits the proliferation of cholangiocarcinoma cells. However, in some respects, our study is different from the previous study as follows. 1) Metformin was, for the first time, found to have anticancer activity also in extrahepatic bile duct cancer cells. 2) Bile duct cancer cells with mutations in cancer-related genes were resistant to the inhibitory effects of metformin on proliferation compared to cells without mutations in these genes. 3) Metformin inhibited bile duct cancer cell proliferation by inhibiting the IGF-1R/IRS-1 pathway as well as AMPK/mTOR pathway in bile duct cancer cells. 4) The inhibitory effect of metformin on bile duct cancer cell proliferation was attenuated in high glucose conditions. 5) The antineoplastic effect of metformin in the bile duct cancer cell was demonstrated by invasiveness studies through the wound healing assays as well as the molecular studies.
In normal cells including podocytes and cardiomyocytes, metformin has cytoprotective effects by preventing apoptosis under several pathological conditions, such as hyperglycemia, ischemia and hypoxia. This antiapoptotic action is suggested to be mainly mediated by activated Akt which is regarded as an additional indicator for cell viability [28, 38, 39]. In comparison, metformin treatment in malignant cells has been suggested to have apoptotic and antineoplastic effects by activating AMPKThr172 with subsequent downstream inhibition of the growth promoting PI3K/Akt/mTOR pathway [20, 40, 41]. Some studies have suggested that Akt-induced phosphorylation of AMPKSer485 can inhibit AMPKThr172 activation, reducing the effects of metformin [41, 42]. Other studies have reported that the antineoplastic effect of metformin was not observed in high glucose conditions in pancreas, lung, and colon carcinoma cells [20, 43]. Similarly, we found that the growth inhibitory effects of metformin are associated with the activation of AMPKThr172 in extrahepatic bile duct cancer cells. In hyperglycemic conditions, we observed that the efficacy of metformin on antiproliferative and proapoptotic activity was reduced. The experiment for expressional change of TSC-2 and mTOR following siRNA transfection to knock down gene and protein expression of AMPKα (involved in phosphorylation at threonine-172) or AMPKα1 (involved in phosphorylation at serinet-485), the AMPKα silencer inhibited the metformin-induced TSC-2 activation and mTOR inactivation in FG levels and the AMPKα1 silencer maintained metformin-induced TSC-2 activation and mTOR inactivation in HG conditions, which suggests that AMPKThr172 and AMPKSer485, depending on the glucose level, have a crucial role in modulating bile duct cancer cell proliferations.
Increased IGF-1 induced by hyperinsulinemia has been suggested to have critical role in the link between type 2 diabetes and cancer [44]. Activation of the IGF-1R causes sequential activation IRS-1, which in turn activates the PI3K/Akt pathway, resulting in activation of mTOR, which is involved in protein synthesis and cell growth [45]. In a study using pancreatic cancer cells, metformin inhibited IGF-1R phosphorylation at normal glucose levels, causing a down-stream suppression of IGF-1 stimulated Akt phosphorylation. In high glucose conditions, IGF-1-induced Akt and AMPKSer485 phosphorylation appeared to correlate with a further reduction of the already-impaired AMPKThr172 phosphorylation by metformin. [20]. Our study also showed that IGF-1R silencing induced TSC-2 activation and mTOR inactivation without metformin treatment in FG level, but induced constant TSC-2 activation and mTOR inactivation in hyperglycemic condition. We concluded that IGF-1R pathway is another crucial pathway in modulating bile duct cancer cell proliferations in both normal fasting and hyperglycemic conditions.
In a meta-analysis study to investigate the association between metformin and overall survival in patients with pancreatic cancer and concurrent type 2 diabetes, a borderline significant relative survival benefit was found in metformin-treated patients compared with non-metformin treated patients [46]. Disappointingly, in phase 2 studies of metastatic or advanced pancreatic cancer to determine whether metformin has an actual anticancer effect or a synergistic effect when used with existing anticancer agents, addition of metformin at the dose commonly used by diabetes patients did not improve outcome [47, 48]. However, since all of the patients had metastatic or advanced cancer, and metformin was used at the general dose in the treatment of diabetes, metformin dose might be not sufficient to produce anticancer effects. Additionally, it should be considered that tumor cells express a variety of cell-surface transport proteins which allow metformin to get into the cells [49]. To date, there has been no clinical phase 2 or 3 study regarding the antitumor effect of metformin in bile duct cancer. In consideration of the limitations in the phase 2 studies of pancreatic cancers, we anticipate well-designed future studies with prospective, controlled, clinical trials to reveal the actual effects of metformin on the anticancer effect in bile duct cancer.
In this study, antiproliferative actions by metformin-induced apoptosis were documented in extrahepatic bile duct cancer cells harboring wild-type and mutant copies of cancer-related genes for the first time. In addition, we demonstrated that metformin suppresses the proliferation of bile duct cancer cells as a result of inhibiting mTOR induced by AMPK-Thr172 activation and IGF-1R inhibition. We also demonstrated that hyperglycemic conditions impair AMPKThr172 activation, evoke AMPKSer485 phosphorylation, and reduce the efficacy of metformin. Importantly, we confirmed that metformin attenuate the invasiveness of bile duct cancer cells. This study shows that metformin may be an anticancer agent in bile duct cancer alone or in combination with other anticancer agents. In addition, the bile duct cancer cells were found to be more resistant to growth-inhibiting agents such as metformin in a hyperglycemic environment. Accordingly, if patients with a high risk of bile duct cancer develop diabetes, choosing a treatment which includes metformin is recommended to reduce the risk of developing bile duct cancer. In addition, AMPK and IGF-1R play a key role in the proliferation of bile duct cancer cells and, thus, they are expected to be important targets for future development of chemotherapeutic agents of bile duct cancer.
Abbreviations
Akt: protein kinase B; AMPKThr172: AMP-activated protein kinase at threonine-172; AMPKSer485: AMP-activated protein kinase at serine-485; Bcl-2: B-cell lymphoma 2; Bax: bcl-2-like protein; FG: normal fasting glucose; HG: high glucose; ICC: intrahepatic cholangiocarcinoma; IGF-1R: insulin-like growth factor 1 receptor; IL-6: interleukin-6; IRS-1: insulin receptor substrate 1; LKB1: liver kinase B1; mTOR: mammalian target of rapamycin; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; SiRNA: small interfering ribonucleic acid; TSC-2: tuberous sclerosis complex 2.
Acknowledgements
This study was supported by Hallym University Academic-Industrial Cooperation Program (H20140422, 2014). This study was presented in the poster session of United European Gastroenterology Week 2017.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Berdah SV, Delpero JR, Garcia S, Hardwigsen J, Le Treut YP. A western surgical experience of peripheral cholangiocarcinoma. The British journal of surgery. 1996;83:1517-21
2. Song GW, Lee SG, Lee YJ, Park KM, Hwang S, Kim KH. et al. [Analysis of survival and factors affecting the survival after surgical resection of peripheral cholangiocarcinoma: 318 cases in single institute]. The Korean journal of hepatology. 2007;13:208-21
3. Shaib YH, El-Serag HB, Davila JA, Morgan R, McGlynn KA. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case-control study. Gastroenterology. 2005;128:620-6
4. Shaib YH, El-Serag HB, Nooka AK, Thomas M, Brown TD, Patt YZ. et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a hospital-based case-control study. The American journal of gastroenterology. 2007;102:1016-21
5. Welzel TM, Graubard BI, El-Serag HB, Shaib YH, Hsing AW, Davila JA. et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based case-control study. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2007;5:1221-8
6. Grainge MJ, West J, Solaymani-Dodaran M, Aithal GP, Card TR. The antecedents of biliary cancer: a primary care case-control study in the United Kingdom. British journal of cancer. 2009;100:178-80
7. Yamamoto S, Kubo S, Hai S, Uenishi T, Yamamoto T, Shuto T. et al. Hepatitis C virus infection as a likely etiology of intrahepatic cholangiocarcinoma. Cancer science. 2004;95:592-5
8. Donato F, Gelatti U, Tagger A, Favret M, Ribero ML, Callea F. et al. Intrahepatic cholangiocarcinoma and hepatitis C and B virus infection, alcohol intake, and hepatolithiasis: a case-control study in Italy. Cancer causes & control: CCC. 2001;12:959-64
9. Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology. 2011;54:463-71
10. Donadon V, Balbi M, Mas MD, Casarin P, Zanette G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver international: official journal of the International Association for the Study of the Liver. 2010;30:750-8
11. Lee MS, Hsu CC, Wahlqvist ML, Tsai HN, Chang YH, Huang YC. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC cancer. 2011;11:20
12. Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482-8
13. Lai SW, Chen PC, Liao KF, Muo CH, Lin CC, Sung FC. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. The American journal of gastroenterology. 2012;107:46-52
14. Lee JH, Kim TI, Jeon SM, Hong SP, Cheon JH, Kim WH. The effects of metformin on the survival of colorectal cancer patients with diabetes mellitus. International journal of cancer Journal international du cancer. 2012;131:752-9
15. Eisenreich A, Leppert U. Update on the protective renal effects of metformin in diabetic nephropathy. Curr medl chem. 2017;24:3397-412
16. El-Serag HB, Johnson ML, Hachem C, Morgana RO. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology. 2009;136:1601-8
17. Chiu HF, Ho SC, Chen CC, Yang CY. Statin use and the risk of liver cancer: a population-based case-control study. The American journal of gastroenterology. 2011;106:894-8
18. Lee J, Hong EM, Jang JA, Park SW, Koh DH, Choi MH. et al. Simvastatin Induces Apoptosis and Suppresses Insulin-Like Growth Factor 1 Receptor in Bile Duct Cancer Cells. Gut and liver. 2016;10:310-7
19. Chaiteerakij R, Yang JD, Harmsen WS, Slettedahl SW, Mettler TA, Fredericksen ZS. et al. Risk factors for intrahepatic cholangiocarcinoma: association between metformin use and reduced cancer risk. Hepatology. 2013;57:648-55
20. Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC cancer. 2013;13:235
21. Ku JL, Yoon KA, Kim IJ, Kim WH, Jang JY, Suh KS. et al. Establishment and characterisation of six human biliary tract cancer cell lines. British journal of cancer. 2002;87:187-93
22. Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Archives of physiology and biochemistry. 2008;114:63-70
23. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. The New England journal of medicine. 2003;348:1625-38
24. Becker S, Dossus L, Kaaks R. Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Archives of physiology and biochemistry. 2009;115:86-96
25. van Kruijsdijk RC, van der Wall E, Visseren FL. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009;18:2569-78
26. Papanas N, Maltezos E, Mikhailidis DP. Metformin: diamonds are forever. Expert opinion on pharmacotherapy. 2009;10:2395-7
27. Ravindran S, Kuruvilla V, Wilbur K, Munusamy S. Nephroprotective Effects of Metformin in Diabetic Nephropathy. Journal of cellular physiology. 2017;232:731-42
28. Langer S, Kreutz R, Eisenreich A. Metformin modulates apoptosis and cell signaling of human podocytes under high glucose conditions. Journal of nephrology. 2016;29:765-73
29. Provinciali N, Lazzeroni M, Cazzaniga M, Gorlero F, Dunn BK, DeCensi A. Metformin: risk-benefit profile with a focus on cancer. Expert opinion on drug safety. 2015;14:1573-85
30. Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B. et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer prevention research. 2010;3:1451-61
31. Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer research. 2009;69:6539-45
32. Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:2505-11
33. Bao B, Wang Z, Li Y, Kong D, Ali S, Banerjee S. et al. The complexities of obesity and diabetes with the development and progression of pancreatic cancer. Biochimica et biophysica acta. 2011;1815:135-46
34. Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer prevention research. 2010;3:1060-5
35. Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Molecular cancer therapeutics. 2010;9:1092-9
36. Li D. Metformin as an antitumor agent in cancer prevention and treatment. Journal of diabetes. 2011;3:320-7
37. Ling S, Feng T, Ke Q, Fan N, Li L, Li Z. et al. Metformin inhibits proliferation and enhances chemosensitivity of intrahepatic cholangiocarcinoma cell lines. Oncology reports. 2014;31:2611-8
38. Eisenreich A, Langer S, Herlan L, Kreutz R. Regulation of podoplanin expression by microRNA-29b associates with its antiapoptotic effect in angiotensin II-induced injury of human podocytes. Journal of hypertension. 2016;34:323-31
39. Boltzen U, Eisenreich A, Antoniak S, Weithaeuser A, Fechner H, Poller W. et al. Alternatively spliced tissue factor and full-length tissue factor protect cardiomyocytes against TNF-alpha-induced apoptosis. Journal of molecular and cellular cardiology. 2012;52:1056-65
40. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast cancer research and treatment. 2010;123:271-9
41. Ning J, Xi G, Clemmons DR. Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology. 2011;152:3143-54
42. Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. The Journal of biological chemistry. 2005;280:32081-9
43. Algire C, Amrein L, Bazile M, David S, Zakikhani M, Pollak M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011;30:1174-82
44. Magruder JT, Elahi D, Andersen DK. Diabetes and pancreatic cancer: chicken or egg? Pancreas. 2011;40:339-51
45. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nature reviews Cancer. 2008;8:915-28
46. Zhang JW, Sun Q. Metformin may improve the prognosis of patients with pancreatic cancer. Asian Pacific journal of cancer prevention: APJCP. 2015;16:3937-40
47. Reni M, Dugnani E, Cereda S, Belli C, Balzano G, Nicoletti R. et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:1076-85
48. Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, Beeker A. et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. The Lancet Oncology. 2015;16:839-47
49. Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handbook of experimental pharmacology. 2011:105-67
Author contact
![]() Corresponding author: Jin Lee, M.D., Division of Gastroenterology, Hallym University Dongtan Sacred Heart Hospital, 7 Keunjaebong-Gil, Hwasung, Gyeonggi-Do, 18450, Korea. Tel: +82-31-8086-2450, FAX: +82-31-8086-2029, email: jinleeor.kr
Corresponding author: Jin Lee, M.D., Division of Gastroenterology, Hallym University Dongtan Sacred Heart Hospital, 7 Keunjaebong-Gil, Hwasung, Gyeonggi-Do, 18450, Korea. Tel: +82-31-8086-2450, FAX: +82-31-8086-2029, email: jinleeor.kr