Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(11):2552-2559. doi:10.7150/jca.28167 This issue Cite
Research Paper
Identifying miRNA-mRNA Pairs and Novel miRNAs from Hepatocelluar Carcinoma miRNomes and TCGA Database
Ping Wu1*, Yunyue Xiao2*, Tao Guo1, Yitao Wang1, Shanshan Liao3, Lang Chen3 ![]() , Zhisu Liu1
, Zhisu Liu1 ![]()
1. Department of Hepatobiliary and Pancreas, Research Center of Digestive Diseases, Zhongnan Hospital of Wuhan University, Wuhan, 430071, P.R. China.
2. Institute of Digestive, Southwest Hospital, Third Military Medical University, Chongqing 400038, P.R. China.
3. Department of Immunology, School of Basic Medical Sciences, Wuhan University, Wuhan, 430071, P.R. China.
* These authors contributed equally to this work.
Received 2018-6-28; Accepted 2019-4-3; Published 2019-6-2
Abstract
Increasing evidence shows microRNAs (miRNAs) are engaged in hepatocelluar carcinoma (HCC). To identify novel feasible miRNA/mRNA pairs involved in hepatocarcinogenesis, an in-depth analysis of miRNomes in human non-tumor liver and HCC samples was carried out in this study. Firstly, differentially expressed miRNAs were obtained from deep sequencing of 15 liver samples, and verified in an independent data from The Cancer Genome Atlas (TCGA) database. Then, differentially expressed mRNA targets were selected from TCGA, and the differential miRNA/mRNA pairs with negative correlations were screened out. Finally, functional enrichment analysis was used to predict the functions of miRNA/mRNA pairs in HCC. In our study, 81 miRNA/mRNA pairs and 7 novel miRNAs were found. We constructed a hub interaction model with 9 miRNA/mRNA pairs to further investigate molecular mechanism of HCC. Survival analysis identified nine genes (hsa-miR-137, hsa-miR-490, BIRC5, TOP2A, CDC25C, IGF2BP1, IQGAP3, NCAPG and VIPR1) with significant influence on prognosis of HCC patients. In conclusion, the miRNA/mRNA pairs identified in our study may have some potential values to be further studied in progression, diagnosis and prognosis of HCC.
Introduction
As one of the most frequently diagnosed cancers, Hepatocelluar carcinoma (HCC) is a highly lethal malignancy. According to the newest statistics of estimated new cancer deaths, HCC is the fourth leading cause of cancer death in United States [1]. MicroRNA (miRNA) is a vital class of conserved noncoding RNAs. Although mature miRNA is very short, only 20-22 nucleotides (nt), it plays important regulatory roles by targeting to mRNAs for cleavage or translational repression. Abnormal miRNAs expression can affect biological behaviour of cancer, including cell proliferation, cell death, angiogenesis, invasion and migration [2]. It is now generally accepted that both of 5'-untranslational region (5'-UTR) and 3'-untranslational region (3'-UTR) of mRNAs can be targets of miRNAs. Complementary base pairing of miRNAs' seed region and 5'-UTR of mRNAs can enhance the stability of mRNAs [3]. If the base pairing occurs to miRNAs and 3'-UTR of mRNAs, it will lead to mRNA-destabilization and promote degradation of mRNAs [4]. A large amount of researches found that this molecular mechanism has profound influence on carcinogenesis and development in HCC [5-6]. Until now, at least 16 miRNAs and 6 clusters of miRNAs were regarded as circulating biomarkers for HCC diagnosis or for prediction of prognosis [7]. Meanwhile, nucleic acid-based drugs such as small interfering RNA (siRNA) and miRNAs can also be used for the HCC treatment [8].
Although tremendous progress has been made in this research field, novel feasible miRNA/mRNA pairs are still required to reveal molecular mechanism of carcinogenesis and development in HCC, especially to find more therapeutic strategies for HCC. Bioinformatics analysis of miRNA and mRNA expression profile can help to find key regulatory miRNA/mRNA pairs. Recently, with the development of high-throughput sequencing technology and bioinformatics analytic method, more and more miRNA/mRNA pairs were discovered to play a vital role in cancer. Xile Zhou et al. found a total of 372 pairs of miRNA-mRNA were found and 108 target genes of miRNA in colorectal cancer, among which miR-149 was less known in colorectal cancer before [9]. Through an in-depth analysis of miRNomes in human normal liver, hepatitis liver, and HCC, Jin Hou et al. found that miR-199a/b-3p can act as a tumor suppressor, inhibits HCC growth via target to PAK4 [10]. Finding more novel miRNAs related to HCC is still necessary. It can provide more possibility approaches for research on HCC.
To discover novel feasible miRNA/mRNA pairs, we screened the differentially expressed miRNAs and novel miRNAs from small RNAs deep sequencing of 15 human liver samples. The differentially expressed miRNAs were verified in expression profile of miRNAs downloaded from The Cancer Genome Atlas (TCGA) database to improve the credibility of it. Then, through analyzing the verified differentially expressed miRNAs and mRNA expression profile downloaded from TCGA, 81 miRNA/mRNA pairs were screened out. A part of them are less known in HCC still now, hoping them will contribute to diagnosis, prognosis or treatment of HCC. In addition, 7 novel miRNAs expressed differentially in HCC are worthy of being studied.
Materials and Methods
Clinical specimens and cell lines
The collection of human liver tissue was approved by the Ethics Committee in Zhongnan Hospital of Wuhan University. Informed consent was obtained from each patient. The methods were performed in accordance with the relevant guidelines and regulations. The 6 HCC tissues and 6 ANTs were obtained from 6 HCC patients; the 3 non-tumor tissues were collected from 3 hepatic hemangioma patients. These 9 patients were operated in Zhongnan Hospital of Wuhan University. All the liver samples were immersed in RNAlaterTM Stabilization Solution (Invitrogen, USA) and stored at -80 ℃ before total RNA was extracted.
miRNomes of 15 liver tissues
The small RNAs of 15 liver tissues were extracted by trizol reagent and purified by the polyacrylamide gel electrophoresis (PAGE) gel. Briefly, after PAGE purification of small RNA molecules (under 30 bases) and ligation of a pair of adaptors to their 3' and 5' ends, the small RNAs were amplified using the adaptor primers to get DNA fragment. Then, the DNA fragments around 80 bp were purified by agarose gels to get cDNA libraries. Quality of the cDNA libraries was checked and sequenced by Agilent 2100 Bioanalyzer and Illumina HiSeq 2000 technology. Finally, miRNomes of 15liver tissues were obtained.
miRNAseq and RNAseq of TCGA data
The data downloaded from TCGA was accordance with relevant laws, and any necessary approvals and informed consent documents [11]. The miRNAseq and RNAseq level 3 data of HCC were downloaded from TCGA database. Data platforms for miRNAseq and RNAseq were BCGSC_IlluminaHiSeq_miRNAseq and UNC_IlluminaHiSeq_RNAseq. There were 417 samples including 367 HCC samples and 50 non-tumor samples.
Screening of differential miRNAs from miRNAseq
The expression level of miRNAs in miRNAseq was presented using reads of exon model per million mapped reads (RPM), and analysed by limma algorithms to screen differentially expressed miRNAs of group HCC/non-tumor samples [12]. The threshold of differentially expressed miRNAs was set at fold change > 2, adjusted p-value < 0.05.
Identified the differential miRNAs from miRNAseq using miRNomes
The 15 liver samples were divided into two groups, HCC/ANT and HCC/non-tumor. The expression level of miRNAs in miRNomes was presented using reads count and analysed by DEGseq package in R software to screen differentially expressed miRNAs [13]. The threshold of differentially expressed miRNAs was set at fold change > 2, p-value and q-value < 0.05. Then, through comparing the differential miRNAs screened from miRNAseq and miRNomes, the miRNAs changed consistently were selected.
Screening of differential mRNAs from RNAseq
The expression level of mRNAs in RNAseq was presented using fragments per kilobase of exon model per Million mapped fragments (FPKM). The FPKM was normalized to transcripts per kilobase of exon model per million mapped reads (TPM) and analysed by limma algorithms for screening differentially expressed mRNAs in group of HCC/non-tumor samples. The threshold of differentially expressed mRNAs was set at fold change > 2, adjusted p-value < 0.05.
Screening of miRNA/mRNA pairs
First, the target mRNAs of differentially expressed miRNAs were predicted and analysed comparatively by 10 prediction programs of miRwalk 2.0 database, respectively DIANA-mT, miRanda, miRDB, miRWalk, RNAhybrid, PICTAR4, PICTAR5, PITA, RNA22 and TargetScan [14]. The mRNAs predicted by 5 prediction programs or more were selected as target mRNAs. In addition, the target mRNAs of novel miRNAs were predicted by TargetScan, miRanda, PITA and RNAhybrid all together. Then, Pearson correlation tests of the differentially expressed miRNAs and mRNAs were calculated respectively. Finally, the miRNA/mRNA pairs with negatively regulated and targeted relationship were obtained.
Analysis of the function of miRNA/mRNA pairs
To find the function of miRNAs-mRNAs pairs, we mapped the mRNAs into the DAVID database and subjected them to functional enrichment analysis [15]. GO terms and KEGG pathways with p-value < 0.05 were chosen to be analyzed [16]. String online database was used to evaluate the PPI relationships and find hub gene of the differentially expressed mRNAs [17]. According to clinic data of liver cancer downloaded from TCGA database, the miRNAs and mRNAs with negatively regulated and targeted relationship were selected for survival analysis by Kaplan-Meier method. The genes with log rank value < 0.05 had significant influence on prognosis of HCC.
Identification of novel miRNA
The sequence tags length in 49 nt from deep sequencing will go through the data cleaning analysis to get credible clean tags. Then the length distribution of the clean tags and common and specific sequences between samples will be summarized. Then the standard analysis will annotate the clean tags into different categories and take those which cannot be annotated to any category to predict the novel miRNA and seed edit of potential known miRNA. The miRdeep 2.0.0.5 software was used to discover miRNAs [18]. The miRdeep score and true positive rate were selected to evaluate miRNAs' quality.
Results
miRNomes of 15 liver tissues
The flowcharts for screening miRNA/mRNA pairs were shown in Figure 1A. A total of 42,220,814 reads were generated by deep sequencing on average. After low-quality reads, poly A, adapters and contaminated sequences were eliminated, 40,150,896 (95.10 %) clean reads length in 18-30 nt remained for further analysis (Figure 1B). The peak of small RNA length distribution was 22 nt, followed by 23 nt (Figure 1C). As shown in Figure 1D, 52 % unique reads were remained unannotated, 18 %, 11 %, 9 % and 1 % unique reads were annotated to intron and exon, rRNA, tRNA and miRNA respectively. On the other hand, there were 53 %, 26 %, 16 % and 2 % total reads classified to miRNA, unannotated sequence, tRNA and rRNA respectively (Figure 1D). The read counts of known miRNAs and novel miRNAs in miRNomes of 15 liver tissues were shown in Supplementary Table S1 and Supplementary Table S2.
The characteristics of miRNomes. (A) Flowcharts for identification of the miRNA/mRNA pairs. (B) The reads quality of 15 liver samples' cDNA librarys. (C) The frequency percent of small RNA length distribution. (D) The frequency percent of unique and total reads annotation.
Differentially expressed miRNAs of miRNomes and miRNAseq
There were 1883 miRNAs included in 417 samples of TCGA miRNAseq microarray. Having screened differentially expressed miRNAs by limma algorithms, 157 differentially expressed miRNAs with fold change > 2 and adjusted p-value < 0.05 were selected for futher analysis. The 100 top differentially expressed miRNAs were shown in Supplementary Figure S1. In miRNomes of the 15 liver samples, a total of 1887 miRNAs had been detected. 555 miRNAs expressed differentially in group HCC/ANTs, and 734 miRNAs expressed differentially in group HCC/non-tumor with fold change > 2 and adjusted p-value < 0.05. After the miRNAs which didn't change consistently in deep sequencing of miRNomes and miRNAseq, 38 differentially expressed miRNAs were chosen from miRNAseq for further analysis, among which 22 miRNAs were up-regulated while 16 miRNAs were down-regulated in HCC samples (Figure 2).
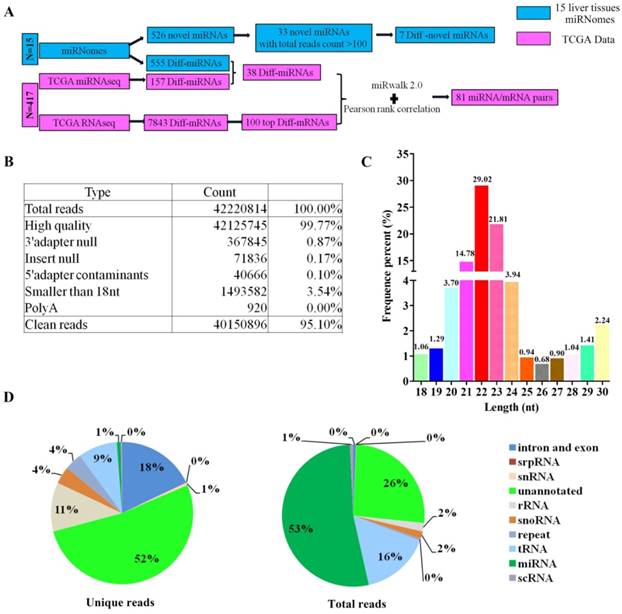
Heat map of the differential miRNAs and mRNAs that were identified to be miRNA/mRNA pairs. The names of miRNA and mRNA were shown on the right longitudinal axis; The information of samples was shown on the upper abscissa axis.
Differentially expressed mRNAs from RNAseq
According to the human liver cancer RNAseq level 3 data downloaded from TCGA database, there were 60484 mRNAs available for screening differentially expressed mRNAs by limma algorithms. 7843 mRNAs were expressed differentially in group HCC/non-tumor samples. The 100 top differentially expressed mRNAs with fold change > 16 and adjusted p-value < 0.05 were selected for further analysis, among which 50 mRNAs were up-regulated while 50 mRNAs were down-regulated in HCC (Supplementary Figure S2).
The miRNA/mRNA pairs
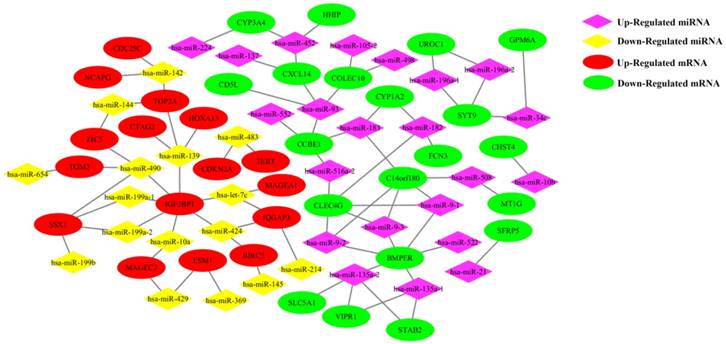
The 38 differentially expressed miRNA and the 100 top differentially expressed mRNAs were subjected to perform Pearson correlation tests. The 38 miRNAs were subjected to predict target mRNAs in miRwalk 2.0 database. The miRNA/mRNA pairs with negatively regulated and targeted relationship were obtained. As a result, 81 miRNA/mRNA pairs including 38 miRNAs and 36 mRNAs were remained (Figure 2). The regulation network of miRNA/mRNA pairs was shown in Figure 3.
The regulation network of miRNA/mRNA pairs.
Function analysis of the differentially expressed genes
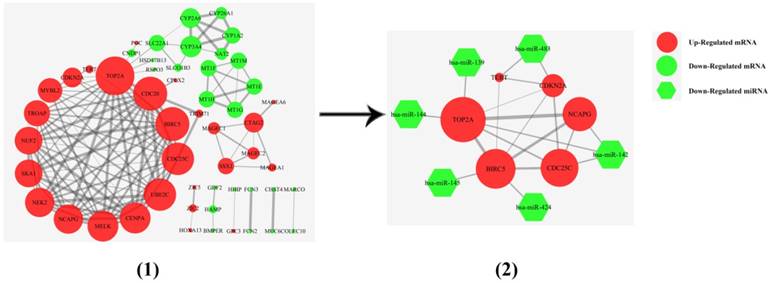
The 36 differentially expressed mRNAs of miRNA/mRNA pairs were mapped into the DAVID database and subjected to functional enrichment analysis, 23 genes were enriched in 31 GO Ontology (GO) term including 24 Biological Process process, 7 Molecular Function process (Supplementary Figure S3). The top 20 of GO term were shown in Supplementary Figure S4. 17 genes were included in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, among which 5 genes participated in cancer pathways, while 5 genes were associated with 6 signaling pathway. CYP1A2 and CYP3A4 were mainly related to metabolism and chemical carcinogenesis pathways (Supplementary Figure S5). A protein-protein interaction (PPI) interaction model was constructed using the 100 top differentially expressed mRNAs. In this model, a hub interaction model with 9 miRNA/mRNA pairs was found (Figure 4).
The PPI interaction model of differential mRNAs. (1) A PPI interaction model was calculated using the 100 top differential mRNAs of TCGA RNAseq. (2) A hub interaction model with 9 miRNA/mRNA pairs. The thickness of the ligature between mRNAs indicated the strength of the interaction. The size of the circle indicated the number of interactive genes.
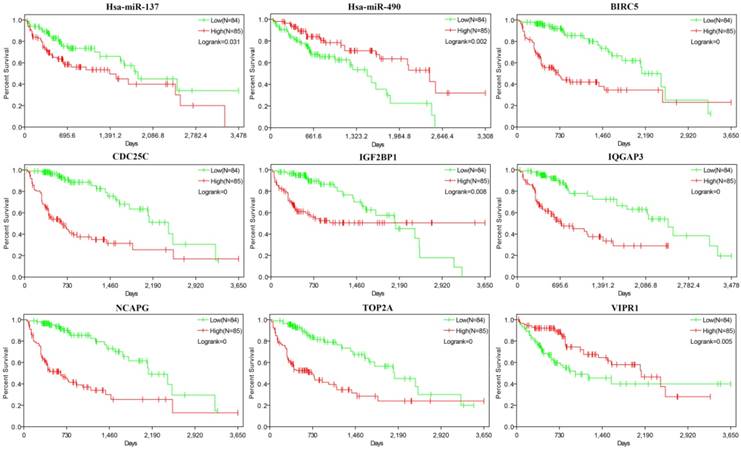
Survival analysis of the miRNAs and mRNAs
367 HCC samples were shared by 3 kinds of data (miRNAseq, RNAseq and clinic data of liver cancer) downloaded from TCGA database. The 38 miRNAs and 36 mRNAs of miRNA/mRNA pairs were chosen for survival analysis by Kaplan-Meier method. As a result, 9 genes (miR-137, miR-490, BIRC5, TOP2A, CDC25C, IGF2BP1, IQGAP3, NCAPG and VIPR1) had significant influence on prognosis (Figure 5). 7 genes (miR-137, CDC25C, IGF2BP1, IQGAP3, TOP2A, BIRC5 and NCAPG) were up-regulated in HCC, and high expression of them was correlated with shorter survival periods of HCC patients. 2 genes (miR-490 and VIPR1) were down-regulated in HCC and low expression of them was correlated with shorter survival periods of HCC patients.
Survival analysis of 9 genes. The abscissa axis and the longitudinal axis showed the survival time and rates respectively, the red curve means expression> median, while the green curve means expression< median.
Identification of 7 novel miRNAs
In total of 526 unique reads accounted for 25204 reads were annotated to novel miRNAs. After the low reads sequences (< 100 reads) were eliminated, 33 unique reads were remained to blast in miRbase. 5 novel miRNAs were down regulated and 2 novel miRNAs were up regulated in HCC (p-value and q-value < 0.05). All of the 7 novel miRNAs didn't match with any miRNA in miRbase, as shown in Table 1.
Information of the 7 novel miRNAs. The log2Ratio of novel miRNAs was calculated by DEGseq in R software (p-value and q-value <0.05).
| Name | miRdeep | Total | Mature miRNA sequence | Precursor position | log2(HCC/ | log2(HCC/ |
|---|---|---|---|---|---|---|
| Score | reads count | Normal) | Adjacent Tumor) | |||
| chr3_31470 | 1.4 | 468 | UAGGAGGGCUGAGAGGGC | chr3:165879677-165879714:+ | -2.8 | -4.75 |
| chr17_20757 | 2.7 | 354 | UAUGGAGUGUGUAUAUAUGUGU | chr17:66884155-66884216:- | -3.43 | -4.31 |
| chr8_95005 | 1.7 | 210 | GUGUGUGUGUGUGUGUAA | chr8:10003296-10003356:+ | -8.24 | -10.12 |
| chr10_1351 | 1.10E+01 | 156 | UAGAGAACCAUGGUCAUAGACU | chr10:107803675-107803737:+ | -4.4 | -3.44 |
| chr17_51011 | 4.10E+01 | 138 | UCCAUGGGCACACCUGAAUGACA | chr17:19480916-19480977:- | 4.55 | 1.89 |
| chr19_52695 | 2.60E+01 | 138 | ACCGUGGGCAUGGGGCCUUGGAAC | chr19:6718426-6718478:- | -2.35 | -4.03 |
| chr13_26323 | 1.5 | 130 | UGAGUGUGUGUGUGUGUGUGUGUGU | chr13:25689510-25689581:- | 4.59 | 6.52 |
Discussion
According to statistic of reads, the quality of small RNA deep sequencing in 15 liver samples was qualified to analyse expression of small RNAs in each sample. The peak of small RNAs length distribution was 22 nt, followed by 23 nt. This feature is consistent with small RNA deep sequencing in animals such as Chinese rare minnow and zebrafish [19-20]. The first largest category of total reads is miRNA and the second largest classification was unannotated. There were more than half of unique reads remained unknown. It suggests that miRNAs are the major member of small RNAs and there is still much space to discover novel small RNAs. A regulatory network made of 81 miRNA/mRNA pairs including 38 miRNAs and 36 mRNAs was found in this study. A number of such miRNA/mRNA pairs showed they may play key roles in regulation of tumor formation and development. For example, the let-7 family was demonstrated it can inhibits tumor cell migration, growth and self-renewal by suppressing expression of IGF2BP1 [21]. In our study, 555 differentia expressed miRNAs were found in 15 liver samples from the patients in Zhongnan Hospital of Wuhan University, while only 157 differentia expressed miRNAs in TCGA miRNAseq containing 417 liver samples. This result are largely due to sampling error. To decrease the sampling error, we selected the intersection of them. So, the final 38 differential miRNAs selected have higher reliability.
In order to find the hub genes in the 81 miRNA-mRNA pairs, the top 500, 200 and 100 differentially expressed genes were subjected to perform PPI network analysis on string online database respectively. It was found that 6 genes (CDKN2A, TERT, TOP2A, BIRC5, CDC25C and NCAPG) related to cell cycle process were always located in the center of PPI network, and had nearly the highest combined scores and degree of nodes. Furthermore, 4 of them (TOP2A, BIRC5, CDC25C and NCAPG) were up regulated in HCC samples and negatively correlated to survival periods of HCC patients. These results indicated that the 6 genes might be the hub regulatory genes in HCC growth. Furthermore, our study suggested that they were negatively regulated and targeted by 6 miRNAs (miR-139, miR-142, miR-144, miR-145, miR-424 and miR-483), making up 9 hub miRNA/mRNA pairs.
The 9 hub miRNA/mRNA pairs were selected to emphatically discuss. The influences of miR-139/TOP2A, miR-142/CDC25C and miR-145/BIRC5 in cancer were verified by some researches. MiR-142 is down-regulated in breast, lung and cervical cancer, and over-expression of miR-142 can lead to cell cycle arrest at G2/M and inhibit cancer cell proliferation by directly regulating CDC25C expression [22]; miR-139 as a tumor suppressor gene affects cell proliferation of cancer by target TOP2A, and high expression of TOP2A is significantly related to shorter survival periods in the luminal type breast cancer patients [23]. BIRC5 is one of the miR-145 target genes. miR-145 can break the cervical epithelial barrier via dysregulation of cell adhesion, apoptosis and proliferation [24]. Although 3 miRNA/mRNA pairs as above mentioned have been reported, their molecular mechanisms are still less known in HCC. Also the influences of miR-142/miR-144/TOP2A, miR-424/BIRC5 and miR-142/NCPAG in HCC remained unknown. In our study, miR-142, miR-144 and miR-424 were all down regulated in HCC patients, and it had been identified by a number of researches [25-27]. Function of TOP2A and BIRC5 in HCC were as mentioned above. Overexpression of NCAPG was associated with poor survival of HCC patients, and knockdown of NCAPG will inhibit cell growth [28]. In conclusion, the 9 hub miRNA/mRNA pairs were all less known in HCC, and the expression trends of these miRNAs and mRNAs were supported by our study and others'. These evidences indicated that the 9 hub miRNA/mRNA pairs may have certain influence on the survival periods of HCC patients together.
According to the statistics of the read positions and frequencies of the reads within the stable hairpin, 526 unique reads were strikingly similar to miRNA and scored by miRdeep. Considered to reliability and research value of novel miRNAs, 33 novel miRNAs with total reads count > 100 were selected. 7 novel miRNAs were changed consistently in group HCC/non-tumor and HCC/ANTs. In terms of the reads, 1887 known miRNA were found in 15 liver samples, but only 767 known miRNAs were counted to meet total reads count > 100. The total reads count of the 7 novel miRNAs were between 130 and 468 respectively. Furthermore, their fold p-values of hairpin were significant, and the length of them also met characteristics of miRNA. These evidences suggest that the 7 novel miRNAs are expressed stably in liver tissues rather than randomly and worthy of being studied.
In a word, this study may provide a number of genes for revealing the miRNA/mRNA regulatory network in development and prognosis of HCC.
Abbreviations
ANT: adjacent tumor; BGI: Beijing Genomics Institute; FPKM: fragments per kilobase of exon model per Million mapped fragments; GO: GO Ontology; HCC: hepatocelluar carcinoma; KEGG: Kyoto Encyclopedia of Genes and Genomes; miRNA: microRNA; nt: nucleotides; PAGE: polyacrylamide gel electrophoresis; PPI: protein-protein interaction; RPM: reads of exon model per million mapped reads; siRNA: small interfering RNA; TCGA: The Cancer Genome Atlas; TPM: transcripts per kilobase of exon model per million mapped reads; 5'-UTR: 5'-untranslational region; 3'-UTR: 3'-untranslational region.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
Study concept and design: Zhisu Liu and Lang Chen; acquisition and analysis of data: Ping Wu, Yunyue Xiao; drafting of the manuscript: Ping Wu, Tao Guo, Yitao Wang and Shanshan Liao; modification of the manuscript: Zhisu Liu and Lang Chen.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7-30
2. Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004
3. Li G, Wu X, Qian W. et al. CCAR1 5' UTR as a natural miRancer of miR-1254 overrides tamoxifen resistance. Cell Res. 2016;26(6):655-73
4. Guo H, Ingolia NT, Weissman JS. et al. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835-840
5. Yuan Q, Loya K, Rani B. et al. MicroRNA-221 overexpression accelerates hepatocyte proliferation during liver regeneration. Hepatology. 2013;57(1):299-310
6. Yang X, Liang L, Zhang XF. et al. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology. 2013;58(1):158-170
7. Klingenberg M, Matsuda A, Diederichs S. et al. Non-coding RNA in hepatocellular carcinoma: Mechanisms, biomarkers and therapeutic targets. J Hepatol. 2017;67(3):603-618
8. Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017;173:106-117
9. Zhou X, Xu X, Wang J. et al. Identifying miRNA/mRNA negative regulation pairs in colorectal cancer. Sci Rep. 2015;5:12995
10. Hou J, Lin L, Zhou W. et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. 2011;19(2):232-243
11. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061-1068
12. Diboun I, Wernisch L, Orengo CA. et al. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics. 2006;7:252
13. Wang L, Feng Z, Wang X. et al. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136-138
14. Dweep H, Gretz N, Sticht C. miRWalk database for miRNA-target interactions. Methods Mol Biol. 2014;1182:289-305
15. Dennis G Jr, Sherman BT, Hosack DA. et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3
16. Kanehisa M, Goto S, Sato Y. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199-D205
17. Jensen LJ, Kuhn M, Stark M. et al. STRING 8-a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412-D416
18. Friedländer MR, Chen W, Adamidi C. et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26(4):407-415
19. Hong X, Qin J, Chen R. et al. Identification and characterization of novel and conserved microRNAs in several tissues of the Chinese rare minnow (Gobiocypris rarus) based on illumina deep sequencing technology. BMC Genomics. 2016;17:283
20. Wei C, Salichos L, Wittgrove CM. et al. Transcriptome-wide analysis of small RNA expression in early zebrafish development. RNA. 2012;18(5):915-29
21. Busch B, Bley N, Müller S. et al. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016;44(8):3845-3864
22. Cao XC, Yu Y, Hou LK. et al. miR-142-3p inhibits cancer cell proliferation by targeting CDC25C. Cell Prolif. 2016;49(1):58-68
23. Hua W, Sa KD, Zhang X. et al. MicroRNA-139 suppresses proliferation in luminal type breast cancer cells by targeting Topoisomerase II alpha. Biochem Biophys Res Commun. 2015;463(4):1077-1083
24. Anton L, DeVine A, Sierra LJ. et al. miR-143 and miR-145 disrupt the cervical epithelial barrier through dysregulation of cell adhesion, apoptosis and proliferation. Sci Rep. 2017;7(1):3020
25. Tsang FH, Au SL, Wei L. et al. MicroRNA-142-3p and microRNA-142-5p are downregulated in hepatocellular carcinoma and exhibit synergistic effects on cell motility. Front Med. 2015;9(3):331-343
26. Yu M, Lin Y, Zhou Y. et al. MiR-144 suppresses cell proliferation, migration, and invasion in hepatocellular carcinoma by targeting SMAD4. Onco Targets Ther. 2016;9:4705-4714
27. Yang H, Zheng W, Shuai X. et al. MicroRNA-424 inhibits Akt3/E2F3 axis and tumor growth in hepatocellular carcinoma. Oncotarget. 2015;6(29):27736-27750
28. Zhang Q, Su R, Shan C. et al. Non-SMC Condensin I Complex, Subunit G (NCAPG) is a Novel Mitotic Gene Required for Hepatocellular Cancer Cell Proliferation and Migration. Oncol Res. 2018;26(2):269-276
Author contact
![]() Corresponding authors: zhisuliuedu.cn; Tel.: +86-27-67812588; langchenedu.cn.
Corresponding authors: zhisuliuedu.cn; Tel.: +86-27-67812588; langchenedu.cn.