Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(5):1162-1169. doi:10.7150/jca.37117 This issue Cite
Research Paper
Modulating cancer stemness provides luminal a breast cancer cells with HER2 positive-like features
Yi Mei2*, Dongyan Cai3, Xiaofeng Dai1 ![]() *
*
1. Wuxi School of Medicine, Jiangnan University, Wuxi, China
2. School of Biotechnology, Jiangnan University, Wuxi, China
3. Department of Oncology, Affiliated Hospital of Jiangnan University, Wuxi, China
*co-first authors
Received 2019-5-29; Accepted 2019-9-22; Published 2020-1-1
Abstract
Breast cancers can be classified into luminal A, luminal B, HER2 positive and triple-negative subtypes, each with a distinct therapeutic response. Tumor stemness drives cancer malignancy that challenges cancer control. Understanding the revolutionary relationships driven by tumor stemness among breast cancer subtypes is fundamental to identifying feasible therapeutic modalities for each breast cancer subtype.
Utilizing the endogenous tRNA-processing system, we established a multiplexing CRISPR/dCas9 system in breast cancer cells, and applied it to a four-gene panel controlling cell potency, i.e., OCT4, KLF, MYC, SOX2. The stable cell strain, OKMS#1 was obtained through concomitantly over-expressing these genes in luminal A breast cancer cells. OKMS#1 cells showed increased invasion, proliferation and cancer stemness, shared similar drug response pattern with HER2 positive cells, and exhibited altered MAPK and enhanced NFkB signaling. This study contributes in providing an efficient multiplexing CRISPR/dCas9 system that enriches our genetic modulation tool box, and suggests that HER2 positive cells are potential progenitors of luminal A cells and that these two breast cancer subtypes may share similar treatment strategies once rewired between the two states. Our results also implicate that triple negative breast cancer cells, though sharing similar cancer stemness with HER2 positive cells, represent a distinct type of disease and require unique treatment solutions.
Keywords: breast cancer, cancer stemness, progenitor cells, CRISPR/dCas9, therapeutic modality
Introduction
Cancer stem cells (CSCs) are immortal cells within a tumor that can self-renew and give rise to the bulk cancer cells [1, 2]. Tumors with more CSCs are phenotypically more malignant and more likely to establish resistance to many treatment modalities [3, 4]. Therefore, targeting the CSC cohort or reducing cancer stemness is an attractive onco-therapeutic strategy.
Breast cancer is not a single disease but comprised of at least four subtypes, i.e., luminal A, luminal B, HER2 positive and triple negative, with distinct clinical outcomes and treatment responses [5, 6]. HER2 positive and triple negative breast tumors are pathologically more malignant than the luminal subtypes, with the tumor grade of the former being typically 2 or 3 and that of the latter being 1 or 2 [6]. Although both HER2 positive and triple negative tumors are enriched with CSCs [5], they have distinct therapeutic responses. That is, while HER2 positive breast cancers can be effectively targeted with Trastuzumab [7] and sometimes with Tamoxifen[8], triple negative cancers lack effective targeted treatment due to its lack of cell surface receptors such as estrogen receptor (ER), progesterone receptor (PR) and human epithelial receptor 2 (HER2). Such discrepancies between phenotypic response and molecular profiling suggest distinct revolutionary paths of both subtypes towards high stemness. Uncovering mechanisms underlying such discrepancies could considerably advance our understandings on the pathogenesis of both diseases that could ultimately lead to the effective control of both HER2 positive and triple negative subtypes. Taking luminal A cells (the least malignant subtype with known cure) as the control, we are motivated to study whether these cells can be switched to the HER2 positive or triple negative subtype if cells' stemness was increased.
OCT4, KLF4, MYC and SOX2 were originally found capable of inducing iPS (induced pluripotent stem) cells [9] and have been widely applied for cell stemness reprogramming. For instance, OCT4 overexpression in normal breast cells could empower cells with tumor-initiating and colonization capabilities [10], and SOX2 over-expression could lead breast cancer cells to a more stem-like state [11]. In compliance with our motivation in exploring subtype rewiring among breast cancers, we are interested in utilizing this four-gene panel coupled with the CRISPR technique in our study.
CRISPR technology offers us a precise genome-editing tool, and the use of the CRISPR-dCas9 system that lacks the shear activity could enable us to modulate the expression of targeted genes in a mild mode. For example, simultaneous suppression of multiple genes has been achieved using CRISPR/Cas9 and CRISPR/dCas9 in Streptomyces for functional gene screening and metabolic engineering editing [12]. Concomitant transcriptional activation or inhibition of multiple genes has been achieved using the CRSIPR/dCas9 system in plants [13]. In 2015, Xie et al. successfully demonstrated in rice that tandemly arranged tRNA-gRNA structures could be efficiently and accurately processed into sgRNAs of the desired targeting sequences in vivo to guide Cas9 in the editing of multiple chromosomal targets [14]. An improved dCas9 system enabling simultaneous and precise in vivo transcriptional activation of multiple genes and long noncoding RNAs (lncRNAs) was established and applied in the nervous system [15]. Despite the intensive efforts on multiplexing CRISPR system design and applications, relatively few study has reported the design and use of multiplexing CRISPR/dCas9 system in breast cancers [16]. We are thus motivated to establish a multiplexing CRISPR/dCas9 system and apply it to study the phylogenetic relationship among breast cancer subtypes driven by cancer stemness.
Materials and Methods
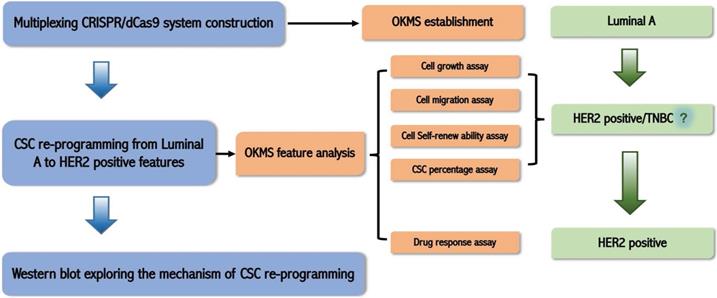
The study design of this work is illustrated in Figure 1.
Schematic illustration of the logic of this study. We firstly established a multiplexing CRISPR/dCas9 system taking advantage of the endogenous tRNA-processing system; then we constructed the OKMS cell line from luminal A cells, found from cell growth, migration, cell self-renew ability, and cancer stem cell (CSC) percentage assays that OKMS cells showed HER2 positive or triple negative breast cancer (TNBC) features, and confirmed from drug response assay that OKMS cells showed HER2 positive properties; we explored the mechanism that led to cancer stem cell (CSC) rewiring using western blot.
Cell culture
Three human breast cancer cell lines, i.e., MCF7 (luminal A), SKBR3 (HER-2 positive) and MDAMB231 (triple negative) were used in this study, which were purchased from American Type Culture Collection (Manassas, VA, USA). MCF7 and MDAMB231 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM, HyClone, American) supplemented with 10% fetal bovine serum (FBS, Lonsera, Shanghai, China) and antibiotics at 37℃ in 5% CO2, respectively, SKBR3 cells were maintained in Roswell Park Memorial Institute (RPMI, HyClone, American) supplemented with 10% FBS and antibiotics at 37℃ in 5% CO2.
Construction of multiplexing sgRNA plasmid
The multiplexing sgRNA (small guide RNA) was constructed taking advantages of the principles of the endogenous tRNA self-shearing system. The constructed multi-gene multi-sgRNA fragment was double-cut using BbsI, resulting in two sticky ends GTGG and GTTT, following ligation with the BbsI double-cut plasmid vector pLenti-U6-sgRNA-PGK-Neo. The sgRNA sequences of OCT4, KLF4, MYC and SOX2 are listed in Table S1.
Cell transfection
The constructed CRISPR plasmid was diluted using 200 μl of serum-free DMEM following cell transfection using LipofectamineTM 2000 (ThermoFisher Scientific, China), with the ratio between the plasmid and LipofectamineTM 2000 being 1:3. The plasmid-reagent mixture was let stand still for 20 min to form chelates. The medium was renewed after 4 h of transfection, and supplemented with 200 mg/ml of G418 and 0.1 mg/ml of puromycin antibiotic after 24 h for screening.
qRT-PCR
After digesting the transfected cells in a 6-well plate with trazol, the total RNA was extracted using the RNA extraction kit (TAKARA, Japan) according to the manufactor's prococol. 1 ug of the extracted RNA was used for reverse transcription to obtain cDNA using TAKARA PrimerScript RT reagent KIT (TAKARA, Japan) as per manufacturer's protocol. The cDNA was diluted to a final concentration of 10 ng/μl, and the SYBR Mix was melted at the room temperature. The PCR reaction mixture was prepared which contains 5 μl SYBR premix Ex Taq II, 0.4 μl forward and 0.4 μl reverse primers, 0.2 μl ROX reference dye and 2μl water. 8 μl PCR reaction mixture was added to the 96-well plate/eight-well strip according to the reaction layout, and 2 μl of the template cDNA solution was added. The 96-well plate or the 8-hole strip cover was sealed with a transparent film, mixed using vortex and centrifuged to ensure that the solution was concentrated at the tube bottom. The PCR reaction was run using ABI 7500 according to the thermal cycling conditions that is consisted of 2 min at 95℃, 5s at 95℃ and 30s at 60℃ for 40 cycles following 15s at 72℃. After the cycling protocol, the final step was applied to all reactions by continuously monitoring fluorescence through the dissociation temperature of the PCR product at a temperature transition rate of 0.3℃ to generate a melting curve. The qPCR primers for each gene are listed in Table S2.
Western blotting
Trypsin-digested cells were centrifuged, re-suspended in pre-cooled phosphate buffered saline (PBS), supplemented with 100 μL cell lysate (containing 1% phenylmethylsulfonyl fluoride (PMSF)) in each well of a six-well plate, placed on ice and lysed for about 10 sec. The mixture was centrifuged at 10000 r·min-1 at 4 °C for 5 min. The supernatant was carefully absorbed, and the concentration of the protein samples was determined according to the BCA kit (ThermoFisher Scientific, China), 5× loading buffer was added to protein samples followed by 5 min boiling. The glue was loaded after cooling. The gel was cut into the appropriate size. A large PVDF film was cut out followed by methanol treatment. The treated film was immersed in the transfer buffer together with the filter paper. The membrane was transferred at a constant flow of 200 mA for 90 min, washed using Tris-buffered saline Tween-20 (TBST) for 3 times. The blocking solution was prepared by supplementing TBST solution with 5% BSA, and used to block the membrane at the room temperature for 120 min. The membrane was washed for 3 times, placed in the primary antibody diluted solution overnight, washed again for 3 times, placed it in the secondary antibody dilution solution for 120 min, and added to the ECL color development solution (Absin, China) for detection.
The secondary antibody and primary antibodies against GAPDH, OCT4, KLF4, MYC, SOX2, ER, HER2 were purchased from Proteintech, and primary antibodies targeting p65, phospho-p65, ERK1/2, phospho-ERK1/2, JNK, phospho-JNK, p38, phospho-p38 MAPK were ordered from Cell Signaling Technology.
Cell migration assay
After cells were plated in a 6-well plate, the medium was aspirated. Cells were drawn in a straight line using a pipette tip, washed three times using PBS, and cultured in a medium supplemented with 1% serum. Photographs were taken at 0h, 8h, 16h, and 24h, respectively.
Cell proliferation assay
Cells were plated in a 96-well plate, with the number of cells per well-being 4000. The medium was refreshed at 24h, 48h, and 72h, followed by addition of 5ul of CCK8 (GLPBIO, USA). The absorbance was measured at 450 nm after incubation at 37 °C for 2 h.
Cell sphericity counting
Cells of the control and the experimental group were resuspended in a serum-free globular medium pre-warmed at 37 °C (concentration: 2×103 cells·mL-1), and spread at 200 μL per well. The 96-well ultra-low adhesion plate was cultured for 7 days in a CO2 incubator, and photographed using the inverted phase contrast microscope (Olympus, Germany) to calculate cell sphericity.
Detection of stem cell percentage
Cells were prepared in 6-well plates with 3×105 cells allocated in each well. The plate was placed in a CO2 incubator for 24 h. After aspirating the medium, cells were washed twice with PBS, followed by digestion with trypsin (excluding ethylenediaminetetraacetic acid (EDTA)). Cells were centrifuged at 1000 r·min-1 for 5 min, the supernatant was discarded and cells were resuspended in PBS. Cells were centrifuged at 1000 r·min-1 for 5 min, the supernatant was discarded and cells were resuspended in the assay buffer of the ALDEFLUOR kit (STEMCELL, Canada) to prepare a cell suspension of 5×105 cells·mL-1. 500 μL of suspension was used as one cell sample and added to each of two tubes. 10 μL of diethylaminobenzaldehyde (DEAB) was added in one tube, 2.5 μL of activated fluoroboron dipyrrole-aminoacetaldehyde-acetic acid diethyl amino malonate (BAAA) was added to both tubes. Both tubes were incubated at 37 °C for 30-40 min in the dark, centrifuged at 1000 r·min-1 for 5 min. The supernatant was discarded and cells were resuspended in 500 μL of assay buffer for each tube to prepare one cell sample. The flow tube was preserved under hypothermic condition, and CSC proportion was measured using a flow cytometer.
Results
Multiplexing CRISPR/dCas9 system construction
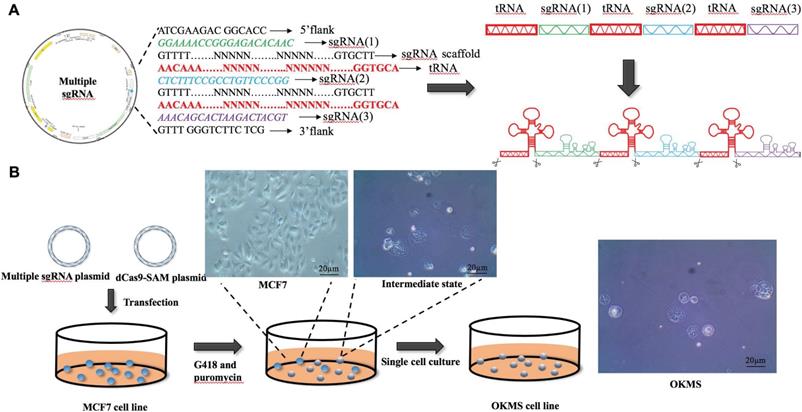
To concomitantly regulate the expression of the four gene panel, i.e., OCT4, KLF4, MYC, SOX2, we established a simple yet robust platform to boost the CRISPR/dCas9 multiplex editing capability taking advantages of the endogenous tRNA-processing system. The endogenous tRNA-processing system precisely cleaves both ends of the tRNA precursor. We engineered a multiplexing sgRNA plasmid with the four genes tandemly arrayed in a tRNA-sgRNA architecture, and transfected it together with the dCas9-SAM (synergistic activation mediator) plasmid (which functions as an engine for activating endogenous gene expression) into the breast cancer cell line MCF7. Through antibiotic screening and single cell culturing, we obtained two stable cell strains with both plasmids successfully transfected (Figure 2B). The results showed that the multiplexing sgRNA plasmid was efficiently and precisely processed into sgRNAs with desired 5' targeting sequences in cells, which directed dCas9 to edit multiple targets simultaneously (Figure 2A). Both qPCR and western blotting revealed concomitant increase in the expression of all four genes in both cell strains (Figures 3A, 3B), demonstrating our success in constructing the multi-gene CRISPR/dCas9 system.
The plasmid design and experimental procedure in the establishment of OKMS cells. A) Design of the multi-sgRNA plasmid. B) Transfection and selection process in the establishment of stable OKMS cells.
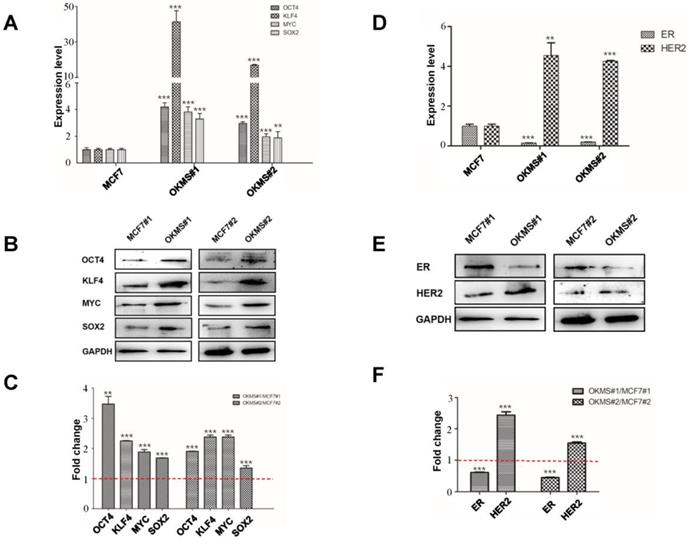
Expression of cell stemness and subtyping markers in stable OKMS cells. A) Expression of cell stemness markers (OCT4, KLF4, MYC and SOX2) at the A) transcriptional and B) translational levels. Expression of subtyping markers (ER, HER2) at the D) transcriptional and E) translational levels. Western blot signalling intensities normalized by that of GAPDH in OKMS cells are plotted in C) for stemness markers (OCT4, KLF4, MYC and SOX2) and in F) for subtyping markers (ER, HER2). Bars represent mean ± SD of fold change from at least three independent experiments, and ** (0.01 <P < 0.05) and *** (P<0.01) represent student t-test p values computed using raw ratios.
Establishment of OKMS cells
A stable cell strain concomitantly over-expressing OCT4, KLF4, MYC, SOX2, namely OKMS, was successfully constructed using the multi-gene CRISPR/dCas9 system. The expression of all genes in the cell strain OKMS#1 was more than three times higher than that of the control (p<0.01), among which KLF4 expression increased over 30 times. The expression of all genes in cells OKMS#2 was enhanced over double-folds as compared with the control (p<0.05) (Figure 3A). The same results were shown at the protein level (Figure 3B). The expressions of canonical breast cancer subtyping markers, ER and HER2, were significantly altered. ER was significantly down-regulated (p<0.05) and HER2 was considerably up-regulated (p < 0.01) at the transcriptional and translational levels, suggesting a switch from the luminal A-like to the HER2 positive-like phenotype. OKMS#1 was selected in the following experiments.
Cell growth and migration behavior of OKMS cells
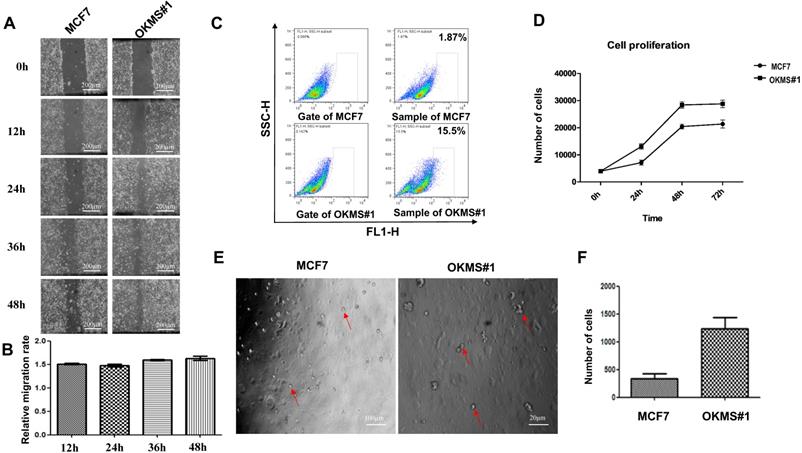
The OKMS#1 cell line underwent a dramatic change in the morphology, i.e., from polygonal to spherical in shape, as compared with the control (Figure 2). Both the growth and migration abilities of OKMS#1 cells were increased up to 1.5 times than that of the control with statistical significance (p < 0.01, Figures 4A, 4B, 4D), where the migration was measured at 12, 24, 36 and 48 hours. The percentage of breast CSCs increased from 1.87% in MCF7 to 15.5% in OKMS#1 cells (Figure 4C, Figure S1). Cell sphericity increased over 2 times in OKMS#1 cells than MCF7 where they were originated from (Figures 4E, 4F). These results suggested that OKMS#1 cells were more malignant than MCF7 cells.
Morphological alterations in stable OKMS cells as compared with MCF7 cells. A) Images measuring cell migration abilities. B) Digital statistics quantitizing cell migration abilities. C) Flow analysis graph showing cell cancer stem cell percentages. D) Growth curves showing cell proliferative abilities. E) Images measuring cell self-renew abilities. F) Digital statistics quantitiziing cell self-renew abilities. Bars represent mean ± SD from at least three independent experiments.
Drug response of OKMS cells
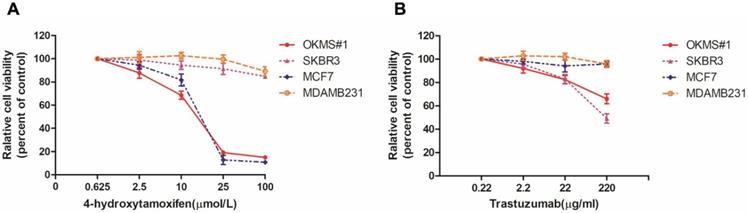
We tested the drug sensitivity of OKMS#1, MCF7, SKBR3, MDAMB231 to two drugs canonically used for breast cancer treatment, i.e., 4-Hydrotamoxifen (a hormonal therapy) and Trastuzumab (targeted therapy against HER2 positive cells). OKMS#1, MCF7 (luminal A) and SKBR3 (HER2 positive) cells shared similar dose response curves and were sensitive to Tamoxifen after 3 days of treatment (Figure 5A). OKMS#1 and SKBR3 responded to Trastuzumab after 3 days of drug administration (Figure 5B). These results revealed that OKMS#1 cells shared similar drug response pattern with the HER2 positive cell line SKBR3.
Drug dose curves of cells in response to Tamoxifen and Trastuzumab. Drug response curves under the treatment of A) Tamoxifen, B) Trastuzumab. MCF7, SKBR3, MDAMB231 are luminal, HER2 positive and triple negative cells, respectively, and OKMS is a stable cell line established from MCF7 by over-expressing the four-gene panel controlling cell stemness (OCT4, KLF4, MYC and SOX2).
Reprogrammed cancer signalings in OKMS cells
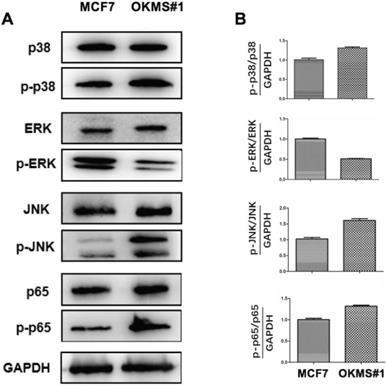
OKMS#1 cells showed decreased ERK phosphoration (around half that of the control), increased JNK phosphoration (over 1.5 folds of the control), and slightly increased p38 and p65 phosphoration (both are around 1.3 folds of the control), suggesting that NFκB, MAPK/JNK, and MAPK/p38 were activated in OKMS#1 and MAPK/ERK was suppressed during the four-gene mediated reprogramming (Figure 6).
Western blot of key signaling molecules in MAPK and NFkB pathways in OKMS cells with MCF7 cells as the control. A) ERK, JNK, p38 are the key signaling molecules of three important MAPK pathways, and p65 is NFkB. B) Western blot signalling intensities normalized by that of GAPDH in OKMS cells.
Discussion
HER2 positive cells are potential progenitor cells of luminal A cells
OKMS#1 cells exhibited increased proliferation (Figure 4D), enhanced migration (Figures 4A, 4B), exemplified CSC percentage and self-renew abilities (Figures 4C, 4E, 4F). These phenotypic changes reflect a phenotype transition from luminal A (MCF7, where OKMS#1 was derived from) to a more malignant state that is similar to HER2 positive or triple negative breast cancer cells. Drug response curves showed that OKMS#1 shared a similar pattern with the HER2 positive cell line SKBR3 (Figure 6) but not triple negative cells. Thus, HER2 positive cells may be potential progenitor cells of luminal A cells, and could be controlled, in principle, using the same therapeutic strategies as the luminal A subtype and eliminated if coupled with appropriate approach rewiring cancer cell stemness.
Triple negative cells form a biologically distinct cell cohort among breast cancer cells and require distinct therapeutic strategies
As aforementioned, HER2 positive cells can ideally be switched to a less stem-like state to achieve desired outcome with hormonal therapies. Tumor subtype state transition has previously been implicated in melanoma through altering Stat3 expression [17]. However, such a strategy may not be feasible to treat triple negative breast cancers, which is phenotypically distinct from the other subtypes that have efficient therapies. This imposes further challenges to the appropriate control of triple negative breast cancers that still lacks safe yet effective treatment approaches [18]. Though being similar on patient survival, HER2 positive and triple negative cancers are distinct in how their CSCs are originated from, which must be reflected in their molecular profiles. Thus, comprehensive decomposition of the molecular features of triple negative cancers and comparison with that of HER2 positive cancers may lead to the identification of the unique path to the malignancy of triple negative breast cancer cells as compared with the other subtypes and, ultimately, the identification of novel approaches for the effective control of such cancers.
Significance
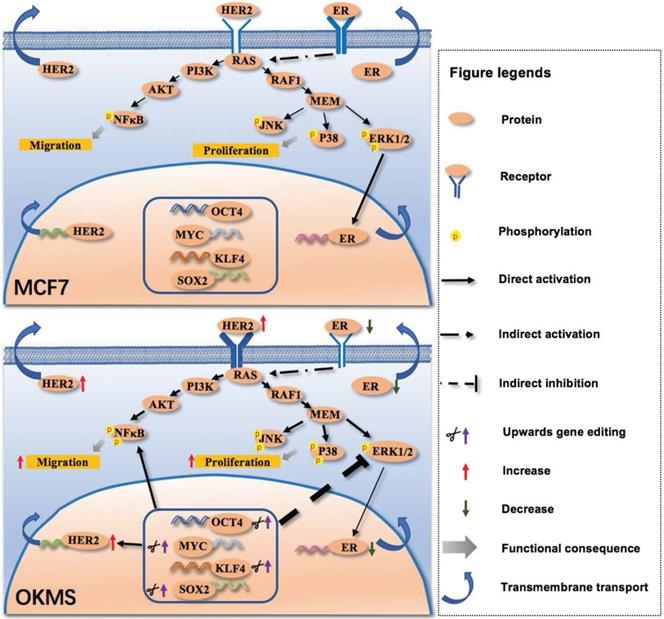
We have successfully designed an efficient multiplexing CRISPR/dCas9 system and applied it to study the phylogenetic relationships among breast cancer subtypes as stratified by cancer stemness. Concomitant up-regulation of OCT4, KLF4, MYC and SOX2 provides luminal A cells with HER2 positive-like features as observed both at the molecular profile and cellular behavior levels. This implicates that cell stemness controls the phenotypic transition between luminal A and HER2 positive breast cancer cells (Figure 7); and triple negative cells, though sharing similar cancer stemness with the HER2 positive subtype, represent a revolutionarily distinct disease from the other breast cancer subtypes that requires distinct therapeutic strategies for effective control.
Signaling network mediated by the four-gene panel controlling cell stemness in the transition of cancer cell states. The four-gene panel controlling cell stemness rewires cells from a luminal A-like feature to a HER2 positive-like feature through collective alterations in MAPK/ERK, MAPK/JNK, MAPK/p38 and NFkB signalings. Once the four-gene panel was activated, HER2 expression was enhanced, leading to increased phosphorylation of NFkB, JNK and p38, and decreased ERK. Elevated NFkB signaling, and MAPK/JNK and MAPK/p38 signalings result in enhanced cell migration and proliferation. Decreased ERK lead to decreased ER expression. Therefore, by concomitantly up-regulating the four-gene panel, HER2 is over-expressed, and ER is suppressed, leading to a state change from exhibiting luminal A like features to HER2 positive like features (namely OKMS cells). Phosphorylation status is shown only for molecules examined in our experiments. The thickness of each line or arrow represents the strength of the regulatory effect, with a more thick line or arrow representing a stronger effect. The thickness of each receptor represents the expression status of each receptor, with a more thick symbol representing a more represented status.
Supplementary Material
Supplementary tables.
Acknowledgements
This study was funded by the National Science and Technology Major project (Grant No. 2018ZX10302205-004-002), the National Natural Science Foundation of China (Grant No. 81972789), Natural Science Foundation of Jiangsu Province (Grant No. BK20161130), the Technology Development Funding of Wuxi (Grant No. WX18IVJN017), the Six Talent Peaks Project in Jiangsu Province (Grant No. SWYY-128), Major Project of Science and Technology in Henan Province (Grant No.161100311400), Research Funds for the Medical School of Jiangnan University ESI special cultivation project (Grant No. 1286010241170320). These funding sources have no role in the writing of the manuscript or the decision to submit it for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rahim F, Arjmand B, Shirbandi K. et al. Stem cell therapy for patients with diabetes: a systematic review and meta-analysis of metabolomics-based risks and benefits. Stem cell investigation. 2018;5:40
2. Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast cancer research: BCR. 2008;10:R25
3. Makena MR, Ranjan A, Thirumala V. et al. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochimica et biophysica acta Molecular basis of disease. 2018
4. Shen C, Hu G, Zhang S. et al. Immunophenotypic characterization of sphere-forming cells derived from the human renal cell carcinoma cell line 786-O. American journal of translational research. 2018;10:3978-90
5. Dai X, Xiang L, Li T. et al. Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes. Journal of Cancer. 2016;7:1281-94
6. Dai X, Li T, Bai Z. et al. Breast cancer intrinsic subtype classification, clinical use and future trends. American journal of cancer research. 2015;5:2929-43
7. Kim J, Pareja F, Weigelt B. et al. Prediction of Trastuzumab Benefit in HER2-Positive Breast Cancers: Is It in the Intrinsic Subtype? Journal of the National Cancer Institute. 2017;109:djw218
8. Ryden L, Landberg G, Stal O. et al. HER2 status in hormone receptor positive premenopausal primary breast cancer adds prognostic, but not tamoxifen treatment predictive, information. Breast cancer research and treatment. 2008;109:351-7
9. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
10. Beltran AS, Rivenbark AG, Richardson BT. et al. Generation of tumor-initiating cells by exogenous delivery of OCT4 transcription factor. Breast cancer research: BCR. 2011;13:R94
11. Corominas-Faja B, Cufi S, Oliveras-Ferraros C. et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell cycle. 2013;12:3109-24
12. Zhao Y, Li L, Zheng G. et al. CRISPR/dCas9-Mediated Multiplex Gene Repression in Streptomyces. Biotechnology journal. 2018;13:e1800121
13. Lowder LG, Paul JW 3rd, Qi Y. Multiplexed Transcriptional Activation or Repression in Plants Using CRISPR-dCas9-Based Systems. Methods in molecular biology. 2017;1629:167-84
14. Xie K, Minkenberg B, Yang Y. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:3570-5
15. Zhou HB, Liu JL, Zhou CY. et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat Neurosci. 2018;21:440-6
16. Feng YZ, Zhang QY, Fu MT. et al. Low expression of PinX1 is associated with malignant behavior in basal-like breast cancer. Oncology reports. 2017;38:109-19
17. Wang Y, Mou Y, Zhang H. et al. Reprogramming Factors Remodel Melanoma Cell Phenotype by Changing Stat3 Expression. International journal of medical sciences. 2017;14:1402-9
18. Hida AI, Sagara Y, Yotsumoto D. et al. Prognostic and predictive impacts of tumor-infiltrating lymphocytes differ between Triple-negative and HER2-positive breast cancers treated with standard systemic therapies. Breast cancer research and treatment. 2016;158:1-9
Author contact
![]() Corresponding author: Xiaofeng Dai, Email: xiaofeng.daiedu.cn, Mobile: +86 18168870169
Corresponding author: Xiaofeng Dai, Email: xiaofeng.daiedu.cn, Mobile: +86 18168870169