Impact Factor ISSN: 1837-9664
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
Genome structure of EBV
Classification and geographical...
Biological activities of...
Epstein Barr nuclear antigens...
Latent membrane protein (LMP)
EBV-microRNAs (EBV-miRNA)
Detection of EBV
Malignancies caused by EBV...
Burkitt's lymphoma (BL)
Hodgkin lymphoma (HL)
Post-transplant...
Nasopharyngeal cancer (NPC)
EBV-associated gastric cancer...
Extranodal nasal-type NK/T-cell...
Breast cancer
Summary and future studies
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(7):1737-1750. doi:10.7150/jca.37282 This issue Cite
Review
Epstein Barr Virus Associated Lymphomas and Epithelia Cancers in Humans
Richmond Ayee1,2, Maame Ekua Oforiwaa Ofori1, Edward Wright3, Osbourne Quaye1,2 ![]()
1. Department of Biochemistry, Cell and Molecular Biology, University of Ghana, Legon, Accra, Ghana
2. West African Center for Cell Biology of Infectious Pathogens (WACCBIP), University of Ghana, Legon, Accra, Ghana
3. Department of Biochemistry, University of Sussex, Brighton, U.K
Received 2019-6-3; Accepted 2019-12-1; Published 2020-1-17
Abstract
Epstein Barr virus (EBV) is a cosmopolitan oncogenic virus, infecting about 90% of the world's population and it is associated to tumors originating from both epithelia and hematopoietic cells. Transmission of the virus is mainly through oral secretions; however, transmission through organ transplantation and blood transfusion has been reported. In order to evade immune recognition, EBV establishes latent infection in B lymphocytes where it expresses limited sets of proteins called EBV transcription programs (ETPs), including six nuclear antigens (EBNAs), three latent membrane proteins (LMP), and untranslated RNA called EBV encoded RNA (EBER), shown to efficiently transform B cells into lymphoblastic cells. These programs undergo different patterns of expression which determine the occurrence of distinct types of latency in the pathogenesis of a particular tumor. Hematopoietic cell derived tumors include but not limited to Burkitt's lymphoma, Hodgkin lymphoma, post-transplant lymphoproliferative disorders, and natural killer (NK)/T cell lymphoma. EBV undergoes lytic infection in epithelia cells for amplification of the viral particle for transmission where it expresses lytic stage genes. However, for reasons yet to be unveiled, EBV switches from the expression of lytic stage genes to the expression of ETPs in epithelia cells. The expression of the ETPs lead to the transformation of epithelia cells into permanently proliferating cells, resulting in epithelia cell derived malignancies such as nasopharyngeal cancer, gastric cancer, and breast cancer. In this review, we have summarized the current updates on EBV associated epithelial and B cell-derived malignancies, and the role of EBV latency gene products in the pathogenesis of the cancers, and have suggested areas for future studies when considering therapeutic measures
Keywords: Epstein Barr virus, lymphomas, epithelial cancers, latency program
Introduction
Human gammaherpes virus 4 (HHV-4) or Epstein Barr virus (EBV) is a ubiquitous oncogenic virus belonging to the family Herpesviridae [1, 2], which is further classified into the subfamilies alpha, beta, and gammaherpes virus. EBV is a classical member of the subfamily Gammaherpesvirinae and among nine viruses that have been identified to solely infect humans [3, 4]. The virus was first discovered and isolated in cells from African Burkitt's lymphoma by Epstein Barr and Achong in 1964 [5, 6], and have been reported to establish latent asymptomatic infection in about 90% of the world's adult human population [7]. Socioeconomic and developmental factors have been shown to influence the age at which primary infection can occur. For instance, in Sub-Saharan African countries where standard of living is poor, primary infection occurs in early childhood and most infected children seroconvert by the age of 3 years, whereas in developed or affluent countries, primary infection is delayed until late childhood or young adulthood [8]. To establish primary infection, the virus is transmitted through oral route where it exhibits dual tropism by infecting two main physiological targets, epithelial cells and B lymphocytes [3]. In addition to infecting the epithelia and B cells, the virus has also been shown to infect unnatural targets such as T lymphocytes and natural killer (NK) cells [9].
Lytic replication of the virus occurs in the epithelial cells, but the virus can establish latency by infecting B cells found in the pharyngeal lymphoid tissues of the Waldeyer's ring [7, 10]. Upon entering the B cells, the viral genome either gets integrated into the host genome and persist as a provirus [11] or remain in the nucleus as a non-integrated circular episome and expresses restricted set of genes that drive latency and survival of the host cell [12, 13]. The expression of the latency stage genes, known as latency programs, in the B cells lead to B cell-derived lymphomas as a result of the transformation of the cells into lymphoblastic lines (Figure 1). The virus can be reactivated from latency in the B cells by a mechanism that is yet to be elucidated. In immunocompetent individuals, viral titres are held in check by EBV specific cytotoxic T cells [14]. Although EBV undergoes lytic replication in the epithelial cells, where lytic stage genes are expressed, the virus can switch to the expression of latency stage genes, and lead to the transformation of the epithelial cells into permanently proliferating cells and resulting in epithelial cell derived malignancies (Figure 1) [15].
Transformation of B lymphocytes and Epithelia cells into malignant cells by Epstein Barr virus (EBV). Epithelia and B lymphocytes are transformed by EBV into malignant cells as a result of expression of EBV latency gene products.
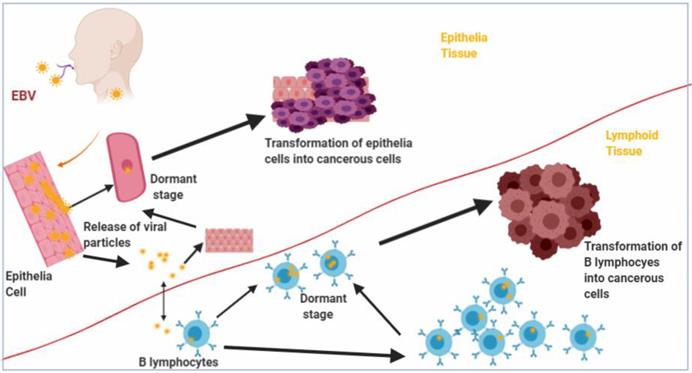
In this review, we have summarized the current updates on EBV associated epithelial and B cell derived malignancies, and the role of EBV latency gene products in the pathogenesis of the cancers. In addition, we suggested areas that can be explored by researchers in future studies when considering therapeutic measures against these malignancies.
Genome structure of EBV
The genome of EBV is a linear, double stranded DNA, approximately 172 Kbp with more than 85 protein coding genes (Open reading frames (ORF)) [16, 17]. The ORFs encode proteins involved in regulation of DNA replication and gene expression, and maintenance of genome integrity in daughter cells [18]. The nomenclatures of the ORFs were determined based on the Bam HI restriction fraction map in which the genes were in decreasing order according to their sizes [19]. The protein coding genes are divided into lytic and latent genes which play structural and non-structural roles. The viral genome is further divided into long and short unique sequence domains which are separated by a series of 0.5 Kbp terminal direct repeats that are found at the end of each sequence domain [20]. The terminal direct repeats increase the coding capacity of the genome and serve as a biomarker to determine the progenitor origin of EBV in the infected cells [17, 21].
Classification and geographical distribution of EBV
EBV can be classified into two main genotypes, A and B, or types 1 and 2, based on sequence divergence in the nuclear proteins, Epstein Barr Nuclear Antigen (EBNA)-1, -2, -3A, -3B, and 3C [22]. However, EBNA-2 is generally used for the classification because the protein has the least percentage of sequence homology between the two genotypes [23, 24]. The two EBV genotypes occur worldwide but vary in geographical distribution; EBV type 1 is found globally but predominant in American, Chinese, European and South-East Asian (SEA) populations whereas type 2 is predominantly found in Africa [25]. The two genotypes also vary in biological properties; EBV type 1 is more efficient in immortalizing B cells and the type 2 has a higher lytic ability [26, 27]. The virus is further classified into seven different strains: China 1 (C1), China 2 (C2), China 3 (C3), Mediterranean with (Med+) or without (Med-) deletions, Alaskan (AL), and North Carolina, based on nucleotide polymorphism, 30 bp deletions, or signature amino acid changes in the LMP1 gene as compared to the sequence of a prototype EBV, B95-8 [22, 28]. The different strains show diversity in tumorigenic activity; a 30 bp deletion at the carboxyl terminal, for example, has a higher tumorigenic activity and poor immunogenicity compared to the undeleted variant [29].
Biological activities of EBV-encoded latency gene products
Establishment of latent infection by EBV has been implicated in several malignancies [30] due to the expression of limited sets of latent proteins, shown to play various biological roles discussed in Table 1.
Biological activities of Epstein Barr virus latency stage gene products and associated cancers
| EBV latency protein | Type of latency | Biological activity | Associated cancersd |
|---|---|---|---|
| EBNA-1a | Latency I, II, III | Segregation of viral genome in progenies, DNA replication, inhibition of MHC class I, enhances p53 degradation | Burkitt lymphoma, Gastric cancer, Breast cancer |
| EBNA-2 | Latency III | Upregulation of host and viral proteins (transactivation), facilitate B cell immortalization | Posttransplant lymphoproliferative disorder |
| EBNA-3 | Latency III | Transcription transactivation of both host and viral proteins, immortalization of B cell | Posttransplant lymphoproliferative disorder |
| EBNA-LPb | Latency III | Transactivation of EBNA-2 to inactivate tumor suppressors, essential for immortalization of B cells | Posttransplant lymphoproliferative disorder |
| LMP-1/2c | Latency II/III | B cell survival, upregulation of antiapoptotic proteins, mimics CD 40 ligand associated signaling, constitutively activate growth and cell survival promoting signaling pathways | Hodgkin lymphoma, Nasopharyngeal cancer, Posttransplant lymphoproliferative disorder, T/NK cell lymphoma, Breast cancer |
| EBV-Micro RNAs | Latency I, II, III | Target host mRNAs involved in apoptosis, proliferation and transformation. Suppress antigen presentation and activation of immune cells | Gastric cancer, T/NK cell lymphoma, nasopharyngeal cancer |
a EBNA-1 is expressed and detected in all EBV associated malignancies. b EBNA-LP is also known as EBNA-5. c LMP-1/2 are both involved in epithelia and B cell tumors, however, LMP 2 is frequently detected in a majority of all tumors as compared to LMP-1. d The associated tumors are not only limited to the ones discussed in this review.
Epstein Barr nuclear antigens (EBNAs)
Six EBNAs (1, 2, 3A, 3B, 3C and LP) are expressed during latent infection in host cells, and their biological functions have been reported by researchers worldwide [31, 32]. Figure 2 shows the biological activities of EBV nuclear antigens (EBNA) in tumorigenesis. The expression of these antigens varies across the latency programs establish by EBV in infected cells. During latency III, all the six EBNAs are expressed, however in latency II and I, only one EBNA (EBNA-1) is expressed, but no EBNA protein is expressed during latency 0 [8, 32]. All the EBNAs are involved in regulation of transcription and also participate in transcriptional activation of viral LMP genes (LMP-1 and -2) in infected cells [33, 34].
Biological activities of EBV nuclear antigens (EBNAs) in tumorigenesis. EBNA-1 inhibits antigen presentation by major histocompatibility complex (MHC) class I; EBNA-1 and EBNA-2 activate the expression of LMP-1; EBNA-2 and EBNA-3 interact with C-myc which constitutively activate cyclin D/E leading to unregulated cell proliferation; EBNA-LP can directly activate cyclin D/E and DNA-dependent protein kinase (DNA-PKCs) to promote cell proliferation; EBNA-LP promotes cell survival by interacting with antiapoptotic protein hematopoietic cell-specific protein 1 (HS-1)-associated protein X-1 (HAX-1).
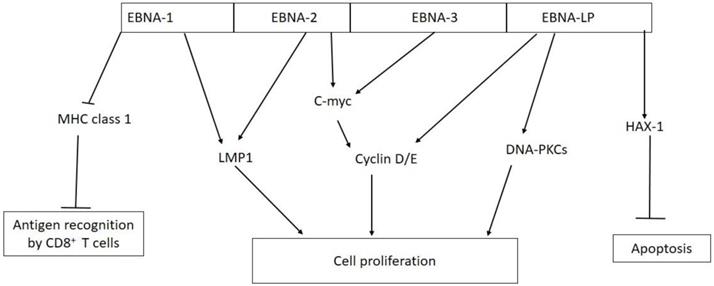
EBNA-1 is a sequence specific DNA-binding protein that interacts with three unique palindromic sequence target sites (family of repeat elements (FR), dyad symmetry element (DS), and sequences found downstream Q promoter (Qp)), which are repeated multiple times on the viral genome [35]. EBNA-1 binds to FR elements, which act as enhancer for viral C promoter, to direct the transcription of all the six EBNAs. Whereas engaging the DS elements by EBNA-1 leads to regulation of S-phase associated viral DNA replication, interaction with Qp down-regulates the transcription of Qp-driven EBNA-1 [36, 37]. EBNA-1 also ensures the segregation of the viral genome in daughter cells and up-regulates LMP promoter, sustaining cell survival or immortalization of infected cells [32]. A more recent study has reported that EBNA-1 derived from nasopharyngeal cancer is required for the maintenance of EBV episome and DNA replication during latent infection [38]. EBNA-1 protein is made up of N and C termini which are separated from each other by variable repeats of glycine-alanine sequences. The amino acid sequence repeats hide EBNA-1 from immune recognition by a cis-acting inhibitor of major histocompatibility complex (MHC) class I and preventing antigen presentation through ubiquitin- proteosome pathway [39, 40].
Unlike EBNA-1 which binds to DNA, EBNA-2 does not interact with DNA but engage cellular transcription factors such as CBF1/RBP-Jk, PU.1, and other proteins to up-regulate the expression of both viral and cellular genes [41]. The cellular genes that are up-regulated by EBNA-2 include CD 23 and c-myc found in B cells. Activation of c-myc leads to expression of proteins such as D-type cyclins and cyclin E, which are associated with cell division [42]. EBNA-2 also interacts with other transcription factors such as Cp, LMP-1, and -2 binding factors for up-regulation of viral genes that are responsible for immortalization of B cells [41, 43, 44].
EBNA-3 family of nuclear proteins consist of three large nuclear phosphoproteins, EBNA-3A, -3B, and -3C [44]. All the EBNA-3 proteins share few sequence homologies at the N terminus region, with the conserved domain used to engage cellular transcription factors such as CBF1/RBP-Jk [45]. By binding to CBF1/RBP-Jk, EBNA-3 acts as a repressor of EBNA-2-mediated transactivation of cellular and viral proteins [46]. In vitro genetic studies have shown that only EBNA-3A and -3C are required for immortalization of EBV-infected cells [47, 48]. Although EBNA-3 B is dispensable, a report has shown that the protein regulates B cell homing by altering the expression of chemokine receptor 4 (CXCR4) [49].
Epstein Barr nuclear antigen leader protein (EBNA-LP) also known as EBNA-5 is a nuclear phosphoprotein that is co-expressed with EBNA-2 upon EBV infection of target cells [50]. The co-expression of EBNA-LP and EBNA-2 has been shown to enhance EBNA-2 transcriptional activation of both cellular and viral proteins [44, 51]. EBNA-LP plays a key role in B cell transformation into a permanently proliferating lymphoblast based on the observation that cells expressing the mutant genes were less transformed as compared to cells with wild type EBNA-LP gene [52, 53]. The leader protein interacts with cellular proteins, some of which are oncogenic and tumor suppressors (pRb, p53, p14ARF, and Fte1/S3a), cell cycle regulators (DNA-PKcs and HA95), and an anti-apoptotic protein (HAX-1) [54-56]. EBNA-LP up-regulates the expression of thymus and activation regulated chemokine (TARC) gene, which has been proposed to play a key role in B cell transformation and survival [57, 58]. Previous studies have reported that EBNA-LP stimulates EBNA-2 transactivation of viral proteins such as latent membrane proteins 1 and -2B (LMP-1 and -2B) in immortalized cells [51, 59, 60].
Latent membrane protein (LMP)
Three latent membrane proteins, LMP-1-2A, and -2B, are expressed by EBV during latency II and III in EBV infected cells [61]. All the three proteins are expressed as cell surface membrane proteins and are required for survival and transformation of the infected cells into permanently proliferating cells (Figure 3).
Biological activities of latent membrane protein-1(LMP-1) in tumorigenesis. EBV LMP-1 activates cellular pathways that lead to tumor invasiveness and metastasis, cell proliferation, and inhibition of apoptosis.
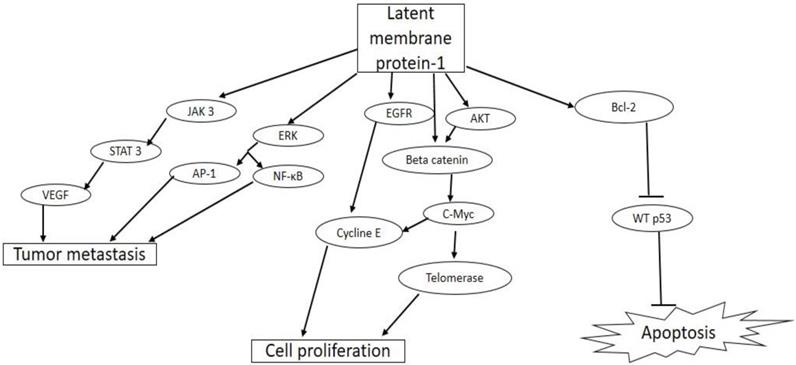
LMP-1 gene is an essential oncogene, which is expressed as a constitutively active receptor in a majority of EBV associated tumor cells [62]. LMP-1 is a 356-amino acid protein with a short cytoplasmic N-terminal domain, six transmembrane spanning domains, and a 200 amino acid long C-terminal cytoplasmic domain [63]. The N-terminal domain tethers the LMP-1 on the surface of the plasma membrane, and oligomerization as well as self-aggregation of the protein is mediated by the six trans-membrane spanning domain. The C-terminal is the biologically active region of LMP-1, with two functional domains, namely, C-terminal activating regions (CTAR)-1 and -2, which are needed for efficient EBV-mediated B cell transformation [64]. Previous reports have shown that these two functional domains interact with intracellular signaling proteins such as tumor necrosis factor (TNF) receptor-associated factor (TRAF)-1, -2, -3, -4, -5, and -6 found in tumor necrosis factor receptor (TNFR) signaling pathway in B cells and epithelia cells [65, 66]. By interacting with these adaptor molecules, CTAR constitutively activates major signaling pathways such as the extracellular signal-regulated kinase (ERK), mitogen-activated protein kinases (MAPKs), PI3K/Akt, AP-1, Jun N-terminal protein kinase (JNK), the p38 and canonical and non-canonical NF-κB, and JAK/STAT pathways [67-69]. The activation of these major signaling pathways results in the up-regulation of expression of antiapoptotic proteins such as A 20, Bcl-2, and down-regulation of tumor suppressor protein, p53, promoting cell growth and survival [70, 71]. LMP-1 activates the Janus kinase 3 (JAK3)/Signal transducer and activator of transcription 3 (STAT3) pathway, which increases the expression of vascular endothelial growth factor (VEGF) and promotes tumor metastasis and invasiveness [72]. LMP-1 also promotes proliferation of EBV infected cells through the activation of epidermal growth factor receptor (EGFR) and AKT/protein kinase B signaling pathways with downstream activation of cyclin E and telomerase, respectively [73]. A more recent study has shown that the activation of AP-1, JAK/STAT, and NF-κB pathways by CTAR resulted in increased expression of programmed cell death protein 1 ligand (PD-L1), a key immune checkpoint suppressor in tumor immunology [74].
LMP-2 is made of two variants, LMP-2A and -2B, which are expressed during latency II and III in EBV infected cells [8]. Structurally, both proteins have 12 trans-membrane domains and a 27 amino acid cytoplasmic C-terminal domain, however, LMP-2A has an additional 119 amino acid cytoplasmic N-terminal domain, which is not found in LMP-2B [75]. The N-terminal cytoplasmic domain of LMP-2A contains immunoreceptor tyrosine-based activation motif (ITAM), which mediate B cell receptor (BCR)-like signaling cascade in EBV infected cells [76, 77]. The BCR-like signaling, mediated by LMP-2A, provides strong survival signal which rescues BCR-negative cells from apoptosis [78, 79] and inhibit signaling that leads to lytic reactivation [80]. LMP-2A has been shown to be essential for activation, proliferation, and survival of EBV infected B cells at early times, after which it is required for long term growth of B cells [81]. Further study has shown that LMP-2A helps EBV infected cells to escape elimination by CD8+ T cells by down regulating (i) the expression of CD8+ T cells specific latent stage genes, especially EBNA-1, (ii) the expression of MHC class 1 proteins to prevent antigen presentation, and (iii) the expression of coactivatory receptor NKG2D to evade T cell recognition [82]. The function of LMP-2B on the other hand, is not clearly known [81], but a previous report has shown that it interacts with LMP-2A to modulate the activities of the latter [75].
EBV-microRNAs (EBV-miRNA)
MicroRNAs (miRNA) are small non-coding RNA molecules, usually 18-24 nucleotides in length, which transcriptionally down-regulate expression of complementary mRNAs [83]. miRNAs have been identified in different organisms such as mammals, plants, algae and worms, and EBV was one of the first viruses that was identified to express these small non-coding RNA molecules [84, 85]. The virus has been found to express 44 miRNAs in all forms of latency and tumor tissues, regulating both cellular and viral genes [86]. The 44 miRNAs are produced from two miRNA coding regions: the BamHI fragment H rightward open reading frame 1 (BHRF1) region which encoded 4 miRNAs, and the BamHI A rightward transcript (BART) region which encodes the remaining 40 miRNAs [87]. Since EBV miRNAs are detected in EBV associated tumors, it is thought that they play important roles in the pathobiology of the life cycle of EBV and tumors associated to EBV [88]. miRNAs help the virus to escape immune surveillance by down-regulating the expression of both immunogenic viral antigens and host immune proteins [83]. For instance, Lung and colleagues in 2009 reported that BART 22, encoded by the BART miRNA region, down-regulated the expression of LMP-2A, an immunogenic viral protein, and enhanced NPC tumorigenesis [89]. Impotin 7 (IPO7), which is a cellular receptor responsible for transporting transcription factors into the nucleus and plays a key role in innate immunity, has been shown to be inhibited by EBV miR-BART3 and miR-BART16 [90, 91], and miR-BART2-5p protects EBV-infected cells from immune recognition by natural killer (NK) cells through the inhibition of major histocompatibility complex (MHC) class I chain-related molecule B (MICB) [92, 93]. Other studies have also demonstrated that EBV miRNA BART 16 disrupt Type I IFN signaling pathway by down-regulating the expression of CREB-binding protein, a transcriptional activator of IFN, and hence suppressing host immune response against the virus [94], and EBV produces BART6-3p which interacts with miR 97, a cellular microRNA, to synergistically suppress the expression of interleukin-6 receptor (IL-6R), which is a receptor for several antiviral cytokines such as IFN-α, IL-12 and IL-27 [95], to promote tumor progression in both epithelia cancers and lymphomas.
Aside targeting cellular cytokines, EBV miRNAs inhibit the destruction of B cell and epithelia cell derived cancers by down-regulating expression of tumor suppressor genes; miR-BHRF1-2 has been reported to silence the expression of PRDM1/Blimp1which is a tumor suppressor in B cells, hence promoting lymphogenesis [96]. The virus also targets the mitogen-activated protein kinase (MAPK) signaling pathway, which suppresses tumor formation by producing BART 22 and shown to inhibit MAP kinase kinase kinase 5 (MAP3K5) expression [97].
Detection of EBV
EBV is implicated in several malignancies ranging from epithelial and lymphoid origins. Biochemical, serological, and molecular methods to detect EBV have increasingly become necessary in the diagnosis and monitoring of patients with EBV-associated diseases [98]. Serological assays, that are used to identify the virus in infected individuals, involve the detection of antibodies against EBV specific antigens such as viral coat antigen (VCA), EBNA-1, early antigen (EA) and viral nucleic acids [99-101]. Immunohistochemistry, which involves the staining of key EBV latency proteins such as LMP-1, LMP-2A, EBNA-1, and -2 in tumor biopsies, is used to confirm the presence of the virus and distinguish between EBV-associated and non EBV-associated tumors [102]. Epstein Barr encoded RNA (EBER) in situ hybridization (EISH) assay is considered as the gold standard for the detection and diagnosis of EBV infection [103]. The EISH technique employ the use of nucleic acid probes, either label or unlabeled, which can hybridize with EBER on paraffin sections of EBV infected tissues [104]. EBER is a small non-coding and non-polyadenylated RNA which is highly expressed in all EBV latently infected cells, irrespective of cell phenotype. Detection and quantification of EBV nucleic acids in body fluids and tissues using polymerase chain reaction (PCR) have also been exploited for diagnosing and monitoring of EBV-associated diseases [105].
Malignancies caused by EBV infection
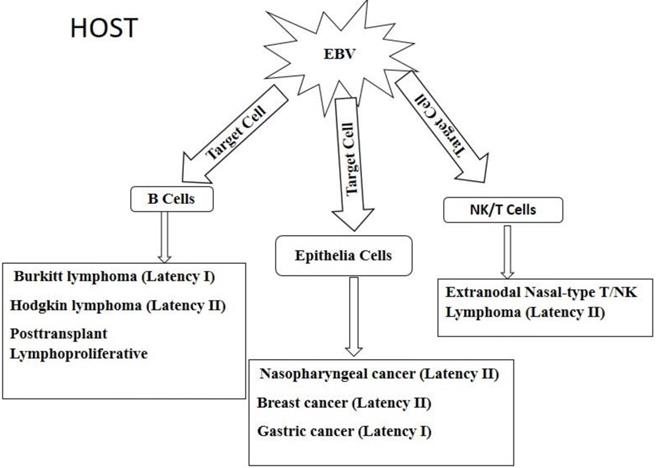
EBV is linked to the pathogenesis of lymphomas such as Burkitt's lymphoma (BL), Hodgkin lymphoma (HL) and post-transplant lympho-proliferative disorder (PTLD). Epithelial malignancies that are associated with EBV in epithelia cells include nasopharyngeal cancer (NPC), gastric cancer (GC) and breast cancer (BC) [34]. In rare cases, EBV has been shown to infect NK and T cells; causing extranodal nasal-type NK/T cell lymphoma [106]. The different malignancies are shown in Figure 4. Upon entering the host, EBV has tropism for two main targets, namely B cell and epithelia cells as well as unnatural targets such as NK/T cells. The virus in the various target cells expresses different patterns of latency genes and these determines the type of cancer that would be developed. B cell derived lymphomas include Burkitt lymphoma, Hodgkin lymphoma, and posttransplant lymphoproliferative disorders which are caused by the expression of latency I, II, and III, respectively, gene products. Infection of epithelia cells by EBV results in cancers such as nasopharyngeal, breast, and gastric cancers, caused by the expression of latency II, II, I, respectively, gene products. The virus can also invade unusual targets such as natural killer (NK) cells and T lymphocytes to cause extranodal nasal-type T/NK lymphoma due to the expression of latency II gene products. Full details of each tumor are further described below.
Diagrammatic view of EBV associated epithelia and hematopoietic cell derived malignancies.
Burkitt's lymphoma (BL)
Burkitt's lymphoma (BL) is a highly aggressive non-Hodgkin B cell neoplasm and is the world's commonest pediatric cancer; endemic in Sub-Saharan Africa [107]. The pathogenesis of BL has been linked to EBV infection as a result of the isolation of the virus from cultured BL cell lines in 1964 [5]. The virus has since been implicated in 95% of BL cases from high risk regions and less than 30% from low risk regions [108]. BL occurs in children living in areas that are both holo-endemic and hyper-endemic for malaria and suggesting that Plasmodium falciparum plays a role in the etiology of the cancer [109, 110]. BL is classified into three different forms based on clinical observations and disease epidemiology [111]. Endemic BL (eBL) has an annual incidence of about 5-10 cases per 100 000 and contributes 50% of all pediatric cancers in malaria endemic regions such as Equatorial Africa and Papua New Guinea, where EBV is found in about 95% of all diagnosed cases [112, 113]. Common sites of tumor occurrence of eBL are the jaws and abdomen [114]. Sporadic Burkitt's lymphoma (sBL), on the other hand, has a wide global distribution, but is at a much lower frequency and mostly diagnose in both children and young adult. In US and Western Europe, sBL is responsible for 30-50% of all pediatric neoplasms and less than 1% in adult [115]. EBV is rarely linked to sBL cases diagnosed in the western world, however in other places like North East Brazil, the frequency of EBV in sBL exceeds 80% [7, 116]. In contrast, immunodeficiency-associated Burkitt's lymphoma (iBL) has been found in HIV carriers who develop the lymphoma before progressing to AIDS. The incidence of iBL is 10 to 100-fold higher than the sporadic form of the disease, and about 30-40% of iBL are positive for EBV [117]. Immune compromise, which occurs as a result of HIV infection, accounts for reactivation of EBV from latently infected B cells and eventually causing rapid progression to iBL [118]. CD4+ T cells prime CD8+ T cells against circulating EBV in immunocompetent individuals, and thereby bringing the virus under immune surveillance. However, in HIV infected individuals the CD4+ T cell count is drastically reduced, and therefore reduces the priming of the EBV specific CD8+ T cells in these individuals [119, 120].
The expression of EBV genes in BL is strictly latent and restricted to the type I latency default programs. Only EBNA-1 and EBER are expressed in EBV positive cells in BL tumors [121]. Whiles some studies have reported the detection of LMP-1 and EBNA-2 in rare cases of eBL and LMP-1 in many sBL cancers, these detections were not observed in all occurrences [122-124]. Even though the role of EBV in the pathogenesis of BL is still unclear, evidence have shown that the expression of EBNA-1 in BL cell lines promotes cell proliferation by inhibiting apoptosis [125]. Recent reports suggest that EBNA-1 inhibits apoptosis in BL cell lines by interacting with host proteins such as survivin, an anti-apoptotic protein and p53-regulator USP7 [38, 126].
All BL tumors, regardless of variants or EBV status, are morphologically and immunophenotypically identical and are related by sharing a general gene expression patterns which resemble that of centroblasts [127, 128]. The BLs, in general, undergo chromosomal translocation of c-myc proto-oncogene on chromosome 8 or on chromosome 14 to one of the three immunoglobin loci, notably the heavy chain immunoglobin (Ig) [129]. This brings the c-myc under the control of highly active Ig gene promoter leading to constitutive expression of c-myc proteins at high levels in BL cells. The expression results in uncontrolled cell growth in BL [130].
Hodgkin lymphoma (HL)
Hodgkin lymphoma (HL) is a lymphoid neoplasm which originates from B cell and characterized by the presence of few multinucleated giant cells known as Hodgkin/Reed Sternberg (HRS) cells, which are surrounded by a non-neoplastic inflammatory infiltrate [131]. The incidence of HL varies depending on geographical distribution, sex, ethnicity, and socioeconomic status; the incidence is higher in developed countries compared to under-developed ones. [132]. There is however lower mortality due to HL in developed countries compared to the less developed ones; an observation that has been ascribed to improved health facilities in the developed countries [133]. About 30-40% of HL cases in North America and Europe were reported to be EBV positive, while in other parts of the world such as Latin America, Asia and Africa, EBV was found in almost 100% of all HL cases [134]. Despite the association of EBV to HL cases, the role of EBV in the pathogenesis of this malignancy remains unknown [135].
There are two variants of HL based on clinical and morphological differences [136]. The first variant, known as nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), is rarely EBV associated [137], hence would not be discussed in this review. The second variant, known as classical Hodgkin lymphoma (CHL), has been reported to account for about 95% of all HL cases and is mostly linked to EBV [138]. Association between EBV and CHL has been suggested due to elevated antibody titres against EBV latent antigens, which proceeded the development of the lymphoma after several years of infection [139]. Detection of EBV latent stage genes and RNA from biopsies of CHL patients was further used to confirm the association between the disease and EBV [140]. CHL can further be divided into four histological subtypes, namely, mixed cellularity (MC), nodular sclerosis (NS), lymphocyte-rich (LR), and lymphocyte depleted (LD) [138]. About 96% of all MC cases are EBV associated, whereas NS is less frequently linked to EBV infection [141]. On the other hand, LR and LD are rarely associated with EBV [142].
Post-transplant lymphoproliferative disorder (PTLD)
Post-transplant lymphoproliferative disorder (PTLD) is a type of malignancy with life-threatening complications in transplant recipients of both solid organ and hematopoietic stem cell allografts [143]. The lymphoma occurs as a result of increase proliferation of B cells after transplant, and it is mostly EBV driven [144]. The genome of the virus has been found in more than 90% of B cells from PTLD patients, either after primary infection, or reactivation of EBV from latently infected cells after treatment with immunosuppressant to avoid allograft rejection [145]. The use of immune suppressive drugs after organ transplant reduces the number and function of T cell to influence the success of the transplant. The use of the immunosuppressant leads to depletion of EBV-specific T cells and disruption of the balance between the immune system and latent virus, and consequently, there is reactivation of EBV from latency [146]. The impairment of EBV-specific T cell mediated immune surveillance results in uncontrolled lymphoproliferative blast, which eventually causes PTLD in transplant recipients [147, 148].
The prevalence of EBV- associated PTLD ranges from 1-20%, with incidence varying according to the type of allograft, age, and pretransplant EBV-serostatus of transplant recipient. PTLD cases are highest (32%) in small intestine transplant recipients, and 3-12% incidence is observed in heart, lung, liver and pancreas transplant recipients. The lowest incidence (1-2%) is reported in renal transplant recipients [146, 149]. At the ages of 5, 18, and 40, about 50, 20, 5-10% of the general population, respectively, have not been exposed to EBV, hence are seronegative. These groups of people develop primary infection after receiving transplant from a donor who is EBV seropositive, predisposing them to PTLD [150, 151]. The overall incidence of PTLD is found to be higher in children than in adults due to increased pretransplant EBV seronegative status among children [146].
Nasopharyngeal cancer (NPC)
Nasopharyngeal cancer (NPC) is a malignant tumor of epithelia squamous cell which arises from the lateral wall of nasopharynx, notably, fossa of Rosenmüller and superior posterior wall [152]. Globally, the disease shows remarkable variation in ethnic and geographical distribution, with about 80% of a total of 65,000 new cases that are reported worldwide, occurring in Southern China and Southeast Asia. [153]. The annual incidence of NPC in Southern China varies between 20-30 per 100,000 person-year among Cantonese living in Hong Kong and Guangdong province, while the incidence in other parts of the world, such as Europe and the United State, are below 1 per 100,000 persons/year [154, 155]. In Africa, specifically, Northern Africa, the incidence of NPC varies between 5 to 7 per 100 000 persons/year [156]. Based on histology, the World Health Organization (WHO) classified NPC into types I, II, and III, which are known as keratinizing squamous cell, differentiated non-keratinizing, and undifferentiated non-keratinizing carcinomas, respectively [157]. An alternatively simpler system of classification has been proposed which groups NPC into two histological variants, namely squamous cell carcinomas (SCCs) and undifferentiated carcinomas of the nasopharyngeal type (UCNT) [158]. In a non-endemic region like North America, about 63% of all NPC cases are UCNT, while in Southern China, about 95% of all the cases are UCNT [159].
EBV, which is the main cause of UCNT, has been classified as group 1 carcinogenic agent by the International Agency for Research and Cancer (IARC) [22]. The viral infection in NPC epithelia cells is clonal in origin; developing from clonal proliferation of single EBV infected epithelia cell. NPC tumors express three EBV latency type II gene products (LMP-1, LMP-2, EBNA-1), which are found in EBV implicated epithelial cancers, in addition to the expression of viral encoded small RNAs such as EBER and microRNAs (miRNAs) [22, 100, 160]. Of all EBV latency gene products, LMP-1 has been found in about two-thirds of NPC cases, indicating the key role this protein plays in tumorigenesis.
As previously mentioned, the infection of epithelia cells by EBV is mainly lytic, and hence default lytic programs are expressed. However, the switch to latency default programs during epithelia infection by EBV represents a key step in the pathogenesis of NPC, although some studies have reported the involvement of both lytic and latent stages EBV genes in transformation of epithelia cells, the role of lytic genes remains unclear [15, 161, 162].
EBV-associated gastric cancer (EBVaGC)
Gastric cancer (GC) is the third leading cause of cancer-related mortality globally, with a worldwide annual incidence of over 950 000 cases. EBV-associated gastric cancer (EBVaGC) accounts for about 10% of all GC reported cases [163, 164]. The incidence of EBVaGC shows variation in geographical distribution across the globe, with a pooled estimate in Europe, Asia, and North and South America, being 9.2%, 8.3% and 9.9%, respectively (Huang et al., 2014), whereas in Africa, a country like Zambia has a frequency of 23% [165]. EBVaGC is classified into three histological subtypes namely: lymphoepithelioma-like carcinoma (LELC)-type, conventional type adenocarcinoma (CA)-type, and carcinoma Crohn's disease- like lymphoid reaction (CLR)-type [166]. The LELC-type is a poorly differentiated carcinoma with dense infiltration of lymphocytes, similar to that of NPC. More than 80% of EBVaGCs show LELC-type morphology [167]. CA-type on the other hand, morphologically resembles EBV negative GCs by infiltration of variable lymphocytes with prominent desmoplasia in the absence of lymphoid follicles [168]. CLR-type is characterized by the presence of three or more lymphoid follicle with active germinal centers located at the advancing edge of the neoplasm [168].
One of the unique features which distinguishes EBVaGC from non-EBVaGC is the monoclonal proliferation of gastric epithelia cells latently infected with EBV and suggesting the presence of the virus in the early stages of tumorigenesis [169]. In contrast to EBV-negative GCs, which mainly occur in the antrum as the predominant pathological site, EBVaGC occurs predominantly in the proximal stomach including the cardia, fundus and body [168]. At early stages of tumorigenesis, EBVaGC forms a well-defined nodular ulcer in the submucosa with less fibrosis as compared to non-EBVaGCs, and this pathological feature is essential for endoscopic submucosal resection of the tumor [170].
The genome of EBV, mainly Latency I default programs (EBER, EBNA-1, and BART), was found in the GC cells and adjacent dysplastic epithelium cells, but could not be found in surrounding lymphocytes, stromal cells, normal mucosa, and intestinal metaplasia, and therefore suggests a key role of EBV in the pathogenesis of GC [171].
Extranodal nasal-type NK/T-cell lymphoma
Extranodal nasal-type NK/T lymphomas are infrequent tumors that are regularly associated with EBV infection [172]. The tumor is characterized by extensive angio-invasion and necrosis in the upper aerodigestive tract, including the nasal cavity, nasopharynx, paranasal sinus and the palate [173]. The disease shows remarkable variation in geographical and sex distribution, with the highest incidence occurring in East Asia, Mexico, and South/Central America [174]. The lymphoma has been shown to be predominant among men than woman, with 2-3:1 ratio of male to female, and the mean age at diagnosis being 50 years [175, 176].
EBV expresses type II latency programs (EBNA-1, LMP-1 and -2) in the extranodal NK/T-cell lymphomas, and the genome of the virus exists as a clonal episome in the infected cells [177]. The expression of these genes has been shown to constitutively activate several signaling pathways including JAK/STAT and NF-κB, to promote cell growth and survival [178]. LMP-1 expression in extranodal NK/T-cell lymphomas has also been reported to promote tumor immune escape through the up-regulation of programmed cell death receptor 1 (PD-1) and PD ligand 1, which are both important immune checkpoint molecules in tumor immunology [179].
Genome wide analysis have reported chromosomal abnormalities, notably, deletions in 6q21 in extranodal NK/T-cell lymphomas [180], which leads to the loss of expression of many tumor suppressor genes such as ATG5, PRDM1, FOXO3, AIM1, and HACE 1. For instance, the loss of FOXO3 and HACE1 in EBV-positive NK/T-cell lymphomas result in apoptosis resistance through the prevention of BIM and PUMA induction, and the suppression of TNF-driven NF-κB activation, respectively [181].
Breast cancer
Breast cancer is the most frequently diagnosed female malignancy globally and accounts for about 25% of incident cancer cases among women [182]. In underdeveloped regions such as sub-Saharan Africa, frequency of breast cancer is relatively lower, characterized by aggressive course and targets female of younger age compared to the Western World [183].
While genetic factors, such as inheritance of breast cancer associated gene 1 and 2 (BRCA1/2) and human epidermal growth factor receptor 2 (HER2), in addition to non-genetic factors, have been shown to influence the development of breast cancer, the etiology remains unknown [184]. Viral agents including EBV, mouse mammary virus (MMV), human papilloma virus (HPV), and cytomegalovirus have been implicated in breast cancer cases [185-187]. Association between EBV and breast cancer have been reported from different parts of the world including Asia [188], Africa [189], and Europe [190], with a prevalence of 30-50%. Subsequently, EBV latency II gene products such as LMP-1, -2, EBNA-, and EBER have been detected in breast cancer cells [102, 191, 192], and a meta-epidemiological study have also shown that EBV infection was highly associated with the risk of breast cancer development [193].
In keeping with the association between EBV and breast cancer, a proposed potential mechanism for the transformation of mammary epithelia cells (MEC) suggests that EBV infects cells that express CD21 as cell surface receptor, leading to tumorigenesis that occurs through LMP-1 mediated activation of c-MET signaling pathway [194].
Summary and future studies
Epstein Barr virus is an oncogenic virus with global distribution and infecting about 90% of the world's population. Humans are the only natural host of EBV, and the virus is transmitted by ingesting infected saliva. In the host, the virus has tropism for two main cells, namely epithelia cells and B lymphocytes, although unnatural targets such as natural killer cells and T lymphocytes have been shown to contain the virus. The presence of EBV in B cells has been linked to lymphomas, which include Burkitt's lymphoma, Hodgkin lymphoma, and posttransplant lymphoproliferative disorders. On the other hand, infection of epithelia cells is implicated in the pathogenesis of epithelia cell derived malignancies such as nasopharyngeal cancer; EBV associated gastric cancer, and breast cancer. The pathogenesis of both B cell and epithelia cell derived neoplasms has been due to the expression of EBV transcription programs or latency stage genes. The programs are divided into latency I (EBNA-1, EBER, and BARF0), latency II (EBNA-1, EBER, LMP-1, and -2), and latency III (EBER, EBNA-1,-2,-3,-4,-5, and LMP-1 and -2A), and their expression varies depending on the differentiation, type, and activation status of the target cell. Latency I programs are implicated in Burkitt's lymphoma and gastric cancer, whereas the expression of latency II default programs is linked to nasopharyngeal cancer, Hodgkin lymphoma, and NK/T-cell lymphomas. The pathogenesis of posttransplant lymphoproliferative disorders has however been associated with the expression of latency III default programs. Tumorigenesis occurs by (1) upregulating the expression of antiapoptotic genes, (2) constitutive activation of major intracellular signaling pathways responsible for cell growth and survival, and (3) creation of tumor microenvironment for malignant cells to escape immune recognition.
EBV have been linked to the pathogenesis of different malignancies, but the mechanism underlying the pathogenesis has not been fully elucidated. Host and environmental factors are thought to play key roles in the malignancies, making it difficult to clearly map out the mechanism involved in the pathogenesis of EBV-associated cancers. With current progress in genomic research, future studies should focus on how host genes and environmental factors interact with viral genes, and how the interaction influences the pathogenesis of EBV-associated malignancies. Due to overlapping symptoms, many EBV implicated cancers are only diagnosed at the advance stage of the disease. Genome-wide association studies and whole genome sequencing of large population of patients are therefore recommended to identify polymorphism in genes which predisposes individuals to the above mentioned EBV-associated cancers. Results from these studies will help identify high risk-populations for further prognostic evolution and increase the chances of detecting the cancers at early stages of development. Future studies must be done on anticancer drugs that selectively regulate the expression of EBV oncogenes involved in antiapoptosis, cell proliferation and invasion. Finally, selective inhibition of various signaling pathways activated by EBV proteins, offers promising anticancer therapy. All the proposed recommended studies will help understand the disease mechanism, and ultimately lead to the development of therapeutic agents that will help slow down disease progression and increase survival.
Abbreviations
BC: Breast cancer; BL: Burkitt's lymphoma; EA: Early Antigen; EBER: Epstein Barr encoded RNA; EBNA: Epstein Barr nuclear antigen; EBV: Epstein Barr Virus; EBVaGC: Epstein Barr associated gastric cancer; GC: Gastric cancer; HHV-4: Human gamma herpes virus 4; HL: Hodgkin lymphoma; IHC: Immunohistochemistry; LMP: Latent membrane protein; NPC: Nasopharyngeal cancer; ORF: Open reading frame; PBMC: Peripheral blood mononuclear cells; PTLD: Post-transplant lymphoproliferative disorder; VCA: Viral coat antigen.
Acknowledgements
Richmond Ayee was supported by a WACCBIP-World Bank ACE M.Phil. fellowship (ACE02-WACCBIP: Awandare) and a DELTAS Africa grant (DEL-15-007: Awandare). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (107755/Z/15/Z: Awandare) and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of the World Bank, AAS, NEPAD Agency, Wellcome Trust or the UK government.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Schafer G, Blumenthal MJ, Katz AA. Interaction of human tumor viruses with host cell surface receptors and cell entry. Viruses. 2015;7:2592-617
2. Chang CM, Kelly JY, Mbulaiteye SM. et al. The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: a need for reappraisal. Virus research. 2009;143:209-21
3. Longnecker RM, Kieff E, Cohen JI. Epstein-barr virus. Fields Virology: Sixth Edition: Wolters Kluwer Health Adis (ESP). 2013
4. Sathiyamoorthy K, Hu YX, Möhl BS. et al. Structural basis for Epstein-Barr virus host cell tropism mediated by gp42 and gHgL entry glycoproteins. Nature communications. 2016;7:13557
5. Epstein MA. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1:702-3
6. Esau D. Viral causes of lymphoma: the history of Epstein-Barr virus and human T-lymphotropic virus 1. Virology: research and treatment. 2017;8:1178122X17731772
7. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Phil Trans R Soc B. 2017;372:20160271
8. Rickinson A. Epstein-Barr Virus and its replication. Virology. 2006;2:2603-54
9. Saha A, Robertson ES. Epstein-Barr virus associated B-cell lymphomas: pathogenesis and clinical outcomes. Clinical Cancer Research. 2011 clincanres. 2578.011
10. Thorley-Lawson DA, Hawkins JB, Tracy SI. et al. The pathogenesis of Epstein-Barr virus persistent infection. Current opinion in virology. 2013;3:227-32
11. Delecluse H, Bartnizke S, Hammerschmidt W. et al. Episomal and integrated copies of Epstein-Barr virus coexist in Burkitt lymphoma cell lines. Journal of virology. 1993;67:1292-9
12. Yates J, Warren N, Reisman D. et al. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proceedings of the National Academy of Sciences. 1984;81:3806-10
13. Kang M-S, Kieff E. Epstein-Barr virus latent genes. Experimental & molecular medicine. 2015;47:e131
14. Hatton OL, Harris-Arnold A, Schaffert S. et al. The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunologic research. 2014;58:268-76
15. Tsao SW, Tsang CM, To KF. et al. The role of Epstein-Barr virus in epithelial malignancies. The Journal of pathology. 2015;235:323-33
16. Santpere G, Darre F, Blanco S. et al. Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1000 Genomes Project. Genome biology and evolution. 2014;6:846-60
17. Bouvard V, Baan R, Straif K. et al. A review of human carcinogens-Part B: biological agents. The Lancet Oncology. 2009;10:321
18. Böhm D. Investigation of the mechanism of Epstein-Barr virus Latent Membrane Protein 1 mediated NF-kB activation: Freie Universität Berlin, 2010.
19. Corvalán AH, Ruedlinger J, de Mayo T. et al. The Phylogeographic Diversity of EBV and Admixed Ancestry in the Americas-Another Model of Disrupted Human-Pathogen Co-Evolution. Cancers. 2019;11:217
20. Arvin A, Campadelli-Fiume G, Mocarski E. et al. Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press. 2007
21. Smatti MK, Al-Sadeq DW, Ali NH. et al. Epstein-Barr Virus Epidemiology, Serology, and Genetic Variability of LMP-1 Oncogene Among Healthy Population: An Update. Frontiers in oncology. 2018:8
22. Banko AV, Lazarevic IB, Folic MM. et al. Characterization of the variability of Epstein-Barr virus genes in nasopharyngeal biopsies: potential predictors for carcinoma progression. PloS one. 2016;11:e0153498
23. Münz C. Epstein Barr virus volume 2: one herpes virus: many diseases. Vol. 391. Springer. 2015
24. Sample J, Young L, Martin B. et al. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. Journal of virology. 1990;64:4084-92
25. Peh S-C, Kim L-H, Poppema S. Frequent presence of subtype A virus in Epstein-Barr virus-associated malignancies. Pathology. 2002;34:446-50
26. Griffiths P. Cytomegalovirus (In: Principles and Practice of Clinical Virology. Eds. Zuckerman AJ, Banatvala JE, Pattison JR, Griffiths PD, Schoub BD): John Wiley and Sons, Ltd, West Suusex, England. 2004
27. Klemenc P, Marin J, Šoba E. et al. Distribution of Epstein-Barr virus genotypes in throat washings, sera, peripheral blood lymphocytes and in EBV positive tumor biopsies from Slovenian patients with nasopharyngeal carcinoma. Journal of medical virology. 2006;78:1083-90
28. Miller WE, Edwards RH, Walling DM. et al. Sequence variation in the Epstein—Barr virus latent membrane protein 1. Journal of General Virology. 1994;75:2729-40
29. da Costa VG, Marques-Silva AC, Moreli ML. The Epstein-Barr virus latent membrane protein-1 (LMP1) 30-bp deletion and XhoI-polymorphism in nasopharyngeal carcinoma: a meta-analysis of observational studies. Systematic reviews. 2015;4:46
30. See HS, Yap YY, Yip WK. et al. Epstein-Barr virus latent membrane protein-1 (LMP-1) 30-bp deletion and Xho I-loss is associated with type III nasopharyngeal carcinoma in Malaysia. World journal of surgical oncology. 2008;6:18
31. AlQarni S, Al-Sheikh Y, Campbell D. et al. Lymphomas driven by Epstein-Barr virus nuclear antigen-1 (EBNA1) are dependant upon MDM2. Oncogene. 2018
32. Kieff E. Epstein-Barr virus and its replication. Fields virology. 2007:2603-54
33. Rowe DT. Epstein-Barr virus immortalization and latency. Front Biosci. 1999;4:D346-D71
34. Thompson MP, Kurzrock R. Epstein-Barr virus and cancer. Clinical Cancer Research. 2004;10:803-21
35. Dresang LR, Vereide DT, Sugden B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. Journal of virology. 2009;83:2930-40
36. Sample J, Henson E, Sample C. The Epstein-Barr virus nuclear protein 1 promoter active in type I latency is autoregulated. Journal of virology. 1992;66:4654-61
37. Frappier L, Goldsmith K, Bendell L. Stabilization of the EBNA1 protein on the Epstein-Barr virus latent origin of DNA replication by a DNA looping mechanism. Journal of Biological Chemistry. 1994;269:1057-62
38. Dheekollu J, Malecka K, Wiedmer A. et al. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr virus episomal latency. Oncotarget. 2017;8:7248
39. Levitskaya J, Coram M, Levitsky V. et al. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 1995;375:685
40. Levitskaya J, Sharipo A, Leonchiks A. et al. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proceedings of the National Academy of Sciences. 1997;94:12616-21
41. Kaiser C, Laux G, Eick D. et al. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. Journal of virology. 1999;73:4481-4
42. Wood CD, Veenstra H, Khasnis S. et al. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife. 2016;5:e18270
43. Radkov SA, Bain M, Farrell PJ. et al. Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21. Journal of virology. 1997;71:8552-62
44. Klein G, Ernberg I. Comparative aspects of EBV, HHV-8, and previously known DNA tumor viruses in experimental systems.Human Herpesviruses. 2007; 29.
45. on the Evaluation IWG. EPSTEIN-BARR VIRUS. 2012.
46. Wang A, Welch R, Zhao B. et al. Epstein-Barr virus nuclear antigen 3 (EBNA3) proteins regulate EBNA2 binding to distinct RBPJ genomic sites. Journal of virology. 2016;90:2906-19
47. Saha A, Robertson ES. Impact of EBV essential nuclear protein EBNA-3C on B-cell proliferation and apoptosis. Future microbiology. 2013;8:323-52
48. Maruo S, Zhao B, Johannsen E. et al. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proceedings of the National Academy of Sciences. 2011;108:1919-24
49. Chen A, Zhao B, Kieff E. et al. EBNA-3B-and EBNA-3C-regulated cellular genes in Epstein-Barr virus-immortalized lymphoblastoid cell lines. Journal of virology. 2006;80:10139-50
50. Alfieri C, Birkenbach M, Kieff E. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology. 1991;181:595-608
51. Harada S, Kieff E. Epstein-Barr virus nuclear protein LP stimulates EBNA-2 acidic domain-mediated transcriptional activation. Journal of virology. 1997;71:6611-8
52. Allan GJ, Inman GJ, Parker BD. et al. Cell growth effects of Epstein—Barr virus leader protein. Journal of general virology. 1992;73:1547-51
53. Mannick J, Cohen J, Birkenbach M. et al. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. Journal of virology. 1991;65:6826-37
54. Dufva M, Olsson M, Rymo L. Epstein-Barr virus nuclear antigen 5 interacts with HAX-1, a possible component of the B-cell receptor signalling pathway. Journal of General Virology. 2001;82:1581-7
55. Han I, Harada S, Weaver D. et al. EBNA-LP associates with cellular proteins including DNA-PK and HA95. Journal of virology. 2001;75:2475-81
56. Kashuba E, Yurchenko M, Szirak K. et al. Epstein-Barr virus-encoded EBNA-5 binds to Epstein-Barr virus-induced Fte1/S3a protein. Experimental cell research. 2005;303:47-55
57. Kanamori M, Watanabe S, Honma R. et al. Epstein-Barr virus nuclear antigen leader protein induces expression of thymus-and activation-regulated chemokine in B cells. Journal of virology. 2004;78:3984-93
58. Imai T, Nagira M, Takagi S. et al. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. International immunology. 1999;11:81-8
59. Nitsche F, Bell A, Rickinson A. Epstein-Barr virus leader protein enhances EBNA-2-mediated transactivation of latent membrane protein 1 expression: a role for the W1W2 repeat domain. Journal of virology. 1997;71:6619-28
60. Peng C-W, Xue Y, Zhao B. et al. Direct interactions between Epstein-Barr virus leader protein LP and the EBNA2 acidic domain underlie coordinate transcriptional regulation. Proceedings of the National Academy of Sciences. 2004;101:1033-8
61. Webster-Cyriaque J, Raab-Traub N. Transcription of Epstein-Barr virus latent cycle genes in oral hairy leukoplakia. Virology. 1998;248:53-65
62. Siegler G, Kremmer E, Gonnella R. et al. Epstein-Barr virus encoded latent membrane protein 1 (LMP1) and TNF receptor associated factors (TRAF): colocalisation of LMP1 and TRAF1 in primary EBV infection and in EBV associated Hodgkin lymphoma. Molecular Pathology. 2003;56:156
63. Liebowitz D, Wang D, Kieff E. Orientation and patching of the latent infection membrane protein encoded by Epstein-Barr virus. Journal of Virology. 1986;58:233-7
64. Huen D, Henderson S, Croom-Carter D. et al. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. 1995;10:549-60
65. Arcipowski KM, Stunz LL, Graham JP. et al. Molecular mechanisms of TNFR-associated factor 6 (TRAF6) utilization by the oncogenic viral mimic of CD40, latent membrane protein 1 (LMP1). Journal of Biological Chemistry. 2011;286:9948-55
66. Xie P, Bishop GA. Roles of TNF receptor-associated factor 3 in signaling to B lymphocytes by carboxyl-terminal activating regions 1 and 2 of the EBV-encoded oncoprotein latent membrane protein 1. The Journal of Immunology. 2004;173:5546-55
67. Lam N, Sugden B. LMP1, a viral relative of the TNF receptor family, signals principally from intracellular compartments. The EMBO journal. 2003;22:3027-38
68. Morris MA, Dawson CW, Young LS. Role of the Epstein-Barr virus-encoded latent membrane protein-1, LMP1, in the pathogenesis of nasopharyngeal carcinoma. Future oncology. 2009;5:811-25
69. Tsao SW, Tramoutanis G, Dawson CW. et al. The significance of LMP1 expression in nasopharyngeal carcinoma. In: Seminars in cancer biology. Elsevier. 2002;12(6):473-87
70. Tao Y, Shi Y, Jia J. et al. Novel roles and therapeutic targets of Epstein-Barr virus-encoded latent membrane protein 1-induced oncogenesis in nasopharyngeal carcinoma. Expert reviews in molecular medicine. 2015:17
71. Hatzivassiliou E, Mosialos G. Cellular signaling pathways engaged by the Epstein-Barr virus transforming protein LMP1. Front Biosci. 2002;7:d319-d29
72. Wang Z, Luo F, Li L. et al. STAT3 activation induced by Epstein-Barr virus latent membrane protein1 causes vascular endothelial growth factor expression and cellular invasiveness via JAK3 And ERK signaling. European journal of cancer (Oxford, England: 1990). 2010;46:2996-3006
73. Chou J, Lin YC, Kim J. et al. Nasopharyngeal carcinoma-review of the molecular mechanisms of tumorigenesis. Head & neck. 2008;30:946-63
74. Fang W, Zhang J, Hong S. et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget. 2014;5:12189-202
75. Rovedo M, Longnecker R. Epstein-barr virus latent membrane protein 2B (LMP2B) modulates LMP2A activity. Journal of virology. 2007;81:84-94
76. Beaufils P, Choquet D, Mamoun RZ. et al. The (YXXL/I) 2 signalling motif found in the cytoplasmic segments of the bovine leukaemia virus envelope protein and Epstein-Barr virus latent membrane protein 2A can elicit early and late lymphocyte activation events. The EMBO journal. 1993;12:5105-12
77. Fruehling S, Longnecker R. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology. 1997;235:241-51
78. Merchant M, Caldwell RG, Longnecker R. The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo. Journal of virology. 2000;74:9115-24
79. Caldwell RG, Wilson JB, Anderson SJ. et al. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405-11
80. Miller CL, Lee JH, Kieff E. et al. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proceedings of the National Academy of Sciences. 1994;91:772-6
81. Wasil LR, Tomaszewski MJ, Hoji A. et al. The effect of Epstein-Barr virus Latent Membrane Protein 2 expression on the kinetics of early B cell infection. PloS one. 2013;8:e54010
82. Rancan C, Schirrmann L, Hüls C. et al. Latent membrane protein LMP2A impairs recognition of EBV-infected cells by CD8+ T cells. PLoS pathogens. 2015;11:e1004906
83. Dong M, Chen J-n, Huang J-t. et al. The roles of EBV-encoded microRNAs in EBV-associated tumors. Critical reviews in oncology/hematology. 2019
84. Alptekin B, Akpinar BA, Budak H. A comprehensive prescription for plant miRNA identification. Frontiers in plant science. 2017;7:2058
85. Pfeffer S, Zavolan M, Grässer FA. et al. Identification of virus-encoded microRNAs. Science. 2004;304:734-6
86. Shinozaki-Ushiku A, Kunita A, Isogai M. et al. Profiling of virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma and their roles in gastric carcinogenesis. Journal of virology. 2015;89:5581-91
87. Barth S, Meister G, Grässer FA. EBV-encoded miRNAs. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2011;1809:631-40
88. Navari M, Etebari M, Ibrahimi M. et al. Pathobiologic roles of epstein-barr virus-encoded microRNAs in human lymphomas. International journal of molecular sciences. 2018;19:1168
89. Lung RW-M, Tong JH-M, Sung Y-M. et al. Modulation of LMP2A expression by a newly identified Epstein-Barr virus-encoded microRNA miR-BART22. Neoplasia (New York, NY). 2009;11:1174
90. Yang IV, Wade CM, Kang HM. et al. Identification of novel genes that mediate innate immunity using inbred mice. Genetics. 2009;183:1535-44
91. Dölken L, Malterer G, Erhard F. et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human γ-herpesviruses by RISC immunoprecipitation assay. Cell host & microbe. 2010;7:324-34
92. Nachmani D, Stern-Ginossar N, Sarid R. et al. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell host & microbe. 2009;5:376-85
93. Bauer S, Groh V, Wu J. et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727-9
94. Hooykaas MJ, Van Gent M, Soppe JA. et al. EBV microrna BART16 suppresses type I ifn signaling. The Journal of Immunology. 2017;198:4062-73
95. Zhang YM, Yu Y, Zhao HP. EBV-BART-6-3p and cellular microRNA-197 compromise the immune defense of host cells in EBV-positive Burkitt lymphoma. Molecular medicine reports. 2017;15:1877-83
96. Ma J, Nie K, Redmond D. et al. EBV-miR-BHRF1-2 targets PRDM1/Blimp1: potential role in EBV lymphomagenesis. Leukemia. 2016;30:594
97. Chen R, Zhang M, Li Q. et al. The Epstein-Barr Virus-encoded miR-BART22 targets MAP3K5 to promote host cell proliferative and invasive abilities in nasopharyngeal carcinoma. Journal of Cancer. 2017;8:305
98. Gulley ML. Molecular Diagnosis of Epstein-Barr Virus-Related Diseases. The Journal of Molecular Diagnostics. 2001;3:1-10
99. Shao JY, Li YH, Gao HY. et al. Comparison of plasma Epstein-Barr virus (EBV) DNA levels and serum EBV immunoglobulin A/virus capsid antigen antibody titers in patients with nasopharyngeal carcinoma. Cancer: Interdisciplinary International Journal of the American Cancer Society. 2004;100:1162-70
100. Gao R, Wang L, Liu Q. et al. Evaluation of seven recombinant VCA-IgA ELISA kits for the diagnosis of nasopharyngeal carcinoma in China: a case-control trial. BMJ open. 2017;7:e013211
101. Chen Y, Zhao W, Lin L. et al. Nasopharyngeal Epstein-Barr virus load: an efficient supplementary method for population-based nasopharyngeal carcinoma screening. PloS one. 2015;10:e0132669
102. Lorenzetti MA, De Matteo E, Gass H. et al. Characterization of Epstein Barr virus latency pattern in Argentine breast carcinoma. PLoS One. 2010;5:e13603
103. Gulley ML, Glaser SL, Craig FE. et al. Guidelines for interpreting EBER in situ hybridization and LMP1 immunohistochemical tests for detecting Epstein-Barr virus in Hodgkin lymphoma. American journal of clinical pathology. 2002;117:259-67
104. Cao P, Zhang M, Wang W. et al. Fluorescence in situ hybridization is superior for monitoring Epstein Barr viral load in infectious mononucleosis patients. BMC infectious diseases. 2017;17:323
105. Gulley ML, Tang W. Using Epstein-Barr viral load assays to diagnose, monitor, and prevent posttransplant lymphoproliferative disorder. Clin Microbiol Rev. 2010;23:350-66
106. Fox CP, Shannon-Lowe C, Rowe M. Deciphering the role of Epstein-Barr virus in the pathogenesis of T and NK cell lymphoproliferations. Herpesviridae. 2011;2:8
107. Rochford R, Moormann AM. Burkitt's lymphoma. Epstein Barr Virus Volume 1: Springer. 2015:267-85
108. Magrath IT. African Burkitt's lymphoma. History, biology, clinical features, and treatment. The American journal of pediatric hematology/oncology. 1991;13:222-46
109. God JM, Haque A. Burkitt lymphoma: pathogenesis and immune evasion. Journal of oncology 2010. 2010
110. Thorley-Lawson D, Deitsch KW, Duca KA. et al. The link between Plasmodium falciparum malaria and endemic Burkitt's lymphoma—new insight into a 50-year-old enigma. PLoS pathogens. 2016;12:e1005331
111. Swerdlow SH, Campo E, Pileri SA. et al. The 2016 revision of the World Health Organization (WHO) classification of lymphoid neoplasms. Blood. 2016;27(20):2375-90
112. Bornkamm GW. Epstein-Barr virus and the pathogenesis of Burkitt's lymphoma: More questions than answers. International journal of cancer. 2009;124:1745-55
113. Lenze D, Leoncini L, Hummel M. et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia. 2011;25:1869
114. Aguilar R, Casabonne D, O'Callaghan-Gordo C. et al. assessment of the combined effect of Epstein-Barr Virus and Plasmodium falciparum infections on endemic Burkitt lymphoma Using a Multiplex serological approach. Frontiers in immunology. 2017;8:1284
115. Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood. 2004;104:3009-20
116. Queiroga EM, Gualco G, Weiss LM. et al. Burkitt lymphoma in Brazil is characterized by geographically distinct clinicopathologic features. American journal of clinical pathology. 2008;130:946-56
117. Gloghini A, Dolcetti R, Carbone A. Lymphomas occurring specifically in HIV-infected patients: from pathogenesis to pathology. In: Seminars in cancer biology. Elsevier. 2013;23(6):457-67
118. Amria S, Cameron C, Stuart R. et al. Defects in HLA class II antigen presentation in B-cell lymphomas. Leukemia & lymphoma. 2008;49:353-5
119. Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leukemia & lymphoma. 2008;49:1028-41
120. Sebelin-Wulf K, Nguyen TD, Oertel S. et al. Quantitative analysis of EBV-specific CD4/CD8 T cell numbers, absolute CD4/CD8 T cell numbers and EBV load in solid organ transplant recipients with PLTD. Transplant immunology. 2007;17:203-10
121. Tsao SW, Tsang CM, Lo KW. Epstein-Barr virus infection and nasopharyngeal carcinoma. Phil Trans R Soc B. 2017;372:20160270
122. Kelly GL, Milner AE, Baldwin GS. et al. Three restricted forms of Epstein-Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells. Proceedings of the National Academy of Sciences. 2006;103:14935-40
123. Niedobitek G, Agathanggelou A, Rowe M. et al. Heterogeneous expression of Epstein-Barr virus latent proteins in endemic Burkitt's lymphoma. Blood. 1995;86:659-65
124. Carbone A, Gloghini A, Zagonel V. et al. Expression of Epstein-Barr virus-encoded latent membrane protein 1 in nonendemic Burkitt's lymphomas. Blood. 1996;87:1202-4
125. Kirchmaier AL, Sugden B. Dominant-negative inhibitors of EBNA-1 of Epstein-Barr virus. Journal of virology. 1997;71:1766-75
126. Frappier L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses. 2012;4:1537-47
127. Hummel M, Bentink S, Berger H. et al. A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. New England Journal of Medicine. 2006;354:2419-30
128. Dave SS, Fu K, Wright GW. et al. Molecular diagnosis of Burkitt's lymphoma. New England Journal of Medicine. 2006;354:2431-42
129. Guikema JE, Schuuring E, Kluin PM. Structure and consequences of IGH switch breakpoints in Burkitt lymphoma. Journal of the National Cancer Institute Monographs. 2008;2008:32-6
130. Allday MJ. How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt's lymphoma? In: Seminars in cancer biology. 2009;19(6):366-76
131. Mirsadraee S, Farzadnia M, Heidary F. et al. A Comparative Immunohisto chemical Diagnosis of Hodgkinâ s lymphoma with Conventional Method. medical journal of mashhad university of medical sciences. 2010;53:214-21
132. Mozaheb Z, Aledavood A, Farzad F. Distributions of major sub-types of lymphoid malignancies among adults in Mashhad, Iran. Cancer epidemiology. 2011;35:26-9
133. Salati M, Cesaretti M, Macchia M. et al. Epidemiological overview of Hodgkin lymphoma across the mediterranean basin. Mediterranean journal of hematology and infectious diseases. 2014:6
134. Glaser SL, Lin RJ, Stewart SL. et al. Epstein-Barr virus-associated Hodgkin's disease: epidemiologic characteristics in international data. International journal of cancer. 1997;70:375-82
135. Massini G, Siemer D, Hohaus S. EBV in Hodgkin lymphoma. Mediterranean journal of hematology and infectious diseases. 2009:1
136. Vockerodt M, Cader FZ, Shannon-Lowe C. et al. Epstein-Barr virus and the origin of Hodgkin lymphoma. Chinese journal of cancer. 2014;33:591
137. Wang S, Medeiros LJ, Xu-Monette ZY. et al. Epstein-Barr virus-positive nodular lymphocyte predominant Hodgkin lymphoma. Annals of diagnostic pathology. 2014;18:203-9
138. Swerdlow SH. WHO classification of tumours of haematopoietic and lymphoid tissues. WHO classification of tumours. 2008;22008:439
139. Levine PH, Ablashi DV, Berard CW. et al. Elevated antibody titers to Epstein-Barr virus in Hodgkin's disease. Cancer. 1971;27:416-21
140. Wu TC, Mann RB, Charache P. et al. Detection of EBV gene expression in Reed-Sternberg cells of Hodgkin's disease. International journal of cancer. 1990;46:801-4
141. Pallesen G, Hamilton-Dutoit SJ, Rowe M. et al. Expression of Epstein-Barr virus replicative proteins in aids-related non-Hodgkin's lymphoma cells. The Journal of pathology. 1991;165:289-99
142. Čičkušić E, Mustedanagić-Mujanović J, Iljazović E. et al. Association of Hodgkin's lymphoma with Epstein Barr virus infection. Bosnian journal of basic medical sciences. 2007;7:58
143. Rasche L, Kapp M, Einsele H. et al. EBV-induced post transplant lymphoproliferative disorders: a persisting challenge in allogeneic hematopoetic SCT. Bone marrow transplantation. 2014;49:163
144. Kim H-J, Ko YH, Kim JE. et al. Epstein-Barr virus-associated lymphoproliferative disorders: review and update on 2016 WHO classification. Journal of pathology and translational medicine. 2017;51:352
145. Green M. Management of Epstein-Barr virus-induced post-transplant lymphoproliferative disease in recipients of solid organ transplantation. American Journal of Transplantation. 2001;1:103-8
146. Nijland ML, Kersten MJ, Pals ST. et al. Epstein-Barr Virus-Positive Posttransplant Lymphoproliferative Disease After Solid Organ Transplantation: Pathogenesis, Clinical Manifestations, Diagnosis, and Management. Transplantation direct. 2016:2
147. Stojanova J, Caillard S, Rousseau A. et al. Post-transplant lymphoproliferative disease (PTLD): Pharmacological, virological and other determinants. Pharmacological research. 2011;63:1-7
148. Snow AL, Martinez OM. Epstein-Barr virus: evasive maneuvers in the development of PTLD. American Journal of Transplantation. 2007;7:271-7
149. Allen U, Preiksaitis J, Practice AIDCo. Epstein-Barr virus and posttransplant lymphoproliferative disorder in solid organ transplantation. American Journal of Transplantation. 2013;13:107-20
150. Weikert BC, Blumberg EA. Viral infection after renal transplantation: surveillance and management. Clinical Journal of the American Society of Nephrology. 2008;3:S76-S86
151. Macsween KF, Crawford DH. Epstein-Barr virus—recent advances. The Lancet infectious diseases. 2003;3:131-40
152. Carle LN, Ko CC, Castle JT. Nasopharyngeal carcinoma. Head and neck pathology. 2012;6:364-8
153. Mimi CY, Yuan J-M. Epidemiology of nasopharyngeal carcinoma. In: Seminars in cancer biology. 2002;12(6):421-9
154. Chang ET, Adami H-O. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiology and Prevention Biomarkers. 2006;15:1765-77
155. Jia W-H, Qin H-D. Non-viral environmental risk factors for nasopharyngeal carcinoma: a systematic review. In: Seminars in cancer biology. 2012;22(2):117-26
156. Hila L, Farah F, Ayari H. et al. Epidemiological study, immunohistochemistry and in situ hybridization studies of nasopharyngeal carcinomas: A Tunisian report. Pathologie Biologie. 2009;57:427-9
157. Shanmugaratnam K. Histological typing of nasopharyngeal carcinoma. IARC scientific publications. 1978:3-12
158. Micheau C, Rilke F, Pilotti S. Proposal for a new histopathological classification of the carcinomas of the nasopharynx. Tumori Journal. 1978;64:513-8
159. Yoshizaki T, Kondo S, Wakisaka N. et al. Pathogenic role of Epstein-Barr virus latent membrane protein-1 in the development of nasopharyngeal carcinoma. Cancer letters. 2013;337:1-7
160. Wu T-C, Mann RB, Epstein JI. et al. Abundant expression of EBER1 small nuclear RNA in nasopharyngeal carcinoma. A morphologically distinctive target for detection of Epstein-Barr virus in formalin-fixed paraffin-embedded carcinoma specimens. The American journal of pathology. 1991;138:1461
161. Lee C-P, Chen J-Y, Wang J-T. et al. Epstein-Barr virus BGLF4 kinase induces premature chromosome condensation through activation of condensin and topoisomerase II. Journal of virology. 2007;81:5166-80
162. Ma S-D, Yu X, Mertz JE. et al. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. Journal of virology. 2012 JVI. 00770-12
163. Shibata D, Weiss L. Epstein-Barr virus-associated gastric adenocarcinoma. The American journal of pathology. 1992;140:769
164. Jácome AAdA, Lima EMd, Kazzi AI. et al. Epstein-Barr virus-positive gastric cancer: a distinct molecular subtype of the disease? Revista da Sociedade Brasileira de Medicina Tropical. 2016;49:150-7
165. Naseem M, Barzi A, Brezden-Masley C. et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer treatment reviews. 2018;66:15-22
166. Shinozaki-Ushiku A, Kunita A, Isogai M. et al. Profiling of virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma and their roles in gastric carcinogenesis. Journal of virology. 2015 JVI. 03639-14
167. van Beek J, Zur Hausen A, Klein Kranenbarg E. et al. EBV-positive gastric adenocarcinomas: a distinct clinicopathologic entity with a low frequency of lymph node involvement. Journal of Clinical Oncology. 2004;22:664-70
168. Song H-J, Kim K-M. Pathology of epstein-barr virus-associated gastric carcinoma and its relationship to prognosis. Gut and liver. 2011;5:143
169. Uozaki H, Fukayama M. Epstein-Barr virus and gastric carcinoma-viral carcinogenesis through epigenetic mechanisms. International journal of clinical and experimental pathology. 2008;1:198
170. Lee JY, Kim K-M, Min B-H. et al. Epstein-Barr virus-associated lymphoepithelioma-like early gastric carcinomas and endoscopic submucosal dissection: case series. World Journal of Gastroenterology: WJG. 2014;20:1365
171. Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein-Barr virus and gastric cancer. International journal of oncology. 2015;46:1421-34
172. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12:358-71
173. Chan J. Extranodal NK/T-cell lymphoma, nasal type. WHO classification of tumours of haematopoietic and lymphoid tissues. 2008:285-8
174. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372:20160271
175. Au W-y, Weisenburger DD, Intragumtornchai T. et al. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood. 2009;113:3931-7
176. Chim C, Ma E, Loong F. et al. Diagnostic cues for natural killer cell lymphoma: primary nodal presentation and the role of in situ hybridisation for Epstein-Barr virus encoded early small RNA in detecting occult bone marrow involvement. Journal of clinical pathology. 2005;58:443-5
177. Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-Positive Lymphoproliferations of B-T-and NK-Cell Derivation in Non-Immunocompromised Hosts. Pathogens. 2018;7:28
178. Jiang L, Gu Z-H, Yan Z-X. et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nature genetics. 2015;47:1061
179. Bi X-w, Wang H, Zhang W-w. et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. Journal of hematology & oncology. 2016;9:109
180. Nakashima Y, Tagawa H, Suzuki R. et al. Genome-wide array-based comparative genomic hybridization of natural killer cell lymphoma/leukemia: different genomic alteration patterns of aggressive NK-cell leukemia and extranodal Nk/T-cell lymphoma, nasal type. Genes, Chromosomes and Cancer. 2005;44:247-55
181. Huang Y, de Leval L, Gaulard P. Molecular underpinning of extranodal NK/T-cell lymphoma. Best practice & research Clinical haematology. 2013;26:57-74
182. Shadmani FK, Mansori K, Khazaei S. et al. Geographic distribution of breast cancer incidence in Iran. Biomedical Research and Therapy. 2017;4:1295-304
183. Fregene A, Newman LA. Breast cancer in sub-Saharan Africa: how does it relate to breast cancer in African-American women? Cancer: Interdisciplinary International Journal of the American Cancer Society. 2005;103:1540-50
184. Sun Y-S, Zhao Z, Yang Z-N. et al. Risk factors and preventions of breast cancer. International journal of biological sciences. 2017;13:1387
185. Salman NA, Davies G, Majidy F. et al. Association of High Risk Human Papillomavirus and Breast cancer: A UK based Study. Sci Rep. 2017:7
186. Pasquereau S, Al Moussawi F, Karam W. et al. Cytomegalovirus, Macrophages and Breast Cancer. The Open Virology Journal. 2017;11:15-27
187. Glenn WK, Heng B, Delprado W. et al. Epstein-Barr virus, human papillomavirus and mouse mammary tumour virus as multiple viruses in breast cancer. PloS one. 2012;7:e48788
188. Joshi D, Quadri M, Gangane N. et al. Association of Epstein Barr virus infection (EBV) with breast cancer in rural Indian women. PLoS One. 2009;4:e8180
189. Hachana M, Amara K, Ziadi S. et al. Investigation of Epstein-Barr virus in breast carcinomas in Tunisia. Pathology-Research and Practice. 2011;207:695-700
190. Marrão G, Habib M, Paiva A. et al. Epstein-Barr virus infection and clinical outcome in breast cancer patients correlate with immune cell TNF-α/IFN-γ response. BMC cancer. 2014;14:665
191. Arbach H, Viglasky V, Lefeu F. et al. Epstein-Barr Virus (EBV) Genome and Expression in Breast Cancer Tissue: Effect of EBV Infection of Breast Cancer Cells on Resistance to Paclitaxel (Taxol). J Virol. 2006;80:845-53
192. Fessahaye G, Elhassan AM, Elamin EM. et al. Association of Epstein - Barr virus and breast cancer in Eritrea. Infectious Agents and Cancer. 2017:12
193. Bae J-M, Kim EH. Epstein-Barr Virus Infection and Risk of Breast Cancer: An Adaptive Meta-Analysis for Case-Control Studies. Archives of Clinical Infectious Diseases. 2016:11
194. Hu H, Luo M-L, Desmedt C. et al. Epstein-Barr virus infection of mammary epithelial cells promotes malignant transformation. EBioMedicine. 2016;9:148-60
Author contact
![]() Corresponding author: Osbourne Quaye, Department of Biochemistry, Cell and Molecular Biology, University of Ghana, Volta Road, P. O. Box LG 54, Legon, Accra-Ghana; Tel: +233-2611-70904.
Corresponding author: Osbourne Quaye, Department of Biochemistry, Cell and Molecular Biology, University of Ghana, Volta Road, P. O. Box LG 54, Legon, Accra-Ghana; Tel: +233-2611-70904.