Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(7):1828-1838. doi:10.7150/jca.35375 This issue Cite
Research Paper
Aminooxyacetic acid (AOAA) sensitizes colon cancer cells to oxaliplatin via exaggerating apoptosis induced by ROS
Taohua Yue1*, Shuai Zuo1*, Dingfang Bu2, Jing Zhu1, Shanwen Chen1, Yongchen Ma1, Ju Ma1, Shihao Guo1, Long Wen1, Xiaoqian Zhang1, Jianwen Hu1, Yurong Wang1, Zihao Yao1, Guowei Chen1, Xin Wang1, Yisheng Pan1, Pengyuan Wang1 ![]() , Yucun Liu1
, Yucun Liu1 ![]()
1. Division of General Surgery, Peking University First Hospital, Peking University, 8 Xi Shiku Street, Beijing, 100034, People's Republic of China.
2. Central laboratory, Peking University First Hospital, Peking University, 8 Xi Shiku Street, Beijing, 100034, People's Republic of China.
* These authors contributed equally to this work, and should be considered as co-first authors.
Received 2019-4-1; Accepted 2019-11-14; Published 2020-1-20
Abstract
Background: As the third confirmed gaseous transmitter, the role of hydrogen sulfide (H2S) in the pathogenesis of multiple types of cancer has been attracting increasing attention. Increased expression of cystathionine β-synthase (CBS) and H2S in colon cancer tissue samples has been validated and tumor-derived H2S, mainly produced by CBS, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Recently, the therapeutic manipulation of H2S has been proposed as a promising anticancer approach. However, the effect of aminooxyacetic acid (AOAA), which has been widely used as an inhibitor of CBS dependent synthesis of H2S, on the chemotherapeutic effect of oxaliplatin (OXA) and the underlying mechanisms remain to be illustrated.
Methods: We examined the expression of CBS in human colorectal cancer specimens and matched normal mucosa by immunohistochemistry. The effect of AOAA on the sensitivity of colon cancer cells to OXA and the level of apoptosis induced by caspase cascade was investigated in both HCT116 and HT29 cell lines utilizing CCK-8 assays, flow cytometry analysis and western blot analysis. The endogenous levels of reactive oxygen species (ROS) were detected fluorescently by DCF-DA, and glutathione (GSH) levels were measured by a Total GSH Detection Kit. Tumor bearing xenograft mouse models and in vivo imaging systems were further used to investigate the effect of AOAA in vivo and immunohistochemistry (IHC) and TUNEL analysis were performed.
Results: In the current study, we confirmed CBS, the main target of AOAA, is overexpressed in human colorectal cancer by immunohistochemistry. The inhibitory effect of AOAA on the synthesis of H2S was validated utilizing fluorescent probe and specific electrode. AOAA significantly reduced the IC50 values of OXA in both colon cancer cell lines. Co-incubation with AOAA elicited increased apoptosis induced by OXA, featured by increased activation of caspase cascade. Besides, AOAA further increased the levels of ROS induced by OXA and attenuated the synthesis of glutathione (GSH), which is a vital antioxidant. Besides, the results of in vivo imaging and following IHC and TUNEL analysis were in accordance with cellular experiments, indicating that AOAA sensitizes colon cancer cells to OXA via exaggerating intrinsic apoptosis.
Conclusion: The results suggested that CBS is overexpressed in colorectal cancer tissues and AOAA sensitizes colon cancer cells to OXA via exaggerating apoptosis both in vitro and in vivo. Decreasing the endogenous level of GSH and consequently impaired detoxification of ROS might be one of the mechanisms underlying the effect of AOAA.
Keywords: colorectal cancer, oxaliplatin, hydrogen sulfide, AOAA, reactive oxygen species, apoptosis.
1. Introduction
Colorectal cancer (CRC) is the third most common diagnosed cancer worldwide and the second leading cause of cancer related mortality. More than half of the patients with CRC will eventually die of this dreadful disease [1]. As the standard chemotherapy regimens, FOLFOX and XELOX still play an irreplaceable role in the treatment of CRC in advanced stages. Oxaliplatin (OXA), a third-generation platinum drug, is a DNA-interacting agent that disrupts DNA replication and transcription and induces ROS production, promoting apoptosis [2,3]. Although OXA improves the objective response and median overall survival rates, the main dose-limiting toxicity, peripheral sensory neuropathy and drug resistance still limit its widespread use. Thus, constant efforts need to be made to improve efficacies and attenuate dose-related side effects of OXA.
As the third confirmed gaseous signaling molecule after nitric oxide (NO) and carbon monoxide (CO), H2S has received increasing attention as an endogenous biological mediator in recent years [4-9]. In mammalian cells, H2S is produced mainly by three key enzymes: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) and 3-mercaptopyruvate thiotransferase (3-MST) [4]. Like the other two gaseous transmitters, NO and CO, the physiological action of H2S is also bidirectional: a low concentration of H2S exerts cytoprotective effects, and a high concentration of H2S shows cytotoxic effects [10]. Abnormal H2S metabolism is implicated in multiple types of cancers [11, 12]. Increased expression of CBS and H2S has been validated in colon cancer tissue samples and cell lines. Silencing CBS or inhibiting its enzymatic activity significantly attenuated the proliferation of colon cancer cells both in vitro and in vivo [13]. Similar effect has been validated in ovarian cancer, breast cancer and lung adenocarcinoma, indicating that the CBS/H2S axis plays an essential role in the pathogenies of multiple types of cancer [14-16].
However, the effect of inhibition of the CBS/H2S axis on the sensitivity of colon cancer cells to OXA has not been illustrated. Increased production of ROS and P53 has been implicated in the apoptosis of colon cancer cells induced by OXA [3, 17-21]. One of the mechanisms underlying the acquired resistance of OXA is increased intracellular GSH and consequently increased detoxification of ROS [22]. While, the CBS inhibitor aminooxyacetic acid (AOAA) is also involved in regulating ROS levels and P53 activation [12, 16].Considering the antioxidant effect of H2S and the pivotal role of increased ROS in mediating the apoptosis induced by OXA, inhibition of H2S synthesis might sensitize colon cancer cells to OXA. Thus, we set out to investigate the effect of AOAA on the chemotherapeutic effect of OXA in colon cancer cells both in vitro and in vivo. The results suggested that AOAA sensitized colon cancer cells to OXA via exaggerating intrinsic apoptosis. Decreasing the endogenous level of GSH and consequently impaired detoxification of ROS might be one of the mechanisms underlying the effect of AOAA.
2. Materials and methods
2.1. Cells, chemicals and reagents
Human colon cancer cell lines HCT116, DLD-1and HT29 and the nonmalignant colonic epithelial cell line NCM460 were obtained from the ATCC (Manassas, VA, USA) and cultured in McCoy's 5A medium (modified), DMEM and RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin, respectively. All cells were cultured in the optimal environment of 37 °C with 5% CO2. Cells treated with DMSO or PBS were used as the negative control (Ctrl). Oxaliplatin, D-Luciferin sodium, DCF-DA, Hoechst 33342 (HO), the Annexin V-FITC PI Apoptosis Detection Kit, the In Situ Cell Death Detection Kit, and the Cell Counting Kit-8 (CCK-8) were all purchased from Sigma-Aldrich (St. Louis, MO, USA). All antibodies were purchased from Cell Signaling Technology (Beijing, China). A new H2S-specific near-infrared fluorescence enhanced probe was donated by Beijing University of Chemical Technology. A Total GSH Detection Kit was purchased from Beyotime Biotechnologies (Jiangsu, China).
2.2. Measurement of cell viability
The CCK-8 assay was used to detect cell viability according to the manufacturer's instructions. Briefly, HCT116 and HT29 cells were cultured until ~80% confluence. HCT116 and HT29 cells were digested completely and added to each well (6,000 cells/well) of a 96-well plate (Corning, USA). According to the protocol provided by the manufacturer, at the end of treatments, add 10% CCK-8 solution to each well of the 96-well plate. Be careful not to introduce bubbles to the wells, since they interfere with the O.D. reading. To obtain a concentration of AOAA that inhibited cellular H2S synthesis but was noncytotoxic to cell survival, cells were treated with gradient concentrations of AOAA for 48 hours. After determining the AOAA concentrations, cells were treated with gradient concentrations of OXA in the presence or absence of this specific concentration of AOAA for 48 hours, and the IC50 values of OXA were measured.
2.3. H2S detection
To determine the inhibitory efficacy of AOAA on cellular H2S synthesis, the probe and a Mettler sulfur ion electrode were applied according to the manufacturer's instructions. For the qualitative detection of endogenous H2S, cells were seeded in a glass-bottom 35 mm plate (~ 2×104 cells per well) (Corning, USA) and first incubated with DMSO or AOAA for 30 minutes, replaced with medium containing the H2S probe (10 µmol/L) for an additional 30 minutes and then washed with PBS twice before fluorescence imaging [23]. To quantify the level of H2S, we measured the H2S content in the supernatants of HCT116 and HT29 cells treated with DMSO and AOAA for 48 hours by the electrode.
2.4. Flow cytometry analysis of apoptosis
An Annexin V-PI Staining Kit was applied to detect the apoptosis of HCT116 and HT29 cells treated with DMSO, AOAA, OXA and AOAA+OXA. Apoptotic cells were examined by flow cytometry according to the manufacturer's instructions (BD Bioscience, USA). The results were presented as the percentage of total cells and were compared to the proportion of four groups of apoptotic cells (early apoptosis + late apoptosis).
2.5. Western blot analysis
The group division was the same as that used in the cell viability and apoptosis assay. Total proteins were separated by 4-12% SurePAGE and transferred onto a PVDF membrane. After blocking in 5% BSA for 1 hour, the bands were incubated with the primary antibodies overnight at 4 °C, followed by incubation with the corresponding secondary antibodies for 1 hour. The membranes were washed with TBST after incubation with each antibody. The specific primary antibodies were used as follows: PARP (1:1000 Dilution; CST, MA, USA), cleaved PARP ( 1:1000 dilution, CST, MA, USA), P53 (1:1000 dilution, CST, MA, USA), cleaved caspase 3 (1:1000 dilution, CST, MA, USA), caspase 9 (1:1000 dilution, CST, MA, USA), Bcl-2 (1:1000 dilution, CST, MA, USA), Bax (1:1000 dilution, CST, MA, USA) and GAPDH (1:1000, CST, MA, USA). GAPDH served as the internal controls. The bands were detected using the Syngene GeneGenius gel imaging system (Syngene, Cambridge, UK). The results were analyzed by ImageJ software.
2.6. Measurement of intracellular ROS
To explore the mechanisms of OXA combined with AOAA in exaggerating apoptosis, a 96-well plate was seeded at 6,000 cells per well. After 24 hours of adherence, five treatments were administered: DMSO, DMSO, AOAA, OXA, and AOAA +OXA. The drug is updated once every 24 hours for a total of 72 hours. Then, either of the DMSO groups was treated with H2O2 (30 mmol/L) for 30 minutes as a positive control. Thereafter, all groups were treated with a mixture of DCF-DA (30 µmol/L) and HO (2.5 µg/ml) for 30 minutes to detect ROS and the corresponding number of viable cells, respectively. After discarding the supernatant and PBS rinse, fluorescence intensity was measured by a microplate reader, and the corresponding excitation/emission wavelengths were 490/530 nm (DCF-DA) and 340/425 nm (HO). The amount of ROS depended on the ratio of DCF-DA/HO signals per well [18].
2.7. Measurement of total intracellular GSH
Reports have indicated that AOAA reduces intracellular cystathionine, a precursor of GSH, thereby decreasing the antioxidant capacity of cells [16]. To determine whether the intracellular increase in ROS was caused by a decrease in the antioxidant GSH, both HCT116 and HT29 cells were treated with DMSO and AOAA. Total GSH detection assays were performed using the Total GSH Detection Kit according to the manufacturer's protocol [24, 25].
2.8. Establishment of a mouse xenograft model of a human colon cancer cell line stably expressing luciferase
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Peking University First Hospital. Athymic nude BALB/c male mice (4 weeks old) purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd., were injected subcutaneously in the right armpit with 8× 105 cells in 100 µl PBS. four to five days later, mice were randomly assigned to one of four groups and subjected to intraperitoneal injection : control (PBS), AOAA (9 mg/kg in PBS, 5 days per week), OXA (5 mg/kg in PBS, once a week), and AOAA + OXA (as before) [13, 14]. D-Luciferin sodium was intraperitoneal administration once every two weeks for in vivo imaging of nude mice (500 nmol/g body weight D-luciferin for an average-sized mouse) [26]. After 28 days, mice were euthanized, and xenografts were harvested and weighed.
2.9. Immunohistochemistry (IHC) and TUNEL assay
To measuring the proliferation of in vivo tumor cells, we detected two biomarkers at the protein level by IHC staining. Briefly, xenografts embedded in paraffin were cut into 4-5 µm sections and fixed onto slides. Then, the slides were deparaffinized, hydrated and microwave treated for antigen retrieval. Antigen detection and the dilution ratios were as follows: (1) Ki67 at a dilution of 1:400 (CST, MA, USA) and (2) PCNA at a dilution of 1:8,000 (CST, MA, USA). Both antibodies were incubated overnight in the dark. The next day, the corresponding secondary antibody, horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (ZSGB-BIO, Beijing, China) was incubated at 37 °C for 30 minutes. Then, the DAB display color was as follows: Ki67, 40 seconds, and PCNA, 1 minutes. Finally, the slides were counterstained with hematoxylin, subjected to gradient alcohol and xylene dehydration, sealed with neutral gum, and counted under a microscope (Olympus, Japan) at a magnification of 20X.
To confirm that the study is clinically relevant, we searched the hospital case management system, and 11 cases of colorectal cancer patients in our hospital recent surgical resection specimens and matched normal mucosa were included in this study. Immunohistochemistry was performed to detect the expression of CBS, the main target of AOAA. Antigen detection and the dilution ratios were 1:100 (CST, MA, USA). The DAB display color was 6 minutes. Other steps are the same as above.
For the measurement of apoptosis, TUNEL assays were performed with an In Situ Cell Death Detection Kit, following the manufacturer's protocol. DNA strand gaps were identified by TUNEL probes [27]. At least 500 cells and four high-powered fields were counted per slide.
In order to take a good contrast picture, we were photographed at 20X and 40X objective lens and applied white balance technology.
2.10. Statistical analysis
All data are presented as the mean ± SEM and were analyzed using GraphPad Prism 7.0 software. Statistical analysis was based on Student's t test. All experiments were repeated at least three times to ensure reproducibility. * p<0.05 represents a significant difference from the Ctrl. #p < 0.05 represents a significant difference from the OXA group.
3. Results
3.1. CBS is overexpressed in colorectal cancer tissues
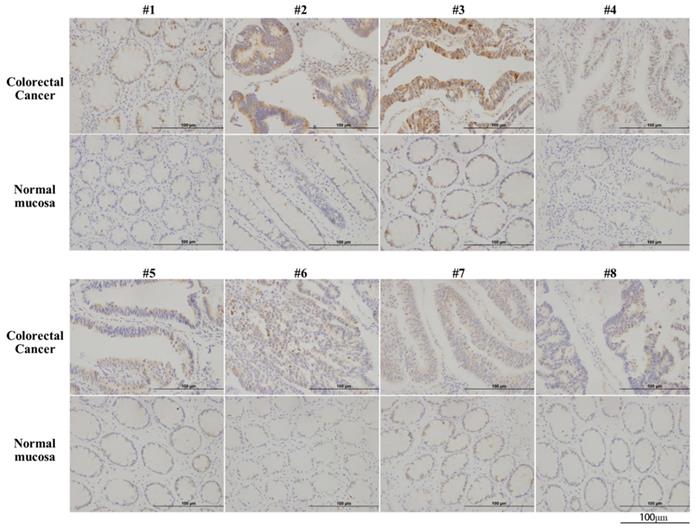
Immunohistochemical analysis of CBS in 11 colorectal cancer tissues and paired normal colon epithelial tissues revealed the significantly upregulated expression of CBS in colorectal cancer tissues (Fig. 1).
CBS is overexpressed in human colorectal cancer. Arabic numerals 1-8 represented pathological specimens from eight different patient sources (scale bar represented 100 µm).
3.2. AOAA inhibited survival of colon cancer cell lines in a concentration-dependent manner
The CCK-8 assay indicated that AOAA inhibited the viability of colon cancer cells in a concentration-dependent manner. AOAA (100 and 200 µmol/L) had no effect on the survival of HCT116 and HT29 cells, respectively (Fig. 2A). In the following in vitro experiments, AOAA has been using these concentrations in the presence or absence of OXA.
AOAA inhibited the synthesis of H2S and decreased the IC50 values of OXA in HCT116 and HT29 cells. A, AOAA (100 and 200 µmol/L) had no effect on the proliferation of HCT116 and HT29 cell. B, The inhibitory effect of AOAA on the intracellular levels of H2S in both cell lines. H2S was probed fluorescently. Scale bar represents 50 µm. C, AOAA (100 and 200 µmol/L) significantly inhibited intracellular H2S synthesis. D, AOAA significantly decreased the IC50 values of OXA in both cell lines (*p < 0.05 vs ctrl).
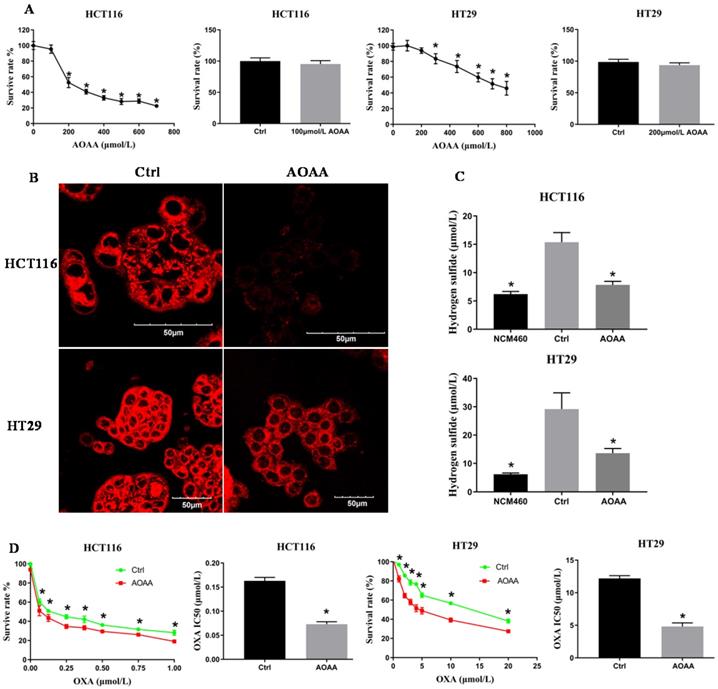
3.3. AOAA significantly decreased the intracellular synthesis of H2S
The H2S content was qualitatively measured by a fluorescent probe. The fluorescence of the AOAA group was significantly weaker than that of the control (Fig. 2B). The H2S content was quantitatively detected by the electrode. AOAA significantly inhibited intracellular H2S synthesis. NCM460 cells, characterized by low expression of H2S, served as a negative control (Fig. 2C).
3.4. AOAA significantly decreased the IC50 values of OXA in HCT116 and HT29 cells
Colon cancer cells were incubated with various concentrations of OXA in the absence or presence of AOAA for 48 hours. Although AOAA (100 and 200 µmol/L) alone had no inhibitory effect on the proliferation of both cell lines, significant decrease in viability was observed in cells treated with OXA in the presence of AOAA compared with cells treated with OXA alone. When OXA was combined with AOAA, the IC50 values of OXA were reduced by 55% and 60.49% in HCT116 and HT29 cells, respectively (Fig. 2D).
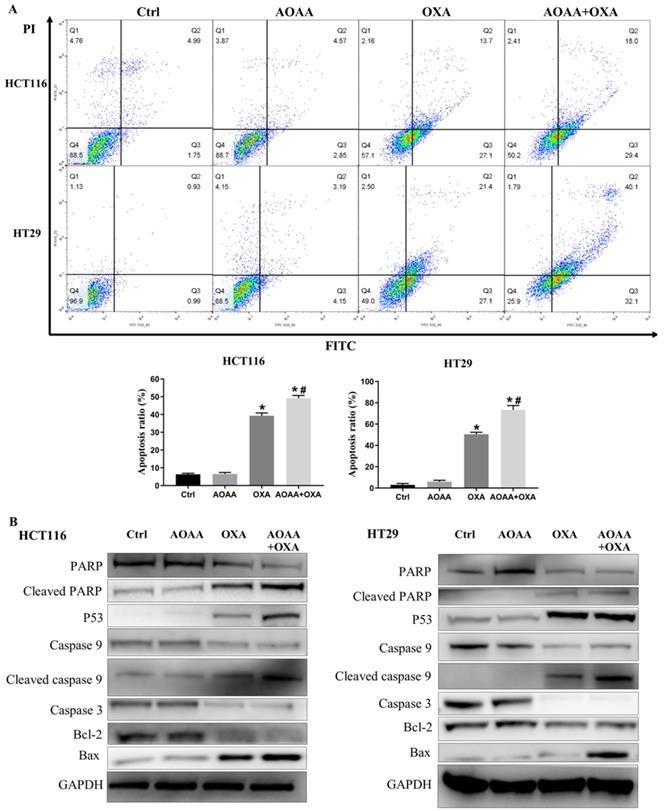
3.5. AOAA significantly exaggerated apoptosis induced by OXA
Flow cytometry analysis suggested that AOAA significantly increased the apoptosis induced by OXA in both cell lines, featured by increased proportion of cells in the phase of early and late apoptosis (Fig. 3A). The OXA-mediated mitochondrial apoptotic pathway is ROS-, P53-mediated and caspase-dependent [18, 19, 21, 28]. Interestingly, AOAA upregulates the level of ROS, the expression of P53 protein and inhibits the survival of cancer cells [16, 19]. We examined whether OXA combined with AOAA synergized to promote caspase-dependent mitochondrial apoptosis. Western blot analysis showed that the AOAA+OXA group exhibited a significant increase in the expression of cleaved caspase-9, cleaved PARP, Bax and P53 and a significant decrease in the expression of Bcl-2, total caspase-9, total caspase-3 and total caspase-PARP (Fig. 3B). Taken together, these data indicated that AOAA and OXA acted synergistically to promote apoptosis through activating apoptotic caspase cascade.
AOAA exaggerated the apoptosis induced by OXA in both cell lines. A, Compared with that in OXA group, the proportion of apoptotic cells (early apoptosis + late apoptosis) increased significantly in the AOAA+OXA group. B, Co-incubation with AOAA significantly increased the expression of proapoptotic proteins and decreased the expression of antiapoptotic proteins induced by OXA (*p < 0.05 vs ctrl. # p < 0.05 vs OXA).
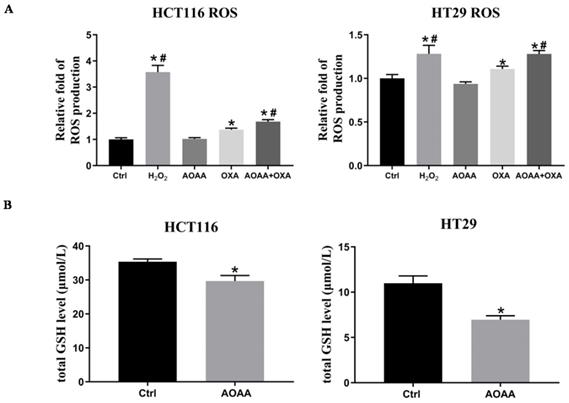
3.6. AOAA significantly increased OXA-induced ROS production and decreased intracellular levels of GSH
Previous studies indicated that OXA-induced apoptosis is ROS-dependent [22]. Thus, the effect of AOAA on intracellular ROS levels were further investigated. As expected, the results suggested that OXA induced a significant increase in ROS production. AOAA alone did not have a significant effect on ROS levels. What's intriguing is that AOAA significantly exaggerated the increased ROS levels induced by OXA, indicating the pivotal role of ROS in mediating the chemosensitizer role of AOAA (Fig. 4A). We also found that AOAA significantly inhibited the synthesis of GSH in HCT116 and HT29 cells, thus impairing detoxification of ROS (Fig. 4B). Considering the role of GSH as an important antioxidant, the inhibition of GSH might be one of the mechanisms underlying the effect of AOAA.
AOAA significantly increased OXA-induced ROS production and decreased intracellular GSH levels. A, AOAA significantly increased OXA-induced ROS production. The amount of ROS was calculated with the ratio of DCF-DA/HO signals as described in the section of materials and methods. B, AOAA significantly inhibited the synthesis of intracellular GSH (*p < 0.05 vs ctrl. # p < 0.05 vs OXA).
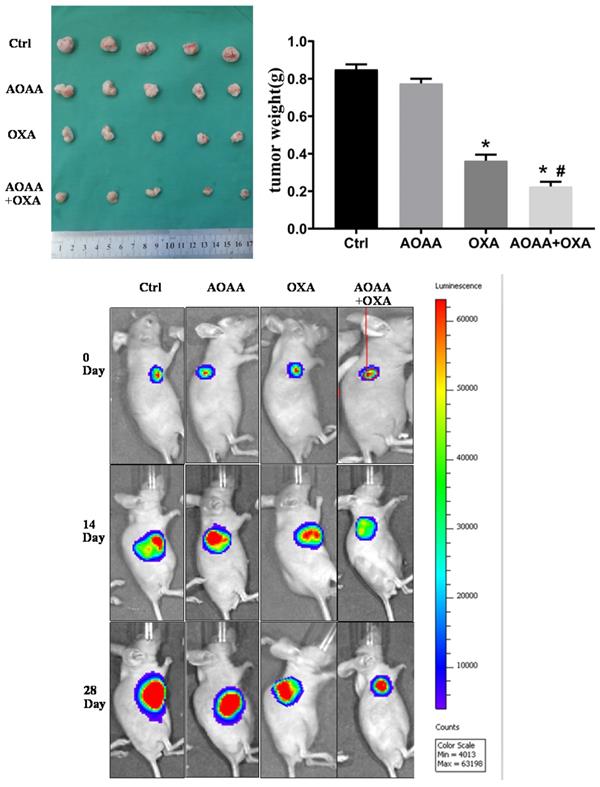
3.7. AOAA enhanced the chemotherapeutic effect of OXA in vivo
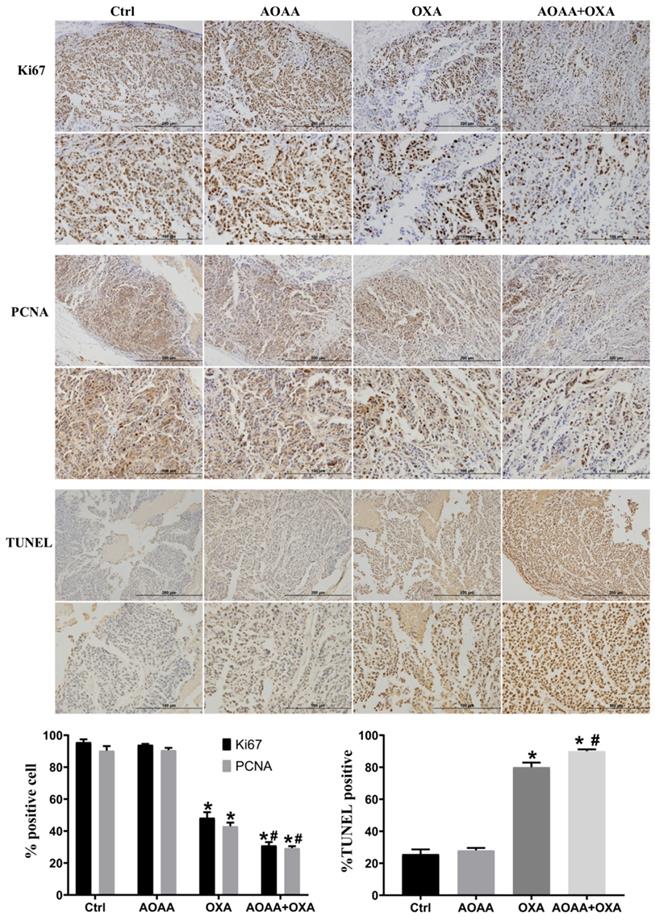
Tumor bearing xenograft mice and in vivo imaging systems were used to investigate the effect of AOAA and OXA in vivo. The results suggested that OXA alone had a significant inhibitory effect on the tumor growth. AOAA alone had no effect on tumor growth. However, AOAA significantly exaggerated the inhibitory effect of OXA, featured by further decreased tumor weight and volume. In vivo imaging of xenograft mice provided visualized evidence, which was also in accordance with the results collected by manual measuring (Fig. 5). To evaluate whether OXA combined with AOAA can synergistically amplify apoptosis in xenografts, IHC and TUNEL assays were performed and the results suggested that, the expression of Ki67 and PCNA decreased significantly and the proportion of TUNEL-positive cells increased significantly in tumors treated with both AOAA and OXA (Fig. 6). These results suggested that AOAA exaggerated the inhibitory effect of OXA in tumor growth in vivo and increased the level of apoptosis induced by OXA.
AOAA sensitizes colon cancer cells to OXA in vivo. Co-treatment with AOAA significantly exaggerated the inhibitory effect of OXA on the tumor growth. (*p < 0.05 vs ctrl. # p < 0.05 vs OXA).
AOAA sensitizes colon cancer cells to OXA in vivo. Compared with the OXA group, the AOAA+OXA group showed a further decrease in the expression of Ki67 and PCNA. TUNEL assays suggested that co-treatment with AOAA significantly increased the apoptosis induced by OXA (scale bar represented 200 and 100 µm) (*p < 0.05 vs ctrl. # p < 0.05 vs OXA).
4. Discussion
Increased expression of CBS has been revealed in colorectal cancer tissues and increased tumor-derived H2S has been validated to stimulate bioenergetics, cell proliferation, and angiogenesis in colon cancer. Inhibiting synthesis of H2S has also been exploited as a promising therapeutic target for cancer and multiple inhibitors of the endogenous synthesis of H2S have been developed. However, the effect of inhibiting endogenous production of H2S on the chemotherapeutic effect of OXA has not been illustrated. Considering the irreplaceable role of OXA in the chemotherapy of colorectal cancer, investigating the potential role of inhibiting H2S as a sensitizer of OXA may shed light on the chemoresistance of colorectal cancer. The current study indicated that inhibition of H2S synthesis utilizing AOAA, a classic inhibitor of CBS, significantly sensitized colon cancer cells to OXA both in vitro and in vivo. AOAA significantly exaggerated the apoptosis induced by OXA and increased the intracellular levels of ROS. Besides, AOAA significantly decreased intracellular levels of GSH, which is an important antioxidant. Together, these results indicated that AOAA could serve as a potential sensitizer of OXA via exaggerating apoptosis in colon cancer cells. Decreased intracellular GSH and the consequently impaired detoxification of ROS might be one of the mechanisms underlying the effect of AOAA.
There are two main apoptotic pathways: the intrinsic mitochondrial apoptosis pathway and the extrinsic death receptor pathway [29]. Both modes of apoptosis are involved in chemotherapeutic drugs related cancer cell death. Our results indicated that OXA combined with AOAA induces apoptosis synergistically through the mitochondrial apoptotic pathway. Caspases are essentially a cysteine proteases family, which play a key role in inducing apoptosis by activating downstream substrates [30]. Specifically, OXA combined with AOAA activates caspase-3, caspase-9, and PARP. Previous studies have shown that Bcl-2 family proteins also regulate mitochondrial apoptosis pathways [31, 32]. We also found that combination treatment significantly upregulated pro-apoptotic Bcl-2 family proteins (Bax), but downregulate anti-apoptotic Bcl-2 family proteins (Bcl-2).
ROS is a collective term that includes O2-derived free radicals, such as superoxide anion (O2-) and hydroxyl (HO), and O2-derived nonradicals, such as H2O2. ROS are beneficial or harmful to cells in a concentration-dependent manner [29]. In cancer cells, ROS are upregulated compared to normal cells [33]. However, contrary to tumor-promoting effects, cancer is more sensitive to ROS-induced apoptosis once ROS exceed certain physiological thresholds [17, 29]. Therefore, increasing the intracellular ROS concentration is an effective anticancer strategy [20]. Previous reports have indicated that OXA-induced apoptosis is mediated by increased intracellular levels of ROS and the consequent mitochondrial apoptosis [3, 17-19]. In addition, H2S was highly associated with ROS resistance and AOAA upregulated ROS and P53 by inhibiting H2S synthesis [7, 12, 16]. Our results suggested that, AOAA further increased the intracellular levels of ROS induced by OXA, which might be caused by decreased intracellular GSH levels.
GSH is a major intracellular nonprotein thiol that is one of the main intracellular antioxidants. Studies suggested that GSH could spontaneously (nonenzymatically) inactivates many electrophiles, including ROS and OXA [34, 35]. Previous studies suggested that AOAA reduced intracellular cystathionine, a precursor of GSH, thereby decreasing the antioxidant capacity of cells [12, 16, 36]. Our results suggested that AOAA synergizes with OXA in inhibiting GSH mediated the depletion of ROS and exaggerating apoptosis.
The acquired resistance to OXA and other chemotherapeutic reagents has been an important risk factor for decreased overall survival in colorectal cancer patients. Increased endogenous levels of H2S has been revealed to play an important role in the resistance to 5-FU in colon cancer cells [37]. The potential role of H2S in the resistance to OXA and the potential role of inhibiting synthesis of H2S in restoring sensitivity to OXA in colon cancer cells remains to be illustrated, which will also be the focus of our future work.
In conclusion, this study demonstrates that CBS is overexpressed in human colorectal cancer and AOAA sensitizes colon cancer cells to OXA via increasing apoptosis both in vitro and in vivo. The inhibition on the intracellular levels of GSH and the consequent decreased detoxification of ROS might be one of the mechanisms underlying the effect of AOAA.
Abbreviations
H2S: Hydrogen sulfide; CRC: Colorectal cancer; CBS: Cystathionine β-synthase; OXA: Oxaliplatin; AOAA: Aminooxyacetic acid; ROS: Reactive oxygen species; GSH: Glutathione; PBS: Phosphate buffer saline; DMEM: Dulbecco's Modified Eagle Medium; ATCC: American tissue culture collection; DMSO: Dimethyl sulfoxide; FBS: Fetal bovine serum; CCK-8: Cell counting kit-8; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; IC50: Half maximal inhibitory concentration; IHC: Immunohistochemistry; PCNA: Proliferating cell nuclear antigen; TUNEL: Terminal dexynucleotidyl transferase (TdT)-mediated dUTP nick end labeling.
Acknowledgements
The authors wish to thank Professor Ding-fang Bu from the Central Laboratory of Peking University First Hospital for his excellent technical assistance.
Funding
This work was supported by a grant from the National Natural Science Foundation of China (No. 81770522).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68:394-424
2. Woynarowski JM, Faivre S, Herzig MC, Arnett B, Chapman WG, Trevino AV. et al. Oxaliplatin-induced damage of cellular DNA. Molecular pharmacology. 2000;58:920-7
3. Santoro V, Jia R, Thompson H, Nijhuis A, Jeffery R, Kiakos K. et al. Role of Reactive Oxygen Species in the Abrogation of Oxaliplatin Activity by Cetuximab in Colorectal Cancer. Journal of the National Cancer Institute. 2016;108:djv394
4. Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nature reviews Drug discovery. 2016;15:185-203
5. Farrugia G, Szurszewski JH. Carbon monoxide, hydrogen sulfide, and nitric oxide as signaling molecules in the gastrointestinal tract. Gastroenterology. 2014;147:303-13
6. Shaposhnikov M, Proshkina E, Koval L, Zemskaya N, Zhavoronkov A, Moskalev A. Overexpression of CBS and CSE genes affects lifespan, stress resistance and locomotor activity in Drosophila melanogaster. Aging. 2018;10:3260-72
7. Mironov A, Seregina T, Nagornykh M, Luhachack LG, Korolkova N, Lopes LE. et al. Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:6022-7
8. Meng G, Ma Y, Xie L, Ferro A, Ji Y. Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. British journal of pharmacology. 2015;172:5501-11
9. Szabo C, Papapetropoulos A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacological reviews. 2017;69:497-564
10. Szabo C, Ransy C, Modis K, Andriamihaja M, Murghes B, Coletta C. et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. British journal of pharmacology. 2014;171:2099-122
11. di Masi A, Ascenzi P. H2S: a "double face" molecule in health and disease. BioFactors (Oxford, England). 2013;39:186-96
12. Hellmich MR, Coletta C, Chao C, Szabo C. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxidants & redox signaling. 2015;22:424-48
13. Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A. et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12474-9
14. Szczesny B, Marcatti M, Zatarain JR, Druzhyna N, Wiktorowicz JE, Nagy P. et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Scientific reports. 2016;6:36125
15. Sen S, Kawahara B, Gupta D, Tsai R, Khachatryan M, Roy-Chowdhuri S. et al. Role of cystathionine beta-synthase in human breast Cancer. Free radical biology & medicine. 2015;86:228-38
16. Bhattacharyya S, Saha S, Giri K, Lanza IR, Nair KS, Jennings NB. et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PloS one. 2013;8:e79167
17. Liang W, Cai A, Chen G, Xi H, Wu X, Cui J. et al. Shikonin induces mitochondria-mediated apoptosis and enhances chemotherapeutic sensitivity of gastric cancer through reactive oxygen species. Scientific reports. 2016;6:38267
18. Lim SC, Choi JE, Kang HS, Han SI. Ursodeoxycholic acid switches oxaliplatin-induced necrosis to apoptosis by inhibiting reactive oxygen species production and activating p53-caspase 8 pathway in HepG2 hepatocellular carcinoma. International journal of cancer. 2010;126:1582-95
19. Chen HY, Islam A, Yuan TM, Chen SW, Liu PF, Chueh PJ. Regulation of tNOX expression through the ROS-p53-POU3F2 axis contributes to cellular responses against oxaliplatin in human colon cancer cells. Journal of experimental & clinical cancer research: CR. 2018;37:161
20. Laurent A, Nicco C, Chereau C, Goulvestre C, Alexandre J, Alves A. et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer research. 2005;65:948-56
21. Thangam R, Senthilkumar D, Suresh V, Sathuvan M, Sivasubramanian S, Pazhanichamy K. et al. Induction of ROS-dependent mitochondria-mediated intrinsic apoptosis in MDA-MB-231 cells by glycoprotein from Codium decorticatum. Journal of agricultural and food chemistry. 2014;62:3410-21
22. Martinez-Balibrea E, Martinez-Cardus A, Gines A, Ruiz de Porras V, Moutinho C, Layos L. et al. Tumor-Related Molecular Mechanisms of Oxaliplatin Resistance. Molecular cancer therapeutics. 2015;14:1767-76
23. Zhang K, Zhang J, Xi Z, Li LY, Gu X, Zhang QZ. et al. A new H2S-specific near-infrared fluorescence-enhanced probe that can visualize the H2S level in colorectal cancer cells in mice. Chemical science. 2017;8:2776-81
24. Mehmood T, Maryam A, Tian X, Khan M, Ma T. Santamarine Inhibits NF-small ka, CyrillicB and STAT3 Activation and Induces Apoptosis in HepG2 Liver Cancer Cells via Oxidative Stress. Journal of Cancer. 2017;8:3707-17
25. Chowdhury AA, Chaudhuri J, Biswas N, Manna A, Chatterjee S, Mahato SK. et al. Synergistic apoptosis of CML cells by buthionine sulfoximine and hydroxychavicol correlates with activation of AIF and GSH-ROS-JNK-ERK-iNOS pathway. PloS one. 2013;8:e73672
26. Iwano S, Sugiyama M, Hama H, Watakabe A, Hasegawa N, Kuchimaru T. et al. Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science (New York, NY). 2018;359:935-9
27. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. The Journal of cell biology. 1992;119:493-501
28. Um HJ, Kwon TK. Protective effect of melatonin on oxaliplatin-induced apoptosis through sustained Mcl-1 expression and anti-oxidant action in renal carcinoma Caki cells. Journal of pineal research. 2010;49:283-90
29. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free radical biology & medicine. 2010;48:749-62
30. Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M. et al. Apoptosis and cancer: mutations within caspase genes. Journal of medical genetics. 2009;46:497-510
31. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483-7
32. Li X, Zhu F, Jiang J, Sun C, Wang X, Shen M. et al. Synergistic antitumor activity of withaferin A combined with oxaliplatin triggers reactive oxygen species-mediated inactivation of the PI3K/AKT pathway in human pancreatic cancer cells. Cancer letters. 2015;357:219-30
33. Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:4309-14
34. Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochemical pharmacology. 2003;66:1499-503
35. Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer treatment reviews. 2005;31:90-105
36. Franco R, Panayiotidis MI, Cidlowski JA. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. The Journal of biological chemistry. 2007;282:30452-65
37. Untereiner AA, Pavlidou A, Druzhyna N, Papapetropoulos A, Hellmich MR, Szabo C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochemical pharmacology. 2018;149:174-85
Author contact
![]() Corresponding authors: Pengyuan Wang, Division of General Surgery, Peking University First Hospital, Peking University, 8 Xi Shiku Street, Beijing, 100034, People's Republic of China. E-mail address: pengyuan_wangedu.cn; Yucun Liu, Division of General Surgery, Peking University First Hospital, Peking University, 8 Xi Shiku Street, Beijing, 100034, People's Republic of China. E-mail address: yucun_liucom
Corresponding authors: Pengyuan Wang, Division of General Surgery, Peking University First Hospital, Peking University, 8 Xi Shiku Street, Beijing, 100034, People's Republic of China. E-mail address: pengyuan_wangedu.cn; Yucun Liu, Division of General Surgery, Peking University First Hospital, Peking University, 8 Xi Shiku Street, Beijing, 100034, People's Republic of China. E-mail address: yucun_liucom