Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(9):2735-2746. doi:10.7150/jca.57334 This issue Cite
Review
A snapshot of the PD-1/PD-L1 pathway
Chinmoy Ghosh, Gary Luong, Yue Sun ![]()
Philips Institute for Oral Health Research, School of Dentistry and Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23298, USA.
Received 2020-12-19; Accepted 2021-1-23; Published 2021-3-5
Abstract

Cancer cells can evade the attack from host immune systems via hijacking the regulatory circuits mediated by immune checkpoints. Therefore, reactivating the antitumor immunity by blockade of immune checkpoints is considered as a promising strategy to treat cancer. Programmed death protein 1 (PD-1) and its ligand programmed death-ligand 1 (PD-L1) are critical immune checkpoint proteins that responsible for negative regulation of the stability and the integrity of T-cell immune function. Anti-PD-1/PD-L1 drugs have been developed for immune checkpoint blockade and can induce clinical responses across different types of cancers, which provides a new hope to cure cancer. However, the patients' response rates to current anti-PD-1 or anti-PD-L1 therapies are still low and many initial responders finally develop resistance to these therapies. In this review, we provides a snapshot of the PD-1/PD-L1 molecular structure, mechanisms controlling their expression, signaling modulated by PD-1/PD-L1, current anti-PD-1/PD-L1 therapies, and the future perspectives to overcome the resistance.
Keywords: immune checkpoint, PD-1, PD-L1, PD-L2, T cell, cancer immunotherapy
Introduction
Over the past ten years, immune checkpoint blockade has revolutionized the treatment for many malignant cancers, providing a new hope to heal cancer patients. Immune checkpoints refer to a set of immune-regulatory pathways maintaining self-tolerance, preventing autoimmunity, and mitigating collateral tissue damages [1-3]. Cancer cells can use immune checkpoint pathways to escape from the anti-tumor immune attack [4, 5]. Therefore, immune checkpoint blockade can remove the inhibitory signals to unleash the antitumor immune response [2, 6].
T-cell activation is a key step in the immune response initiation and regulation. When confronted by an antigen, effective activation of a naive T-cell and the subsequent immune response requires co-stimulation with “two-signal” from the antigen-presenting cells (APCs) [7, 8]. The first signal confers specificity from antigen recognition, provided by the interaction between antigenic major histocompatibility complex (MHC) and T cell receptor (TCR). The second antigen-independent signal is the “costimulatory signal,” delivered by costimulatory molecules expressed on APCs to T-cell. If T- cells receive only antigen-specific TCR stimulation in the absence of co-stimulation, they will become unresponsive (anergic) to subsequent antigenic challenge [9, 10]. In a further study, negative costimulatory (i.e., coinhibitory) signals are also found to exist. Immune checkpoint proteins deliver coinhibitory signals to negatively modulate the T-cell activation, which is critical to maintaining self-tolerance and contributes to poor anti-tumor T-cell efficacy [11-13].
Currently, the most understood pathway for T cell co-stimulation is the B7-1, B7-2/CD28 superfamily [14-16]. CD28 is a receptor constitutively expressed at the surface of T-cells. B7-1 (CD80) and B7-2 (CD86) are the ligands of CD28 that expressed at the APCs. The binding of B7-1/B7-2 with CD28 provides the primary costimulatory signal from APCs to stimulate T-cell activation [17]. Cytotoxic T lymphocyte antigen-4 (CTLA-4) is a CD28 homologue that mediates a negative regulatory effect on T-cell activation [18]. The expression of CTLA-4 at T-cells is induced by T-cell activation [19]. CTLA-4 binds to B7-1/B7-2 with a much higher affinity (10-20 fold) than CD28 [20]. The CTLA-4-B7-1/B7-2 binding mediates coinhibitory signal to prevent T-cell activation [21]. Therefore, the immune system functions by maintaining an intricate balance between CD28/costimulation-mediated T cell activation and CTLA-4 immune checkpoint-mediated inhibition.
Programmed cell death 1 (PD-1) and its ligand Programmed death-ligand 1 (PD-L1) are identified as another set of the immune checkpoint that mediates coinhibitory signals to T-cell activation [22, 23]. PD-1 and PD-L1, together with CTLA-4, are used as important drug targets to develop immune checkpoint blockade therapies. Since 2011, starting with the FDA approval of ipilimumab (an anti-CTLA4 monoclonal antibody) for immunotherapy, immune checkpoint inhibitors targeting the PD-1/PD-L1 axis were also approved to treat a broader range of cancers. The function of PD-1/PD-L1 and CTLA-4 in anti-tumor immune responses are largely distinct [24]. CTLA-4 functions at the early stage of T-cell immune response primarily in lymph nodes, whereas PD-1 functions at the later stage of T-cell immune response primarily in peripheral tissues [24].
In this review, we will focus on PD-1/PD-L1 immune checkpoints. We will provide a snapshot of the PD-1/PD-L1 molecular structure, basic biological function, and the usage of anti-PD-1/PD-L1 therapies to treat cancer.
Molecular Structure and background of the PD-1/PD-L1 pathway
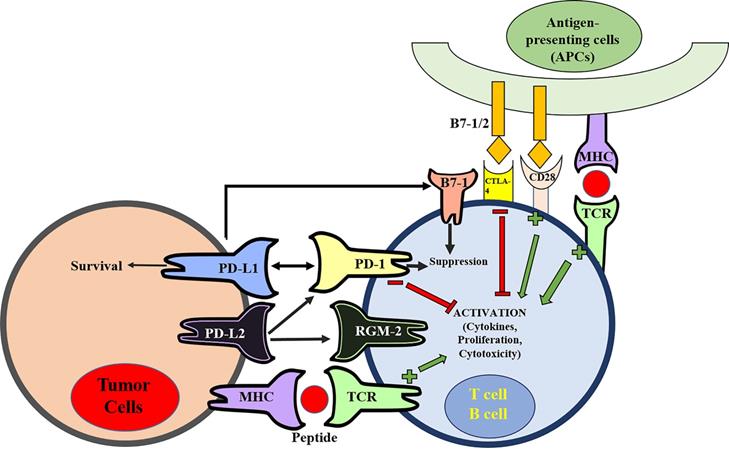
In 1992, Tasuku Honjo and his colleagues at Kyoto University discovered PD-1, a membrane protein in T-cells that was involved in the cellular process of apoptosis [25]. Several other following studies tried to reveal the patterns of PD-1's molecular interaction but it was not until 1999, when a similar B7 homolog, now known today as PD-L1, was observed as an inhibitor of human T-cell responses in-vitro [26]. These two critical studies were merged one year later in the year 2000, when Wood Freeman showed that PD-1 is a binding and functional partner of PD-L1 [23]. The specific function of PD-1 and PD-L1 was unveiled via establishment of gene-knockout mouse strains [2]. PD-1-deficient mice developed autoimmune diseases that varied depending on the genetic nature of the mice. Nishimura et al. reported that PD-1 deficiency in C57BL/6 mice led to the occurrence of lupus-like arthritis and glomerulonephritis with IgG3 and C3 deposits [3], whereas the deficiency in BALB/c mice caused fetal dilated cardiomyopathy with collateral production of autoantibodies [4, 5]. PD-L1 deficient mice were prone to autoimmune diseases [27]. Consistently, the interaction of PD-1 and PD-L1 played a dominant role in the suppression of T-cell responses in vivo [26, 28, 29]. With this insight, by 2001, another B7 homolog (PD-L2) similar in function to PD-L1 was found to also deliver a suppressive immune response by binding to PD-1 [30]. Including PD-1, PD-L2 has the ability to interact with other membrane receptors to mediate distinct biological functions [31]. PD-L2 can bind with repulsive guidance molecule family member 2 (RGM-2), a molecule that is enriched in lung macrophages. The PD-L2-RGM-2 interaction is required for maintaining the respiratory tolerance [32]. Mutagenic study and molecular modeling of PD-L1 and PD-L2 behavior revealed their interaction with B7-1 (CD-80) membrane proteins on activated T-cells, producing the inhibitory immune signal [33, 34]. This finding came as a surprise, because B7-1 was previously believed to only act as a functional ligand for CD28 and CTLA-4 [35, 36]. Therefore, at least five membrane proteins (PD-1, PD-L1, PD-L2, RGM-2, and B7-1) are involved in the PD-1/PD-L1-related pathway (Figure 1). Further studies will be required to understand the relative contributions of these molecules during activation or suppression of T cells.
The pathway of programmed cell death (PD) and Cytotoxic T lymphocyte antigen-4 (CTLA-4) in suppression of T-cell activation. The MHC-TCR interaction together with B7-1/2-CD28 interaction stimulate T-cell activation. On the contrary, CTLA-4 binds to B7-1/2 and mediates inhibitory signal to prevent T-cell activation. There are at least five interacting molecules in the PD pathway: PD-1, PD-L1, PD-L2, B7-1 (CD80), and RGM-2. PD-L1 and PD-L2 are ligands of PD-1, and the PD-L1/PD-L2 binding with PD-1 leads to suppression of T-cell activation. PD-L1 also interacts with B7-1 (CD80) on activated T-cells to inhibit T-cell activity. PD-L2 has its second receptor RGM-2. The PD-L2-RGM-2 interaction mediates respiratory tolerance.
PD-1 is a 288 amino acids protein consisting of an N-terminal IgV domain, a transmembrane domain, a cytoplasmic tail with two tyrosine-based signaling motifs and a 20-amino acids sidechain separating the IgV domain from the plasma domain. PD-1 recruits SH2/SHP3 domain containing proteins with an amino acid sequence (VDYGEL) in the N-terminal domain and a sequence (TEYATI) in the C-terminal domain [37-39]. Both sequences form a tyrosine-based immunoreceptor switch motif (ITSM) that is essential for the inhibitory function of PD-1 [40]. The ligands of PD-1 (PD-L1 and PD-L2) are both type 1 transmembrane glycoproteins containing IgC and IgV domains. They share a 40% amino acid identity, while PD-L1/PD-L2 share a 20% similarity with other B7 homologs. A comparative study between human and mouse PD-L1/PD-L2 orthologs displayed a 70% similarity [39, 41].
PD-1 is present on the surface of B-cells, T-cells and natural killer (NK) cells [42, 43], primarily regulating effector T-cell activity within tissues and limiting their lytic activity in tumors [44]. Similarly as found in T- and B-cells, PD-1 is upregulated in dendritic cells (DCs) by various inflammatory stimuli [45]. Deficits of PD-1 on DCs enhance anti-bacterial capability [44]. Although the PD-1 blockade is more commonly known as an enhancer of effector T cells in tissues and the tumor microenvironment, it may also increase NK cell activity and antibody production through direct and indirect activation of PD-1+ B-cells [46].
Previous studies have shown that tumor-associated PD-L1 facilitates apoptosis of activated T-cells [47] and stimulates IL-10 production in human peripheral blood T-cells to promote immune suppression [26]. The effects of PD-L1 on immune suppression are known to be much more complex. In addition, to induce apoptosis of T-cells and induction of IL-10, PD-L1 may also induce dysfunction of T-cells through a variety of mechanisms [45]. The expression of PD-L1 in tumor cells facilitates apoptosis of activated T-cells via causing T-cells dysfunction and anergy [26, 47-51]. In a mouse model of chronic lymphocytic infection, repetitive antigen exposure induced T-cell exhaustion with decreased effector T-cell (Teff) function. Anti-PD-L1 mAb administration reversed this exhaustion and restored Teff function [52]. This further supports the function of PD-L1 in down-regulating T-cell activation and in modulating Teff cells. In mouse tumor models, PD-L1+ tumor cells are considerably more resistant to CD8+ cytolytic T cell (CTL)-mediated destruction than their PD-L1-negative parental cells [53, 89]. Ablation of the PD-L1-PD-1 interaction by neutralizing antibodies could restore CTL-mediated killing of tumor cells, suggesting that PD-L1-PD-1 interaction forms a barrier between tumor cells and CTL [53]. These PD-L1 functions result in a “molecular shield” on cancer cells that prevents effector immune cells from killing cancer cells [53]. Including binding with PD-1, PD-L1 can also interact with B7-1 on the T-cell membrane. B7-1 not only functions as a ligand of CD28 to stimulate T-cell costimulatory signals, but it could also behave as a receptor of PD-L1 to deliver T-cell inhibitory signals [33, 34]. The relevance of this PD-L1-B7-1 interaction in tumor immune resistance, however, has not yet been determined.
PD-1/PD-L1-related cell signaling and the control of PD-1/PD-L1 expression
As the key regulator of immune tolerance and immune exhaustion, the expression of PD-1 is tightly controlled [54]. On naïve T-cells, PD-1 is only expressed in a low basal level [55]. Initial immune stimulation can induce PD-1 expression on T-cells, B-cells, macrophages, and DCs [54]. The gene Pdcd1 encodes PD-1. A number of transcription factors including NFATc1, FoxO1, AP-1, Notch, STAT3, STAT4, ISGF3, and NF-κB activate the transcription of PD-1 [54]. On the contrary, T-bet and Blimp-1 are two inhibitory factors that block the PD-1 transcription [54]. On CD8+ T-cells, PD-1 transcription can be stimulated by TCR signaling [52]. Stimulation of TCR activates NFATc1 and AP-1 [56-58]. These two transcription activators then bind to the cis-regulatory elements of the Pdcd1 gene to activate PD-1 transcription [54].
As the ligands of PD-1, PD-L1 and PD-L2 are encoded by the CD274 and PDCD1LG2 genes separately [59]. PD-L1 expression was found in tumor cells, epithelial cells, immune cells, and endothelial cells [8], while PD-L2 expression was primarily observed in antigen-presenting cells (APCs). The PD-L1 expression in cancer can be up-regulated by genetic aberration, transcription control, and post-transcriptional modulation, which contribute to cancer evasion from immune attack [60]. PD-L1 genetic copy number gains and amplifications have been found in some types of cancers such as triple-negative breast cancer (TNBC), classical Hodgkin lymphoma (cHL), primary mediastinal B-cell lymphoma (PMBCL), and squamous cell carcinomas of the vulva and cervix [60].
Unlike other members of the CD28 protein family, PD-1 creates a signal only when cross-linked together with a B or T-cell antigen receptor [23, 30]. Considered to be a negative regulator of the immune response, PD-1-mediated signaling inhibits T-cell glucose consumption, cytokine production, and cell proliferation by preventing the expression of transcription factors such as GATA-3, T-bet, and Eomesodermin [30, 51, 61]. PD-1 ligation also diminishes the phosphorylation of CD3, ZAP70, and protein kinase Cθ [62]. In B-cells, PD-1 ligation inhibits Ca2+ mobilization and phosphorylation of Igβ, Syk, PLC-γ2, and Erk1/2. These effects are dependent on SHP-2 recruitment by the ITSM motif of PD-1 [51]. It is worthy to note that CD28 stimulation or IL-2 signaling can override PD-1-mediated inhibition. The effects of PD-1-inhibited ERK activation was found to be nullified through IL-2, IL-7, or IL-15 activation [63].
The recruitment of SHP-1 and SHP-2 proteins via the cytoplasmic tail of PD-1 has been thoroughly documented in human T-cells and B-cells [51, 64]. TCR stimulation results in the phosphorylation of tyrosine residues within the ITIM and ITSM motifs in the PD-1 cytoplasmic tail, which recruits SHP-1 and SHP-2 and causes the subsequent dephosphorylation of signaling molecules within proximity downstream of CD28 and the TCR. Therefore, SHP-1 and SHP-2 must play a role in the suppression of T-cell activation. Studies regarding positional mutagenesis have suggested that the ITSM motif is critical for the inhibitory function of PD-1 [65]. Specifically, the ITSM tyrosine (Y248) of PD-1 binding with SHP-2 is mandatory for PD-1-mediated inhibition of PI3K/Akt activation [65]. SHP-2 can interact with phosphorylated ITSM-Y248 residues on two PD-1 molecules to induce PD-1 dimerization, which also promotes the SHP-2 activation [66]. SHP-1 is expressed primarily in hematopoietic cells [67]. A gene knockout study conducted on SHP-1 deficient mice showed signs of prolonged phosphorylation of the TCR/CD3 complex leading to increased activation of Lck, Fyn, and other proximal TCR signaling proteins [51, 68, 69]. Live-cell imaging studies indicate that SHP-2 dephosphorylates PD-1 upon TCR-mediated activation. The same study also found that PD-1 is part of a dynamic T-cell receptor micro-cluster that accumulates at a central supramolecular activation cluster (c-SMAC) [51]. Another study conducted on site-directed mutagenesis constructs in Jurkat T-cells expressing mutagenized PD-1, established that only mutated PD-1 Y248 avoided interaction with SHP-2, and that both Y248 and Y223 are actively involved in the inhibitory effects of PD-1 on IL-2 production [47]. Although many in vitro studies indicate the important role of SHP-2 in PD-1-mediated T-cell suppression, recent in vivo study using T cell-specific SHP-2-deficient mice suggests that SHP-2 is dispensable for T-cell exhaustion and for PD-1 signaling [70]. Consistently, another recent research indicates that PD-1 can suppress T-cell signaling by a mechanism independent of both SHP-1 and SHP-2 [71]. These reports suggest that redundant mechanisms other than SHP-1 and SHP-2 may exist to mediate the immune inhibitory function downstream of PD-1.
PD-1 ligation also controls the T-cell cycle. p27kip1, a member of the Kip/Cip family of Cdk inhibitors, abundantly presents in T-cells and interacts with Cdk2 proteins. Ubiquitin-dependent degradation of p27kip1 is required to initiate the cell cycle and the subsequent entry into the S phase by allowing the activation of Cdk2. The Skp1-Cullin-F-box (SCF) family of ubiquitin ligases, SCFskp2, primarily mediates this event [56]. TCR/CD3 and CD28 co-stimulation regulates the transcriptional activation of Skp2, the substrate recognition subunit of SCFskp2 ubiquitin ligase, and this process requires simultaneous activation of PI3K/Akt and Ras/MEK/Erk pathways [57]. Ligation of PD-1 during T-cell stimulation causes abrogated expression of Skp2, resulting in elevated p27kip1 levels and inhibition of Cdk2 [58, 59]. The deactivation of Cdk2 disables Rb phosphorylation, consequently affecting its interaction with chromatin remodeling proteins. Cdk2 deactivation also prevents the phosphorylation of checkpoint inhibitor Smad3, upregulating its transcriptional activity [59-61] and increasing the presence of G1 phase Cdk inhibitor, p15INK4B, as well as the loss of Cdk-activating phosphatase Cdc25A [58, 62, 63]. Therefore, PD-1 ligation prevents T-cells entry into the S phase.
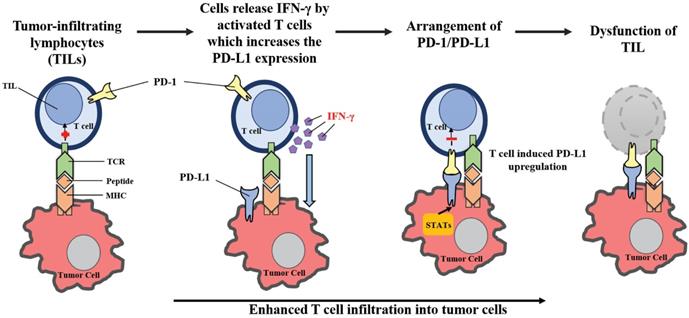
By upregulating PD-L1 expression following cancer-induced immune response, the PD-1/PD-L1-mediated evasion of tumor immunity can be described as an “adaptive resistance” (Figure 2). While PD-L1 is absent in most normal tissues, its expression can be stimulated by the presence of IFN-γ in virtually any nucleated cell [28, 29, 72, 73]. IFN-γ is produced primarily by inflammatory cells of hematopoietic origin, especially by T cells. As a result, in the case of cancer-induced immune response, PD-L1 levels in cancer cells can be upregulated from the exposure to IFN-γ. Tumor-infiltrating lymphocytes (TILs) may develop adaptive resistance through recognition of tumor-specific antigens. These antigens may also be exploited by tumor stromal cells and infiltrating hematopoietic cells, including DCs, neutrophils, macrophages and lymphocytes [2]. Upon specific binding of a T cell receptor, TILs release IFN-γ which upregulates PD-L1 expression within the surrounding cells [74]. While IFN-γ may enhance TIL effector function through differentiation, the stimulation of antigen presentation in IFN-γ can also downregulate the activation of T-cells by inducing PD-L1 expression [75, 76]. On the cell surface, PD-L1 may bind to PD-1 and B7-1, paralyzing T-cells [45]. The primary function of PD-L1 upregulation is to prevent instances of inflammation from occurring, and to limit tissue damage. PD-L1 expression within the tumor microenvironment acts as a negative feedback loop to inhibit tumor immunity. Research utilizing laser-captured microdissection and qPCR detected IFN-γ in the presence of TILs and PD-L1+ cells, but was not found within PD-L1- tumors [2]. In a mouse tumor model, mAb-mediated neutralization of IFN-γ eliminated PD-L1 upregulation in the tumor microenvironment, proving that IFN-γ is a major inducer of PD-L1 in vivo [77]. Therefore, the “adaptive resistance” mechanism model helps explain how cancer escapes the immune response, despite endogenous antitumor immune responses. Furthermore, this model helps explain why various cancer immunotherapy approaches fail to control tumor growth or cannot reach the maximal effect.
Adaptive resistance to tumor immunity mediated by PD-1/PD-L1. Following activation in lymphoid organs, tumor-specific Teffs enter the tumor site to develop TILs. Upon recognition of tumor antigens, TILs release cytokines such as IFN-γ, which stimulates the expression of PD-L1 in the tumor microenvironment. By binding to PD-1, PD-L1 provides a suppressive signal to T-cells and an anti-apoptotic signal to tumor cells, leading to T-cell dysfunction and tumor survival.
Although IFN-γ is one of the primary drivers of PD-L1 upregulation, it has been observed in a small fraction of human cases that tumors lacking TILs also express large amounts of PD-L1 without the presence of IFN-γ [78, 79]. Upregulation of PD-L1 in cancer cells can be attributed to oncogenic signaling pathways that involve the deletion or silencing of phosphatase tensin homologs (PTEN), constitutive anaplastic lymphoma kinase (ALK), and EGFR [2, 80]. Although rare in occurrence, intrinsic oncogenic induction of PD-L1 is only present in roughly 1% of patients with melanoma, but may vary up to 12% in patients diagnosed with lung cancer [78, 79].
The expression of PD-1 or PD-L1 can also be regulated by noncoding RNAs such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) [81]. miRNAs are a class of small single-stranded non-coding RNAs averagely containing about 22 nucleotides that plays important roles in modulating gene expression. By binding with the 3' untranslational region (3' UTR) of target mRNAs, miRNAs can induce the mRNAs degradation and translational repression. In this way, miRNAs down-regulate their target genes expression. Some miRNAs have been found to target to PD-1 or PD-L1 and regulate their expression levels in cancer cells. For example, miR-28, miR-4717, miR-155, miR-33a, miR-138, and miR-374b modulate PD-1 expression [81]. miR-570, miR-513, miR-34, miR-155, miR-140-3p, miR-152, miR-25-93-106b, miR-200, and miR-34 regulate PD-L1 expression [81]. In cancers, structural variations such as single nucleotide polymorphisms (SNPs) at the miRNAs binding sites of PD-L1 3' UTR can disrupt miRNAs interaction with PD-L1 mRNA, which leads to elevated PD-L1 expression [82]. miRNAs can also indirectly modulate PD-1/PD-L1 expression by modulating their upstream or downstream pathways. miR-197 and miR-3127-5p target to STAT3, a regulator of PD-L1 expression, to indirectly modulate PD-L1 levels [83, 84]. miR-21 downregulates JAK2 and STAT1 to inhibit IFN-γ-induced STAT1 signaling [85]. In this way, miR-21 decreases PD-1 expression in macrophage. By targeting to phosphatase inhibition and tensin homolog (PTEN), MiR-20b, miR-21, and miR-130b enhance the expression of PD-L1 [86].
lncRNAs are a diverse class of transcribed RNAs with a length of more than 200 nucleotides that are not translated into proteins. lncRNAs regulate gene expression via diverse mechanisms [87]. The lncRNA small nucleolar RNA host gene 20 (SNHG20) promotes PD-L1 expression in esophageal squamous cell carcinoma (ESCC) [88]. Actin filament-associated protein one antisense RNA 1(AFAP1-AS1), another lncRNA, was found to be co-expressed with PD-1 in nasopharyngeal carcinoma (NPC) [89]. High expression of AFAP1-AS1 and PD-1 was strongly correlated with distant metastasis and poor prognosis that reveals a novel marker and candidate target for clinical trials [89].
In cancers, other types of noncoding RNAs were also found to be involved in modulating PD-1/PD-L1. A study in colorectal cancer (CRC) highlights the role of circular RNA in accommodating PD-L1 expression, revealing that circular RNA has_circ_0020397 promotes the expression of PD-L1 by inhibiting miR-138 activity [90].
As of 2009, a statistical analysis of renal cancer predicted a worse prognosis relative to PD-L1- tumors [91]. Following the study, analysis of various tumors indicated that PD-L1 expression levels might help predict patient survival [73, 92-96]. However, considering the broad range of reported outcomes, it is worthy to note that factors ranging from cancer type, stage assessed, IHC technique and treatment history may contribute to variable results associated with the patients' prognosis. Although most studies on PD-1 ligand expression have been focused primarily on PD-L1, PD-L2 is also reported to be positively expressed in various tumors. PD-L2 is upregulated in certain B cell lymphomas, such as primary mediastinal B-cell lymphoma, follicular cell B-cell lymphoma and characteristic in Hodgkin's disease [97]. These upregulations are commonly associated with gene amplification or rearrangement with the class II major histocompatibility complex (MHC) transactivator (CIITA) locus, a transcriptionally active region in B-cell lymphomas [97].
Anti-PD-1/PD-L1 therapies in the treatment of cancer
The important role of PD-1/PD-L1 in tumor evasion was first validated in animal models. The treatment with anti-PD-L1 monoclonal antibodies can inhibit the growth of J558L cells, a PD-L1-expressing BALB/c mouse myeloma, in the in vivo mouse models [98]. In PD-1 deficient mice, the metastasis of B16 melanoma from the spleen to the liver was significantly inhibited [99]. These results support that the blockade of PD-1 or PD-L1 has potential to be an effective strategy to treat cancer.
In recent years, more and more therapeutic agents that target PD-1/PD-L1 pathways are developed [81]. In Table 1, the anti-PD-1 or PD-L1 drugs currently approved by FDA or under clinical trials are summarized. In recent years, anti-PD-1/PD-L1 immunotherapy has shown positive results in clinical applications by inducing regression of tumor growth and metastasis [45]. Clinical benefits include possessing durable effects, tolerable toxicity, application to a broad spectrum of cancer types, and effectiveness in solid tumors [100].
Summary of the clinical development of therapeutic agents that target PD pathways in clinical trials
| Target | Ligand | Biological function | Therapeutic agent | Class | State of clinical development |
|---|---|---|---|---|---|
| PD-1 | PD-L1, PD-L2 | Negative T-cell costimulation (primarily at priming); attenuate peripheral activity, preserve T-cell function in the context of chronic antigen | Niovolumab [MDX-1106 (also known as BMS-936558) | Human IgG4 | Phase I/II trials in patients with melanoma, renal cell carcinoma, Hodgkin's lymphoma, Head and neck cancer, urothelial carcinoma, hepatocellular carcinoma and lung cancers. |
| Pembrolizumab (MK3475) | Humanized IgG4 | Phase I trial in multiple cancers like melanoma and for metastatic nonsmall-cell lung cancer (NSCLC), gastric cancer. | |||
| CT-011 | Humanized IgG1k | Phase I/II trial in multiple cancers | |||
| AMP-514 (MEDI0680) | PD-L2 IgG2a fusion protein | Phase I/II trial in multiple cancers | |||
| PDR001 | Humanized IgG4 | Phase I/II trial in multiple cancers | |||
| Cemiplimab | Human IgG4 | Pre-Registered in Metastatic CSCC; NSCLC; Cervical Cancer | |||
| Tislelizumab | Humanized IgG4 | Phase III trial in unresectable HCC | |||
| Sintilimab (IBI-308) | Humanized IgG4 | Phase II trial in NSCLC | |||
| Spartalizumab | Humanized IgG4/k | Phase II trial in advanced melanoma | |||
| Atezolizumab | Humanized IgG1k | Phase II trial in NSCLC | |||
| Camrelizumab | Humanized IgG4 | Phase II trial in Non-squamous NSCLC and squamous esophageal Cancer NSCLC | |||
| SHR-1210 | Humanized IgG4 | Phase II trial in Gastric Cancer | |||
| Cetrelimab (JNJ-63723283) | IgG4 | Phase I/II trial in Multiple Myeloma; Castration-Resistant Prostatic Neoplasm | |||
| TTI-622 | Humanized IgG4 | Phase I trial in Lymphoma; Myeloma | |||
| HLX10 | Humanized IgG4 | Phase I trial in Solid Tumor | |||
| PF-06801591 | Humanized IgG4 | Phase I trial in Solid Tumor; Prostatic Cancer; Melanoma; Ovarian Cancer; Sarcoma; Hodgkin lymphoma | |||
| PD-L1 | PD-1, B7-1 (CD80) | Attenuate T-cell activity in inflamed peripheral tissues | Durvalumab (MEDI4736) | Humanized IgG1 | It is approved by FDA for treatment of metastatic urothelial carcinoma |
| Atezolizumab (MPDL-3280A) | Human IgG1 | FDA approved for treatment of metastatic NSCLC | |||
| Avelumab (MSB0010718C) | Fully Humanized IgG1 | FDA approved for treatment of metastatic Marked cell carcinoma (MCC) | |||
| MDX-1105/BMS-936559 | Human IgG4 | Phase I/II trial in multiple cancers | |||
| M-7824 | Fully Humanized IgG1 | Phase II trial in Advanced solid tumors | |||
| CX-072 | Humanized IgG4 | Phase II trial in Solid tumors; Lymphomas | |||
| MSB-2311 | Humanized IgG4 | Phase II trial in Solid tumors | |||
| FS-118 | Human IgG1 | Phase II trial in Advanced cancer | |||
| LY-3300054 | Human IgG1 | Phase I trial in Solid tumors |
In clinical trials involving anti-PD-1, tumor regression was observed in patients with melanoma, renal cell carcinoma, non-small cell lung cancer, and bladder cancer [101, 102]. A large phase I clinical trial with the anti-PD-1 antibody MK-3475 was recently shown to lead to response rates of around 38% in patients who had advanced melanoma [103]. Another subsequent study reported an overall response rate of 26% in patients who continue to suffer from melanoma after being treated with ipilimumab (a monoclonal antibody of CTLA-4) [6, 104]. The phase III trial of nivolumab, a different anti-PD-1 antibody also showed benefits in patients who had metastatic melanoma without a BRAF mutation [105]. In this trial, compared with patients treated with dacrabazine chemotherapy, patients treated with nivolumab showed a better objective response rate (40% vs 13.9%) and a better one year overall survival rate (72.9% vs 42.1%) [105]. This showed an overall improvement in survival for patients treated with nivolumab as compared to patients treated with docetaxel chemotherapy. In December 2014, the FDA approved nivolumab as a treatment for patients with metastatic melanoma and in the following year, approved nivolumab clinical application for patients with previously treated advanced or metastatic non-small cell lung cancer [6].
A number of companies developing and testing antibodies targeting PD-1 or PD-L1 currently have no valid information on their clinical performance. Theoretically it is possible that a PD-1 antibody would prevent PD-1 from interacting with both PD-L1 and PD-L2 but not the interaction between PD-L1 and B7-1 [2]. Most PD-L1 antibodies block the interaction between PD-L1 and B7-1 and between PD-L1 and PD-1 but would not block PD-1 from interacting with PD-L2. Therefore, it is possible that PD-1 and PD-L1 antibodies will not have redundant activity depending on which interactions dominate in a particular cancer.
The response rates to current anti-PD-1 or anti-PD-L1 therapies are still very limited. Therefore, it is urgently required to identify biomarkers that can indicate the patients' response. Initial phase I trials with anti-PD-1 therapy (nivolumab) reported that PD-L1 expression in tumor cells, measured on pretreatment archival samples by immunohistochemical (IHC) methods, could potentially serve as a predictive biomarker, broadening the range of treatable patients [95]. Patients with PD-L1 positive tumors (≥ 5% staining for PD-L1 on tumor cells) had an objective response rate of 36% (9 of 25 patients) whereas patients with PD-L1 negative tumors did not show any objective clinical responses (0 of 17 patients) [6]. Although PD-L1 expression in tumor tissues has been correlated to higher response rates, its clinical benefits have yet to be proven. Recent studies suggest that the PD-L1 expression status of tumor-infiltrating immune cells (TIICs) may also serve as a biomarker in predicting immunotherapy response in different patient subgroups. In a phase I study of anti-PD-L1 mAb (Atezolizumab), urothelial bladder cancer patients with tumors containing PD-L1-positive TIICs shows a higher response rate to this immunotherapy [106].
Combination therapy for clinical benefits against cancer
Combination therapy has been an option to improve the response rate of cancer patients to anti-PD-1/PD-L1 treatment. The main principle for most combination therapies is to boost the efficiency of PD-1/PD-L1 blockade by either improving tumor antigen presentation or rescuing dysfunctional immune effector cells. Combining anti-PD-1/PD-L1 therapy with other immune checkpoint inhibitors, cancer vaccines and immune-stimulatory agents, chemotherapy, and radiotherapy has shown some success in various types of cancers [107, 108].
Preclinical studies in murine models indicate that CTLA-4 and PD-1 regulate distinct inhibitory pathways consisting of non-overlapping mechanisms of action, suggesting combination therapy with both might be more effective than either one alone [109, 110]. Early studies that used conventional KO mice demonstrated that the major role of CTLA-4 lies in regulating the T-cell response to self-antigens because CTLA-4-KO mice spontaneously produce massive infiltrating T-cells to normal organs. These tissue-infiltrating T-cells are highly active and eventually result in death by causing damage to normal tissue without specific antigen exposure [11, 111]. Consistently, anti-CTLA-4 mAb can induce various types of autoreactive T-cell responses in various mouse models [112, 113]. Mice with Treg selective ablation of CTLA-4 reiterate the majority of autoimmune phenotypes found in conventional KO mice, including tissue-infiltrating T-cells, harming normal tissues and organs [114]. Therefore, the physiological function of CTLA-4 appears to be suppressing T-cell responses to self-antigens while controlling Treg activity.
In contrast to CTLA-4 KO mice, PD-L1 KO mice do not spontaneously develop inflammation within their organs, and experience no substantial changes to life span [115]. However, PD-1 KO mice typically develop strain-specific autoimmune diseases in the span of a few months [116]. Both PD-1 and PD-L1 KO mice are prone to autoimmune diseases because severe symptoms develop when they are immunized with autoantigens [115]. As PD-L1 has a limited distribution in normal tissues and is largely an inducible molecule controlled by IFN-γ, its major physiological functions lie in mediating negative feedback in tissue inflammation. In the context of tumor growth, which is often promoted by chronic inflammation, anti-PD-1/PD-L1 therapies selectively modulates inflammatory T-cell responses at the tumor site and inhibits PD-L1 induced tumor function. Thus, the primary physiological function of the PD-1/PD-L1 pathway is to control ongoing inflammatory responses and prevent the spread of inflammation, rather than the systematic regulation of autoreactive T-cell responses. These attributes place anti-PD therapy in a different category scientifically and practically from anti-CTLA-4 therapy.
Substantial data suggests the possible benefits by combining anti-CTLA-4 and anti-PD-1/PD-L1 therapies. Anti-CTLA-4 has been found to accelerate T-cells infiltration into tumors, which resulted with an increase in the number of T-cells and a concomitant increase in IFN-γ production [117]. This can induce expression of PD-L1 in the tumor microenvironment, with subsequent inhibition of anti-tumor T-cell responses. However, it can enhance the chance of benefit from anti-PD-1 and anti-PD-L1 therapies. Thus, the combinative treatment with anti-CTLA-4 plus anti-PD-1 or anti-PD-L1 should lead to the creation of an immunogenic tumor microenvironment with clinical benefits for patients regardless of their tumor original PD-L1 expression levels. From a recent phase I clinical trial, data indicated the administration of anti-CTLA-4 (ipilimumab) and anti-PD-1 (nivolumab), in patients with metastatic melanoma resulted in similar response rates in the setting of concurrent therapy regardless of PD-L1 expression in pretreatment tumor tissues [118]. For patients with PD-L1-positive tumors, the objective response rate was 46% (6 of 13 patients), similar to a response rate of 41% (9 of 22 patients) in patients with PD-L1-negative tumors. For a combinatorial study with anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab) in patients with metastatic renal cell carcinoma (mRCC), similar data was reported [6].
Conventional cancer therapies such as radiotherapy and chemotherapy can induce tumor cell death to promote tumor antigens release. Furthermore, they can activate T-cells and enhance T-cells migrate into tumor tissues [119]. Thus, combination of conventional therapies with immune checkpoint therapies should create an “immunogenic” tumor microenvironment effective in patients. A number of ongoing trials combining radiation therapy and anti-PD-1/PD-L1 mAbs provide valuable information regarding schedule, safety, and efficacy in these combinations for future studies [120].
In radiotherapy, cancer cells are generally killed by radiation. However, accumulating evidence suggests it also has systemic effects such as reverting tumor suppressive barriers in the tumor microenvironment, and indirectly stimulating the immune system by releasing tumor antigens [121, 122]. In clinical trials and preclinical models, radiotherapy has shown synergy with various immunotherapeutic treatments, providing primary rationale to combine radiotherapy with other immunotherapies [123]. Combining radiotherapy with PD-1/PD-L1 blockade therapy in pre-clinical studies activated cytotoxic T lymphocytes (CTLs) and reduced myeloid-derived suppressor cells [123]. These results propelled two open phase III trials of Nivolumab combination with radiotherapy for NSCLC (NCT02768558) and glioblastoma (NCT02617589), currently still ongoing [121].
Chemotherapy eradicates cancer cells by targeting its DNA synthesis and replication process. Recent reports however suggest that it also has several off‐target effects. One particular effect is the priming of tumor‐specific T-cells by increasing the presence of tumor antigens following cell death, and subverting immunosuppressive factors [124]. In consideration of these chemotherapeutic off-target tendencies, appropriate combination of chemotherapy drugs with PD‐1/PD-L1 blockade could augment the efficacy of the anti-PD-1/PD-L1 therapies, particularly in less immunogenic, chemo‐sensitive tumors. A recent phase II clinical study reported that pembrolizumab combined with pemetrexed/carboplatin enhanced the efficacy of chemotherapy alone for the treatment of metastatic non-squamous NSCLC, and has been approved by FDA (KEYNOTE‐021) [121, 125].
Other combinational therapies under development utilize the blockade of multiple inhibitory pathways, such as LAG-3 [126], TIM-3 [127], VISTA [128], BTLA [129], and oncolytic virus [130]. The development of these therapies remain critical in inducing immune antitumor responses in cancer patients, even for individuals who have been diagnosed with non-immunogenic or PD-L1-negative tumors. Although major histocompatibility complex II (MHC-II) has been previously reported to be the ligand of the coinhibitory receptor LAG3, LSECtin has also been reported to be an additional ligand [131, 132]. LSECtin is expressed by liver and tumor cells and may account for the biological role of LAG3 in CD8+ and natural killer (NK) cells, as neither cell type interacts with MHC-II. Further complexity has been observed in the context of the coinhibitory receptor TIM3, as four more ligands have been reported to date: Galectin-9, PtdSer, HMGB1, and CEACAM1 [132, 133]. The underlying mechanisms of ligand regulation, however, whether they affect each other's binding, and whether each ligand leads to unique downstream signaling events remains unclear. Furthermore, although TIM3 is thought to be a primary marker of T-cell activation and exhaustion, TIM3 also functions to attenuate NK cell cytotoxicity [134]. This suggests that other costimulatory molecules like TIM3 may have significant biological functions in a multitude of other cell types. VISTA, another suppressor of T-cell activation, presents yet another ambiguity. Studies have indicated VISTA is most likely both a ligand with an unknown receptor on APCs (homologous to PD-L1) and a receptor on T-cells with an unknown ligand [134, 135]. Furthermore, the biological roles of several B7 ligand family proteins, including their counter-receptors, also remain to be understood. B7-H3 is believed to have both costimulatory and coinhibitory functions, likely dependent on the context of expression [136, 137], but both the receptor and molecular interactions of B7-H3 in posttranscriptional regulation remain unknown.
Oncolytic viruses (OVs) replicate selectively in cancer cells and kill tumor cells without inducing damage to normal tissue [138]. Known to be effective for advanced melanoma, OVs promote innate and adaptive antitumor immunity [139]. Development of therapy combining T‐VEC, oncolytic virus therapy, and pembrolizumab is currently undergoing a phase I clinical trial [140, 141]. Recent data indicates objective tumor response compared to applying pembrolizumab alone (NCT02263508) but further trials are necessary to establish this as a standard option [142].
A key factor in melanoma proliferation is BRAF mutation. The small molecule inhibitors targeting mutant BRAF induces a significant clinical response in specific patients, but remain limited in other cases [143]. It is believed that PD‐L1 upregulation is associated with the development of resistance to BRAF inhibitors, providing a rationale for possible combinative therapies. This insight has paved the way for a fruitful approach of combining PD‐1/PD-L1 blockade with BRAF inhibition in preclinical models. Whilst several clinical trials are ongoing (NCT01656642 and NCT02224781), the success rates have not been promising (NCT02027961) [144, 145]. Vascular endothelial growth factor (VEGF) is an angiogenic factor that influences growth and survival in the vascular endothelium. Although VEGF inhibitors are used for preventing angiogenesis and/or normalization of vascular permeability in tumor microenvironment, VEGF inhibition also promotes the differentiation and function of immune cells [146]. Moreover, certain types of VEGF receptors are also expressed by DCs, macrophages and lymphocytes, evoking immune suppression [147]. Initial studies of the relationship between VEGF signaling and the immune system suggest the combination of PD‐1/PD-L1 blockade with VEGF inhibition have the potential to enhance the efficiency of PD‐1/PD-L1 blockade [148].
Acknowledgements
This work was supported by the American Cancer Society (Research Scholar Grant RSG-17-064-01-TBE).
Author Contributions
C.G. and G.L. contributed to conceptualization and writing of the manuscript. Y.S. helped finalizing the presented idea and supervised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
2. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
3. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450-61
4. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39
5. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. 2018;18:269-82
6. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56-61
7. Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042-9
8. Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. 1999;96:185-90
9. Podojil JR, Turley DM, Miller SD. Therapeutic blockade of T-cell antigen receptor signal transduction and costimulation in autoimmune disease. Adv Exp Med Biol. 2008;640:234-51
10. Appleman LJ, Boussiotis VA. T cell anergy and costimulation. Immunol Rev. 2003;192:161-80
11. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP. et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985-8
12. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141-51
13. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A. et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319-22
14. Karandikar NJ, Vanderlugt CL, Bluestone JA, Miller SD. Targeting the B7/CD28:CTLA-4 costimulatory system in CNS autoimmune disease. J Neuroimmunol. 1998;89:10-8
15. Oosterwegel MA, Greenwald RJ, Mandelbrot DA, Lorsbach RB, Sharpe AH. CTLA-4 and T cell activation. Curr Opin Immunol. 1999;11:294-300
16. Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225-52
17. Green JM, Noel PJ, Sperling AI, Walunas TL, Gray GS, Bluestone JA. et al. Absence of B7-dependent responses in CD28-deficient mice. Immunity. 1994;1:501-8
18. Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229:126-44
19. Iida T, Ohno H, Nakaseko C, Sakuma M, Takeda-Ezaki M, Arase H. et al. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J Immunol. 2000;165:5062-8
20. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL. et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366:76-9
21. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459-65
22. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565-94
23. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34
24. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39:98-106
25. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887-95
26. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365-9
27. Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M. et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691-6
28. Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336-47
29. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467-77
30. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261-8
31. Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med. 2003;197:1083-91
32. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM. et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211:943-59
33. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111-22
34. Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM. et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010;116:1291-8
35. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561-9
36. Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci U S A. 1990;87:5031-5
37. Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. 1999;17:875-904
38. Neel BG, Gu H, Pao L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284-93
39. Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M. et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337-47
40. Sidorenko SP, Clark EA. The dual-function CD150 receptor subfamily: the viral attraction. Nat Immunol. 2003;4:19-24
41. Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci U S A. 2008;105:10483-8
42. Fanoni D, Tavecchio S, Recalcati S, Balice Y, Venegoni L, Fiorani R. et al. New monoclonal antibodies against B-cell antigens: possible new strategies for diagnosis of primary cutaneous B-cell lymphomas. Immunol Lett. 2011;134:157-60
43. Terme M, Ullrich E, Aymeric L, Meinhardt K, Desbois M, Delahaye N. et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011;71:5393-9
44. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015-29
45. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384-91
46. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L. et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206-10
47. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793-800
48. Goldberg MV, Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM. et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood. 2007;110:186-92
49. Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U. et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003;170:3637-44
50. Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H. et al. Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood. 2007;110:180-5
51. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front Immunol. 2016;7:550
52. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682-7
53. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111:3635-43
54. Bally AP, Austin JW, Boss JM. Genetic and Epigenetic Regulation of PD-1 Expression. J Immunol. 2016;196:2431-7
55. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H. et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765-72
56. Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181:4832-9
57. Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc Natl Acad Sci U S A. 2012;109:15419-24
58. Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476-89
59. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677-704
60. Zerdes I, Matikas A, Bergh J, Rassidakis GZ, Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene. 2018;37:4639-61
61. Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, Kaja MK. et al. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006;25:2623-33
62. Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D. et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J Immunol. 2003;170:711-8
63. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229:114-25
64. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201-17
65. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98:13866-71
66. Patsoukis N, Duke-Cohan JS, Chaudhri A, Aksoylar HI, Wang Q, Council A. et al. Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun Biol. 2020;3:128
67. Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G. et al. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science. 1996;272:1173-6
68. Lorenz U, Ravichandran KS, Burakoff SJ, Neel BG. Lack of SHPTP1 results in src-family kinase hyperactivation and thymocyte hyperresponsiveness. Proc Natl Acad Sci U S A. 1996;93:9624-9
69. Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. 2006;177:5990-6
70. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F. et al. Shp-2 Is Dispensable for Establishing T Cell Exhaustion and for PD-1 Signaling In vivo. Cell Rep. 2018;23:39-49
71. Xu X, Hou B, Fulzele A, Masubuchi T, Zhao Y, Wu Z. et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J Cell Biol. 2020 219
72. Flies DB, Sandler BJ, Sznol M, Chen L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J Biol Med. 2011;84:409-21
73. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL. et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37
74. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749-95
75. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163-89
76. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT. et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116
77. Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran QA, Lane AP. et al. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;33:280-9
78. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ. et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84-8
79. Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M. et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014;94:107-16
80. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL. et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355-63
81. Ding L, Lu S, Li Y. Regulation of PD-1/PD-L1 Pathway in Cancer by Noncoding RNAs. Pathol Oncol Res. 2020;26:651-63
82. Wang W, Sun J, Li F, Li R, Gu Y, Liu C. et al. A frequent somatic mutation in CD274 3'-UTR leads to protein over-expression in gastric cancer by disrupting miR-570 binding. Hum Mutat. 2012;33:480-4
83. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F. et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. 2015;23:717-27
84. Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L. et al. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J Cell Mol Med. 2018
85. Xi J, Huang Q, Wang L, Ma X, Deng Q, Kumar M. et al. miR-21 depletion in macrophages promotes tumoricidal polarization and enhances PD-1 immunotherapy. Oncogene. 2018;37:3151-65
86. Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y. et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014;75:348-53
87. Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206
88. Zhang C, Jiang F, Su C, Xie P, Xu L. Upregulation of long noncoding RNA SNHG20 promotes cell growth and metastasis in esophageal squamous cell carcinoma via modulating ATM-JAK-PD-L1 pathway. J Cell Biochem. 2019
89. Tang Y, He Y, Shi L, Yang L, Wang J, Lian Y. et al. Co-expression of AFAP1-AS1 and PD-1 predicts poor prognosis in nasopharyngeal carcinoma. Oncotarget. 2017;8:39001-11
90. Zhang XL, Xu LL, Wang F. Hsa_circ_0020397 regulates colorectal cancer cell viability, apoptosis and invasion by promoting the expression of the miR-138 targets TERT and PD-L1. Cell Biol Int. 2017;41:1056-64
91. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327-37
92. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K. et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104:3360-5
93. Liu Y, Zeng B, Zhang Z, Zhang Y, Yang R. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol. 2008;129:471-81
94. Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N. et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11:2947-53
95. Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS. et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004;101:17174-9
96. Wu C, Zhu Y, Jiang J, Zhao J, Zhang XG, Xu N. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 2006;108:19-24
97. Ghebeh H, Mohammed S, Al-Omair A, Qattan A, Lehe C, Al-Qudaihi G. et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: correlation with important high-risk prognostic factors. Neoplasia. 2006;8:190-8
98. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293-7
99. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133-44
100. Bishayee A, Block K. A broad-spectrum integrative design for cancer prevention and therapy: The challenge ahead. Semin Cancer Biol. 2015;35(Suppl):S1-S4
101. Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L. et al. Application of PD-1 Blockade in Cancer Immunotherapy. Comput Struct Biotechnol J. 2019;17:661-74
102. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
103. Spain L, Younger E, Hatipoglu E, Larkin J. Pembrolizumab in the management of metastatic melanoma. Melanoma Manag. 2015;2:315-25
104. Achkar T, Tarhini AA. The use of immunotherapy in the treatment of melanoma. J Hematol Oncol. 2017;10:88
105. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-30
106. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C. et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558-62
107. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134-44
108. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L. et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521-32
109. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;38:255
110. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275-80
111. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541-7
112. Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184:783-8
113. Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333-6
114. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271-5
115. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327-36
116. Yoshida T, Jiang F, Honjo T, Okazaki T. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc Natl Acad Sci U S A. 2008;105:3533-8
117. Wu L, Wu MO, De la Maza L, Yun Z, Yu J, Zhao Y. et al. Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine mesothelioma model. Oncotarget. 2015;6:12468-80
118. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM. et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-33
119. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32:1267-84
120. Tang C, Wang X, Soh H, Seyedin S, Cortez MA, Krishnan S. et al. Combining radiation and immunotherapy: a new systemic therapy for solid tumors? Cancer Immunol Res. 2014;2:831-8
121. Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD-1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med. 2018;283:110-20
122. Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10:718-26
123. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105:256-65
124. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21:15-25
125. Langer CJ, Socinski MA, Patel JD, Sandler AB, Schiller JH, Leon L. et al. Isolating the Role of Bevacizumab in Elderly Patients With Previously Untreated Nonsquamous Non-Small Cell Lung Cancer: Secondary Analyses of the ECOG 4599 and PointBreak Trials. Am J Clin Oncol. 2016;39:441-7
126. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E. et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171:1393-405
127. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175-86
128. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y. et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577-92
129. Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P. et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010;120:157-67
130. Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK. et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014;22:1949-59
131. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. 2017;276:80-96
132. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y. et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. 2018;9:176-89
133. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20:173-85
134. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8:1069-86
135. Ito H, Seishima M. Regulation of the induction and function of cytotoxic T lymphocytes by natural killer T cell. J Biomed Biotechnol. 2010;2010:641757
136. Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol. 2005;6:223
137. Schildberg FA, Hagmann CA, Bohnert V, Tolba RH. Improved transplantation outcome by epigenetic changes. Transpl Immunol. 2010;23:104-10
138. Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2:295-300
139. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014-22
140. Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum Vaccin Immunother. 2018;14:839-46
141. Johnson DB, Puzanov I, Kelley MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7:611-9
142. Dang TO, Ogunniyi A, Barbee MS, Drilon A. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev Anticancer Ther. 2016;16:13-20
143. Kudchadkar R, Paraiso KH, Smalley KS. Targeting mutant BRAF in melanoma: current status and future development of combination therapy strategies. Cancer J. 2012;18:124-31
144. Pelster MS, Amaria RN. Combined targeted therapy and immunotherapy in melanoma: a review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther Adv Med Oncol. 2019;11:1758835919830826
145. Karachaliou N, Gonzalez-Cao M, Sosa A, Berenguer J, Bracht JWP, Ito M. et al. The combination of checkpoint immunotherapy and targeted therapy in cancer. Ann Transl Med. 2017;5:388
146. Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets. 2010;11:1000-17
147. Li YL, Zhao H, Ren XB. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med. 2016;13:206-14
148. Kudo M. Scientific Rationale for Combined Immunotherapy with PD-1/PD-L1 Antibodies and VEGF Inhibitors in Advanced Hepatocellular Carcinoma. Cancers (Basel). 2020 12
Author contact
![]() Corresponding author: Yue Sun. Philips Institute for Oral Health Research, School of Dentistry and Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23298, USA. E-mail: ysun4edu.
Corresponding author: Yue Sun. Philips Institute for Oral Health Research, School of Dentistry and Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23298, USA. E-mail: ysun4edu.