Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(15):4729-4738. doi:10.7150/jca.50868 This issue Cite
Research Paper
Data mining analysis of the prognostic impact of N6-methyladenosine regulators in patients with endometrial adenocarcinoma
Junyu Zhai1,2#, Shang Li1,2#, Yu Li3, Yanzhi Du1,2 ![]()
1. Center for Reproductive Medicine, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200135, China.
2. Shanghai Key Laboratory for Assisted Reproduction and Reproductive Genetics, Shanghai 200135, China.
3. Department of Gynecology and Obstetrics, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China.
#These authors contributed equally to this work.
Received 2020-7-20; Accepted 2021-5-23; Published 2021-6-5
Abstract

We reanalyzed the expression of 16 acknowledged N6-methyladenosine (m6A) RNA regulators in 406 endometrial adenocarcinoma patients and 19 controls using The Cancer Genome Atlas (TCGA) dataset, and further verified our results using Gene Expression Omnibus (GEO) dataset and real-time quantitative polymerase chain reaction. Thirteen m6A RNA methylation regulators were differentially expressed between patients with endometrial adenocarcinoma and controls. FTO, RBM15, and YTHDF1, were identified as independent prognostic markers and closely associated with International Federation of Gynecology and Obstetrics grade in endometrial cancer patients. GEO dataset also verified the differential expression of FTO and RBM15 between patients with endometrial adenocarcinoma and hyperplasia. Functional enrichment and ingenuity pathway analysis network suggested that FTO and RBM15 contributed to the survival of patients with endometrial adenocarcinoma via the regulation of connective tissue development, catabolic process, RNA stability, oxidative demethylation, temperature homeostasis, and energy metabolism through IGF1, IRS1, RBM24, LARP1, and CBFA2T3. The decreased FTO expression and increased RBM15 expression in endometrial adenocarcinoma from our validation cohort was consistent with in silico analysis using TCGA and GEO datasets. In conclusion, m6A methylation regulators, especially FTO, RBM15, and YTHDF1, are critical in the progression and prognosis of endometrial adenocarcinoma.
Keywords: N6-methyladenosine, Endometrial Adenocarcinoma, Energy Metabolism, RNA-Binding Protein
Introduction
As the most common gynecologic carcinoma [1], more than 80% of endometrial cancers are adenocarcinomas of endometrium, originating from endometrial cells that grow out of control. Endometrial carcinoma accounts for most cases of endometrial adenocarcinoma. In addition, serous adenocarcinoma, adenosquamous carcinoma, and uterine carcinosarcoma also belong to endometrial cancers [2]. Although a considerable number of studies have investigated the potential mechanisms and biomarkers of endometrial cancers [3, 4], the etiology remains unclear. Novel methods for early detection and prognostic prediction for patients, especially those with advanced or recurrent endometrial cancers are essential to improve long-term outcomes of patients.
Aberrant gene expression, induced by epigenetic alterations, contributes to the development of endometrial cancers [5]. Preliminary data suggests that DNA methylation biomarkers are useful in early diagnosis and risk prediction for high-risk populations of endometrial cancers [5, 6]. DNA hypermethylation can be reversed by epigenetic inhibitors, leading to high expression of silenced genes and reversal of malignant biological behavior [7]. Thus, epigenetics provides a potential target in the clinical prediction and management of endometrial cancer.
In addition to DNA methylation, the role of N6-methyladenosine (m6A) in cancers has recently attracted the attention of more and more researches. As the most abundant RNA modification in eukaryotic cells, m6A modifies approximately 0.1-0.4 % of all adenosines in RNA, accounting for about 50 % of total methylated ribonucleotides [8]. m6A is a dynamic and reversible epigenetic modification, whose dynamic methylation and biological function are controlled by “writers” “erasers”, and “readers” that promote, remove, and exert the functions of m6A-modified sites, respectively [8]. “Writers” are composed of several methyltransferase including methyltransferase-like 3 (METTL3), METTL14, METTL16, Wilms' tumor 1-associated protein (WTAP), RNA-binding motif protein 15 (RBM15), KIAA1429, and zinc finger CCCH-type containing 13 (ZC3H13), etc. [9, 10]. “Erasers” are demethylases that catalyze m6A demethylation, among which fat mass and obesity-associated protein (FTO) is firstly identified [11], followed by α-ketoglutarate-dependent dioxygenase homolog 5 (ALKBH5) [12]. Interaction with variable “readers” is required in the m6A modification to exert different downstream biological functions. YT521-B homology (YTH) domain family members are the main “readers” which regulate nuclear export, RNA splicing, decay, and translation [13, 14]. Besides, certain heterogeneous nuclear ribonucleoprotein (HNRNP) family members and insulin-like growth factor 2 mRNA binding proteins (IGF2BPs) could also recognize the m6A modification site and mediate the functions of m6A [15, 16].
Accumulating evidence has revealed the crucial role of m6A modification in numerous physiological processes, especially tumor initiation and progression, of glioblastoma, acute myeloid leukemia, hepatocellular and breast carcinoma [17, 18]. Recently, decreased m6A RNA methylation level caused by METTL14 R298 mutation or decreased METTL3 expression has been reported to promote tumorigenicity and cell proliferation via activation of AKT signaling pathway in endometrial cancer [18]. This research identifies the function of m6A RNA methylation in the pathogenesis of endometrial cancer. However, a comprehensive and systematic analysis of the expression of m6A RNA methylation regulators in endometrial cancer as well as their prognostic value is currently lacking.
Herein, we reanalyzed the expression of 16 acknowledged m6A RNA regulators in 406 endometrial adenocarcinoma patients and 19 controls using The Cancer Genome Atlas (TCGA) dataset to stratify the prognosis of endometrial adenocarcinoma. Subsequently, microarray data from the publicly available Gene Expression Omnibus (GEO) dataset and real-time quantitative polymerase chain reaction (RT-qPCR) using clinical samples were utilized to verify the differential expression of key m6A methylation regulators in patients with endometrial adenocarcinoma.
Materials and Methods
Datasets
The expression data and corresponding clinical characteristics for 406 patients with endometrial adenocarcinoma and 19 controls were downloaded from TCGA dataset (http://cancergenome.nih.gov/). We screened suitable clinical samples from the TCGA database with the terms “corpus uteri”, “adenocarcinoma”, and “TCGA” as the inclusion criteria; thereby 406 patients and 19 controls from studies meeting the database requirements were included in our further analysis. Controls were from tumor adjacent normal endometrium or women without endometrial cancer. The only common information for these enrolled patients were age, International Federation of Gynecology and Obstetrics (FIGO) grade, and survival information, so all patients' ages and tumor FIGO grades were sorted and listed in Table S1.
For further validation, the expression data from 64 endometrial adenocarcinoma and 33 endometrial hyperplasia tissues was downloaded from GSE 106191 of the GEO database (https://www.ncbi.nlm.nih.gov/geo/). All 33 endometrial hyperplasia tissues were adjacent to endometrial adenocarcinoma. The clinical parameters of these endometrial adenocarcinoma and hyperplasia patients have been presented previously [19]. Gene expression dataset was downloaded from the Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2).
Analysis of m6A RNA methylation regulators
According to published literature [20], we assembled 18 m6A RNA methylation regulators. We further selected 16 regulators among them due to available data from GSE 106191 and TCGA database. We analyzed the expression of these 16 genes between women with and without endometrial adenocarcinoma from TCGA and GEO datasets independently using the Wilcoxon test in R software. Correlations between m6A methylation regulators were identified using Spearman correlation in the “Corrplot” package of R software. P < 0.001 was considered as significantly correlated to each other.
Bioinformatic analysis of TCGA dataset
Univariate Cox regression analyses of the expression of m6A RNA methylation regulators in endometrial adenocarcinoma using TCGA dataset to determine their prognostic value. Four genes were identified related with patients' survival (P < 0.1), and were further selected for functional analysis and potential risk signature using least absolute shrinkage and selection operator (LASSO) Cox regression algorithm. Eventually, three genes and their coefficients were identified, selecting the best penalty parameter λ associated with the smallest 10-fold cross validation within TCGA database [21]. A risk score was calculated for the prognosis of each patient in TCGA dataset using the formula: risk score = ΣCoefi∗xi. Risk scores in patients with different clinical features were compared using one-way analysis of variance or t-test. We applied multivariate Cox regression analysis to verify prognostic value of risk score and clinical features.
According to risk score (higher or lower than the median value), patients with endometrial adenocarcinoma were classified into two groups, namely high-risk and low-risk, respectively. The survival of patients in two groups was compared using Kaplan-Meier method with a two-sided log-rank test. The functional enrichment of differentially expressed genes (DEGs) between two groups was determined using Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment.
Identification of DEGs of GEO dataset
After downloading GSE 106191 from GEO database, the “impute” R software package was utilized to impute missing expression data, while the “limma” R software package was utilized to normalize gene expression and to identify the DEGs between endometrial adenocarcinoma and endometrial hyperplasia groups. P < 0.01 denoted significant difference.
Weighted gene co-expression network analysis (WGCNA) and functional enrichment from GEO dataset
We did WGCNA to analyze the DEGs in GEO dataset and identified the relationships among FTO, RBM15, and their potential target genes in women with endometrial adenocarcinoma. GO analysis was utilized to visualize the potential molecular function and biological significance of DEGs in the same module with FTO and RBM15, whereas KEGG pathway enrichment was used to analyze the potential functions of these genes. The gene ID was set using the “org.Hs.eg.db” of R software and, subsequently, GO and KEGG analyses were carried out with “clusterProfiler”.
Ingenuity pathway analysis (IPA)
IPA was performed to identify the possible target genes of FTO and network, which might participate in the progression of endometrial adenocarcinoma.
Clinical sample collection
In order to validate the in silico analysis results, we collected 30 clinical samples of untreated endometrial adenocarcinoma and controls (N=15 for each group) in Department of Gynecology and Obstetrics, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai Jiao Tong University School of Medicine. The normal endometrial tissues as controls were collected from hysteroscopy biopsy in patients of fibroids, polyps, or infertility. The inclusion criteria of enrolled endometrial adenocarcinoma were as follows: patients were diagnosed with stages I-II endometrial cancer according to the staging system of FIGO 2009 guidelines and the histotype is adenocarcinoma. Patients with other malignancies and those who had received chemotherapy or radiotherapy before surgery were excluded. Informed consent was obtained from all participants. All procedures were reviewed and approved by the ethics committees of Institutional Review Board in Shanghai Sixth People's Hospital affiliated for Shanghai Jiao Tong University. Fresh endometrial tumors and normal endometrial tissues were separately dissected at the time of surgery and immediately transferred to liquid nitrogen for storage. The clinical parameters for recruited patients are presented in Table S2.
Real-time quantitative polymerase chain reaction (RT-qPCR)
Total RNA from endometrial tissues was extracted using a Total RNA Isolation Kit (FOREGENE, Chengdu, China) and then reversely transcribed into cDNA (TAKARA, Dalian, China). The mRNA expression of METTL3, FTO, YTHDF1, and RBM15 was detected using RT-qPCR. Results were analyzed by ΔΔCt method. The ratio of the target gene over β-ACTIN was calculated as the target mRNA level. The primer sequences used for targeting genes were as follows:
METTL3 (human), 5'-TTGTCTCCAACCTTCCGTAGT-3' (forward) and 5'-CCAGATCAGAGAGGTGGTGTAG-3' (reverse).
FTO (human), 5'-ACTTGGCTCCCTTATCTGACC-3' (forward) and 5'-TGTGCAGTGTGAGAAAGGCTT-3' (reverse).
YTHDF1 (human), 5'-GTGCTGATAGATGTTGTTCCCC-3' (forward) and 5'-ATACCTCACCACCTACGGACA-3' (reverse).
RBM15 (human), 5'-GTGAGGACTCGACTTCCCG-3' (forward) and 5'-GCCGCTATCGGTCTTTCCG-3' (reverse).
β-ACTIN (human), 5'-GGGAAATCGTGCGTGACATTAAG-3' (forward) and 5'-TGTGTTGGCGTACAGGTCTTTG -3' (reverse).
Statistical analysis
Results are expressed as mean ± SEM or SD. We used paired Student's t-test or one-way analysis of variance followed by the Newman-Keuls multiple comparison tests. Statistical significance is shown as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Altered expression of m6A RNA methylation regulators in endometrial adenocarcinoma
For the first time, we explored the expression of 16 established m6A methylation regulators, including writers, erasers, and readers, in endometrial adenocarcinoma using TCGA dataset. For writers, the expression of METTL3 and RBM15/15B was significantly increased in patients with endometrial adenocarcinoma versus controls. In contrast, the expression of METTL14, KIAA1429, and ZC3H13 was drastically decreased in patients with endometrial adenocarcinoma (Figure 1A). Furthermore, aberrant expression of erasers was also detected, and both FTO and ALKBH5 were significantly decreased in women with endometrial adenocarcinoma versus controls (Figure 1A). Therefore, altered total m6A level induced by aberrant methylase and demethylase might contribute to the development of endometrial adenocarcinoma. Additionally, the expression of readers, biological function mediators of m6A, was also changed in patients with endometrial adenocarcinoma. The expression of YTHDF1, YTHDF2, and IGF2BP1/2 was increased in women with endometrial adenocarcinoma, whereas a reduction in YTHDC1 was detected (Figure 1A). Since RNA splicing, translation, and decay could be regulated by the combination of dysregulated readers, aberrant m6A methylation site might give rise to dysregulation of target genes in endometrial adenocarcinoma.
Expression and correlation of 16 m6A methylation regulators in patients with endometrial adenocarcinoma and controls. (A) Expression of 16 m6A methylation regulators in patients with endometrial adenocarcinoma (N=406) and controls (N=19) from TCGA dataset. * P < 0.05, ** P < 0.01, *** P < 0.001. (B) Spearman correlation analysis of 16 m6A methylation regulators. P < 0.001 denotes statistical significance.
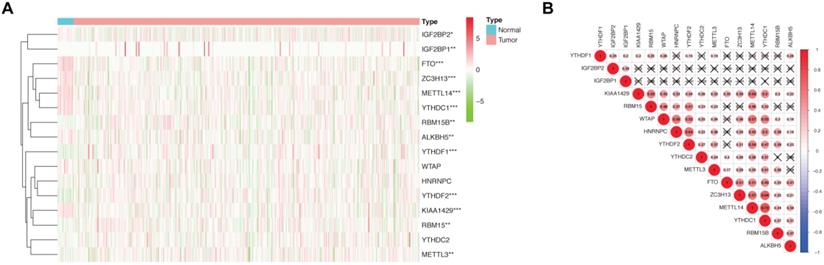
Spearman correlation was subsequently used to clarify the possible co-expression and association between 16 regulators. KIAA1429 was closely related to 13 m6A methylation regulators, while METTL14 and YTHDC1 were associated with 12 regulators (Figure 1B). Combined with the differential expression and correlation results, we hypothesized that these aberrant m6A methylation regulators might contribute to the pathogenesis of endometrial adenocarcinoma.
Prognostic value and risk signature of m6A methylation regulators in endometrial adenocarcinoma
We applied univariate Cox regression analysis to investigate the prognostic value of 16 m6A methylation regulators using their expression in patients with endometrial adenocarcinoma in TCGA dataset (N=406) (Figure 2A). Three of the 16 tested genes (FTO, KIAA1429, and RBM15) were evidently correlated with overall survival (P < 0.05) and had a hazard ratio > 1. We further performed LASSO Cox regression algorithm to predict the clinical outcomes of endometrial adenocarcinoma patients via FTO, KIAA1429, and RBM15. Since the number of genes was insufficient for regression, YTHDF1 (P < 0.1) was also added into the prediction model. That is to say, a total of four genes (P < 0.1 in univariate Cox regression) were applied to LASSO regression. We picked 3 genes to construct the risk signature, and the coefficients of FTO, RBM15, and YTHDF1 were 0.24, 0.13, and 0.02, respectively. The survival curve of high- and low- expression groups for each gene was presented in Figure S1.
Risk signature constructed using m6A methylation regulators. (A) Process of constructing risk signature using 3 m6A methylation regulators. Hazard ratio, 95% confidence intervals analyzed by univariate Cox regression. (B) Kaplan-Meier overall survival curves in patients with endometrial adenocarcinoma (N=406) of TCGA dataset between high-risk and low-risk groups according to risk score using our predictive signature after LASSO regression. (C) Heatmap of the expression of FTO, RBM15, and YTHDF1 between high-risk and low-risk groups. The distribution of clinical features was also presented. (D) The risk score of patients with different FIGO grades. (E-F) Univariate (E) and multivariate (F) Cox regression analyses of correlations between age, grade, risk score, and survival of endometrial adenocarcinoma patients. (G-H) GO and KEGG enrichment of DEGs in patients with endometrial adenocarcinoma between high-risk and low-risk groups. ** P < 0.01, *** P < 0.001, **** P < 0.0001.
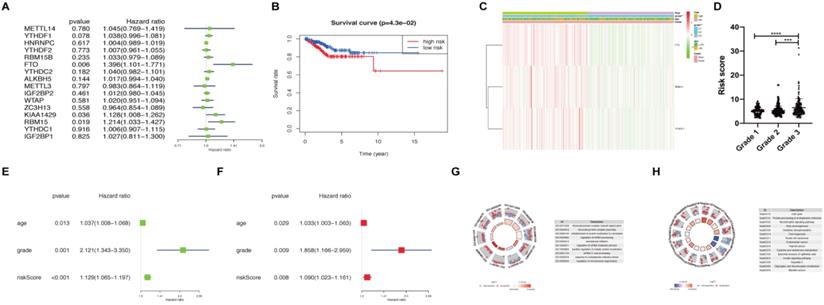
Subsequently, we calculated the risk score for patients with endometrial adenocarcinoma using coefficients of these 3 predicted genes. All patients with endometrial adenocarcinoma were divided into two groups based on risk score, namely as high-risk and low-risk. A distinct difference in overall survival between the two groups was identified in Figure 2B (P < 0.05). We further analyzed the expression of FTO, RBM15, and YTHDF1 in two groups using TCGA dataset (Figure 2C). An apparent difference was observed between two groups with respect to the International Federation of Gynecology and Obstetrics (FIGO) grade (P < 0.01) (Figure 2C). We further analyzed the association between risk scores and age, as well as FIGO grade; the risk score was significantly increased in grade 3 compared with those in grade 1 and grade 2 (P < 0.001) (Figure 2D).
Moreover, univariate and multivariate Cox regression analyses were applied to verify whether the risk signature could be independently identified as the prognostic indicator. Age, FIGO grade, and risk score remained closely correlated with survival no matter by univariate Cox regression analysis (Figure 2E) or multivariate analysis (Figure 2F). Finally, we analyzed DEGs between high-risk and low-risk groups and utilized functional enrichment of GO and KEGG to clarify the potential target functions and pathway of m6A methylation regulators. The biological process enriched from DEGs focused on RNA processing, translation initiation, and protein localization, which are main functions of m6A (Figure 2G). KEGG analysis enriched the altered expression of genes in the cell cycle, oxidative phosphorylation, thermogenesis, carcinogenesis, and protein processing (Figure 2H). Hence, FTO, RBM15, and YTHDF1 may regulate the RNA processing and translation of target genes involved in the cell cycle, thermogenesis and other pathways, leading to the tumor process and prognosis of endometrial adenocarcinoma.
Validation of m6A methylation regulators and potential target networks
The gene expression profiles of 33 hyperplasia tissues and 64 endometrial adenocarcinoma tissues were obtained from GSE 106191. FTO expression was significantly decreased, whereas RBM15 was increased (Figure 3A), similar to results in TCGA dataset. From TCGA and GEO datasets, we found that FTO and RBM15 might take critical roles in the prognosis of endometrial adenocarcinoma. The co-expression of DEGs between women with and without endometrial adenocarcinoma was further detected using WGCNA. We found that both RBM15 and FTO belonged to the gray module (threshold = 0.9; Figure 3B). All 1315 DEGs in the gray module were enriched via GO and KEGG analyses, and we identified 11 biological processes in which FTO and RBM15 were involved. Thus, FTO and RBM15 were suggested to contribute to the progression of endometrial adenocarcinoma through these 11 biological processes, including connective tissue development, reproductive structure development, positive regulation of catabolic process, regulation of multicellular organism growth, regulation of RNA stability, oxidative demethylation, and temperature homeostasis, etc. (Figure 3C and 3D).
Expression of 16 m6A methylation regulators in patients with and without endometrial adenocarcinoma. (A) Expression of 16 m6A methylation regulators in patients with endometrial adenocarcinoma (N=64) and hyperplasia (N=33) from GEO dataset. (B) WGCNA analysis of DEGs between women with and without endometrial adenocarcinoma. (C-D) GO and KEGG enrichment of DEGs belonging to the same module as RBM15 and FTO. GO composed of 11 biological processes in which FTO and RBM15 were involved. (E) The protein-protein interaction network of FTO, RBM15, and other 30 target protein involved in the above 11 biological processes. (F) Identification of the possible target genes of FTO and network that may participate in the progression of endometrial adenocarcinoma via IPA. * P < 0.05, *** P < 0.001. (G) The mRNA expression of METTL3, RBM15, FTO, and YTHDF1 in endometrial adenocarcinoma and control endometrium samples (N=15 for each group). * P < 0.05.
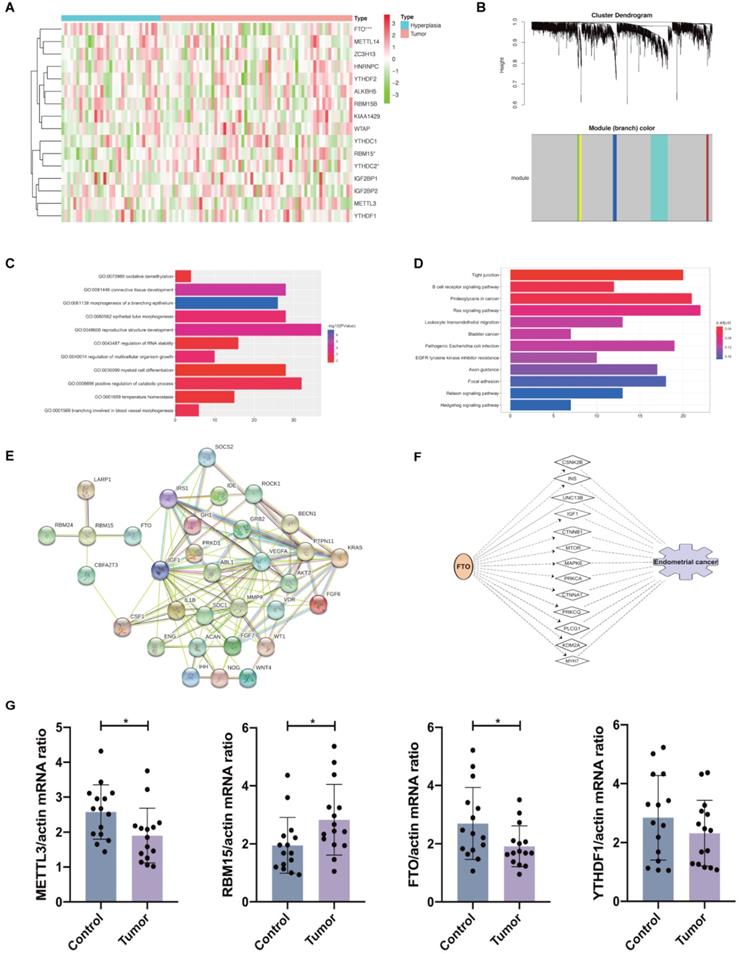
Furthermore, the interactions among 127 DEGs involved in these 11 biological processes were analyzed using STRING. We observed that RBM15 interacted with la ribonucleoprotein 1 (LARP1), RBM24, CBFA2/RUNX1 partner transcriptional co-repressor 3 (CBFA2T3), and FTO; meanwhile, FTO interacted with insulin-like growth factor 1 (IGF1), insulin receptor substrate 1 (IRS1), and RBM15 (Figure 3E). Therefore, we supposed that FTO facilitated the progression of endometrial adenocarcinoma via the functions of IGF1 and IRS1. The potential target genes mediating the regulation of RBM15 and FTO to the progression of endometrial adenocarcinoma have been presented in Table 1. Then, we used IPA to predict the possible target genes and network through which FTO led to the progression of endometrial adenocarcinoma. IGF1 was also shown to mediate the functional role of FTO in endometrial adenocarcinoma (Figure 3F). Hence, our results provided a potential mechanism and network for the regulation of FTO and RBM15 to the progression of endometrial adenocarcinoma.
Potential target genes that mediate the regulation of RBM15 and FTO to the progression of endometrial cancer
| Genes | Biological Process |
|---|---|
| RBM24 | Positive regulation of catabolic process |
| CBFA2T3 | Myeloid cell differentiation |
| LARP1 | Positive regulation of catabolic process |
| IGF1 | Positive regulation of catabolic process/regulation of multicellular organism growth |
| VEGFA | Reproductive structure development/temperature homeostasis/epithelial tube morphogenesis/branching involved in blood vessel morphogenesis/myeloid cell differentiation |
| IRS1 | Positive regulation of catabolic process |
| GRB2 | Reproductive structure development |
| PTPN11 | Reproductive structure development |
| PRKD1 | Positive regulation of catabolic process |
| GH1 | Regulation of multicellular organism growth |
| ACAN | Connective tissue development |
| NOG | Connective tissue development/ morphogenesis of a branching epithelium |
| AKT2 | Positive regulation of catabolic process |
| WT1 | Connective tissue development/ reproductive structure development |
| ABL1 | Epithelial tube morphogenesis |
| SDC1 | Reproductive structure development |
| MMP9 | Myeloid cell differentiation |
| SOCS2 | Regulation of multicellular organism growth |
| CSF1 | Connective tissue development/ epithelial tube morphogenesis |
| ENG | Epithelial tube morphogenesis |
| VDR | Reproductive structure development |
| ROCK1 | Positive regulation of catabolic process |
| BECN1 | Positive regulation of catabolic process |
| FGF6 | Connective tissue development |
| FGF7 | Morphogenesis of a branching epithelium |
| IDE | Positive regulation of catabolic process |
| IHH | Connective tissue development/epithelial tube morphogenesis |
| IL1B | Temperature homeostasis/positive regulation of catabolic process |
| KRAS | Epithelial tube morphogenesis/morphogenesis of a branching epithelium |
| WNT4 | Morphogenesis of a branching epithelium/epithelial tube morphogenesis/reproductive structure development |
All the biological processes were annotated as GO term enrichment analysis from DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/).
Finally, we validated the mRNA expression of METTL3, FTO, RBM15, and YTHDF1 in clinical samples of endometrial tissues from endometrial adenocarcinoma patients and controls. We found that METTL3 and FTO mRNA expression was evidently decreased in endometrial adenocarcinoma while RBM15 mRNA expression was increased with no changes in YTHDF1 expression in cases versus controls (Figure 3G). These results were consistent with previous in silico analysis using TCGA and GEO datasets.
Discussion
As the fourth most common malignancy in females, endometrial cancer presents a high rate of recurrence and poor prognosis in patients with advanced disease [5]. Thus, early diagnosis and the identification of biomarkers associated with the prediction of prognosis are of vital importance in clinical management. Mainly focusing on DNA methylation or histone acetylation, epigenetics takes various roles in progression and diagnosis of endometrial cancer, as well as risk prediction and identification of treatment targets [22, 23]. In the present research, we elucidated that m6A RNA methylation regulators, especially FTO, RBM15, and YTHDF1, were likewise closely related with prognosis of endometrial adenocarcinoma. Tumor grade is the description of a tumor based on the differentiation of its cells. Grade 1 indicates a well-differentiated tumor, while grade 3 is often associated with rapid grow and migration, as well as poor prognosis [24]. According to our risk signature, risk score was positively correlated with tumor grade, further demonstrating the prognostic value of our m6A regulator model in endometrial adenocarcinoma. Moreover, we also validated the roles of FTO and RBM15 in the prognosis of endometrial adenocarcinoma using the GEO dataset and predicted the possible target genes and biological processes of FTO and RBM15, which might contribute to the regulation of these two genes to endometrial adenocarcinoma. Thus, we provided a prediction biomarker and a risk signature that have the potential to predict the prognosis of patients with endometrial adenocarcinoma; this may be beneficial to clinical diagnosis and management.
Reductions in m6A methylation induced by decreased METTL3 and mutation in METTL14 activate the phosphoinositide 3 kinase/protein kinase B (PI3K/AKT) pathway, promoting proliferation and tumorigenicity of endometrial cancer [18]. In our study, the expression of METTL14 and KIAA1429, methylase enzymes promoting methylation of the m6A site, was significantly decreased in endometrial adenocarcinoma. This result was consistent with the reduction of m6A levels in endometrial cancer observed in a previous study [18]. However, decreased FTO expression in the tumor shown in our study was in contrast to its results. We supposed that the type of endometrial cancer used in the research might account for this difference. We selected endometrial adenocarcinoma, while the previous study reported by Liu et al investigated a mix of endometrioid adenocarcinoma, endometrioid, clear-cell carcinoma, and endometrioid with squamous differentiation. Additionally, difference noted in tissues might also be responsible for the discrepancy. The previous study compared the tumor with the adjacent tissue, whereas we chose tissues from patients with and without tumor.
Endometrial cancer is an estrogen-dependent disease; both hormonal dysregulation and obesity can facilitate the development and progression of this disease. Obesity (BMI >30 kg/m2) is considered as the greatest risk factor for patients with endometrial cancer [25]. Weight loss intervention involving metabolic surgery is an alternative to standard surgical treatment [25, 26]. Herein, we built a risk prediction model using FTO, m6A methylation regulator closely associated with body mass, obesity, and energy metabolism [27]. FTO is reported to suppress mitochondrial thermogenesis in the adipocyte precursor cells [28]. According to our prediction model, pathways of thermogenesis were also enriched in DEGs between high-risk and low-risk patients with endometrial adenocarcinoma (Figure 2H). Furthermore, FTO proves to promote glycolysis though PI3K/AKT pathway in breast cancer cells [29] and PI3K/AKT signaling pathway was also enriched in our DEGs. In our results, IGF1 and IRS1, essential factors in the glucose metabolism, thermogenesis, and glucose catabolic process [30], were considered as the potential target genes of FTO in the regulation of endometrial adenocarcinoma (Figure 3E and 3F). Hence, we supposed that the altered expression and variants of the FTO gene contribute to the progression and prognosis of women with endometrial cancer through energy metabolism as well as IGF1 signaling. However, the underlying mechanism warrants further investigation.
Besides of energy disorder, FTO participates in the regulation of prognosis of cancer through several other ways. A previous study demonstrates that reduction of FTO upregulates cyclin D1 RNA methylation, which in turn accelerates cyclin D1 mRNA degradation and results in inhibition of G1 progression and cell cycle [31]. Interestingly, according to the results of our analysis, DEGs between high- and low-risk patients with endometrial cancer were also enriched in cell cycles (Figure 2H). In addition, IPA network also indicated the regulation of lysine demethylase 2A (KDM2A) by FTO (Figure 3F), which may arrest cells in the G2/M phase. Thus, FTO may regulate cell cycle-associated genes in the progression of endometrial cancer. In addition, decreased FTO takes a tumorigenic role in bladder cancer [32] and a highly selective inhibitor of FTO could restrain the cisplatin-induced cytotoxicity in bladder cancer cells [33]. Epithelial-to-mesenchymal transition (EMT) is a vital process that drives, tumor initiation and metastasis, resistance to anoikis, and refractory response to chemotherapy, contributing to the prognosis of patients with cancer [34]. IGF1 signaling contributes to the regulation of EMT [35], and our result also indicated the potential function of IGF1 in the regulatory role of FTO in endometrial cancer. Hence, FTO may be involved in the survival and prognosis of patients by regulating the sensitivity to cisplatin-based chemotherapy and the EMT. The decreased expression of FTO and co-expression of IGF1 and FTO were also validated in the GEO dataset, further demonstrating the reliability of our analysis.
Apart from FTO, RBM15, a writer promoting the methylation of m6A site, also contributed to our risk signature. Our study suggested that RBM15 might give rise to epithelial tube morphogenesis, branching involved in blood vessel morphogenesis, myeloid cell differentiation, and morphogenesis of a branching epithelium. Consistent with our results, increased RBM15 is also associated with prognosis of lung adenocarcinoma [36]. Additionally, YTHDF1 is a reader that can modulate the translation dynamics of m6A-modified mRNA [37] with prognostic value in lung adenocarcinoma [38]. Spearman correlation analysis did not reveal any significant association among the expression of FTO, RBM15, and YTHDF1.
In summary, analysis of TCGA patient dataset, correlation analysis, and construction of a risk signature using m6A methylation regulators revealed their importance in the progression and prognosis prediction of endometrial adenocarcinoma, especially highlighting the roles of FTO, RBM15, and YTHDF1. Functional enrichment and IPA network suggested that FTO and RBM15 contributed to the survival of patients with endometrial cancer via IGF1, IRS1, RBM24, LARP1, and CBFA2T3 under the regulation of connective tissue development, catabolic process, RNA stability, oxidative demethylation, temperature homeostasis, and energy metabolism, etc. Combinational therapy that targets FTO to promote the therapeutic effect and prolong survival may represent a personalized attractive treatment approach for patients with endometrial cancer. Further in vivo and in vitro studies are needed to validate our hypothesis.
Abbreviations
m6A: N6-methyladenosine; METTL3: methyltransferase-like 3; METTL14: methyltransferase-like 14; METTL16: methyltransferase-like 16; WTAP: Wilms' tumor 1-associated protein; RBM15: RNA-binding motif protein 15; ZC3H13: zinc finger CCCH-type containing 13; FTO: fat mass and obesity-associated protein; ALKBH5: α-ketoglutarate-dependent dioxygenase homolog 5; YTH: YT521-B homology; HNRNP: heterogeneous nuclear ribonucleoprotein; IGF2BPs: insulin-like growth factor 2 mRNA binding proteins; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; RT-qPCR: real-time quantitative polymerase chain reaction; LASSO: least absolute shrinkage and selection operator; DEGs: differentially expressed genes; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; WGCNA: weighted gene co-expression network analysis; IPA: ingenuity pathway analysis; FIGO: International Federation of Gynecology and Obstetrics; LARP1: la ribonucleoprotein 1; CBFA2T3: CBFA2/RUNX1 partner transcriptional co-repressor 3; IGF1: insulin-like growth factor 1; IRS1: insulin receptor substrate 1; PI3K/AKT: phosphoinositide 3 kinase/protein kinase B; KDM2A: lysine demethylase 2A; EMT: epithelial-to-mesenchymal transition.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors gratefully acknowledge contributions from the GEO and the TCGA Network. The authors declare using two endometrial cancer public available datasets: Sugiyama Y dataset at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106191, and adenocarcinoma of corpus uterus in TCGA.
Funding
This research was supported by grants from the National Key Research and Development Program of China (No. 2018YFC1003202), National Natural Science Foundation (No. 81971343 and 81901549), and Shanghai Commission of Science and Technology (No. 19410760300 and 20DZ2270900).
Authors' contributions
J.Z., S.L., and Y.D. contribute to the design of the study. J.Z. and S.L. contribute to conducting the experiments and analyzing the data. Y.L. contributes to collecting clinical samples. J.Z., S.L., and Y.D. finish drafting the manuscript and revising it critically for important intellectual content. Y.D. is responsible for the final approval of the version to be published. The authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Creutzberg CL, van Putten WL, Koper PC, Lybeert ML, Jobsen JJ, Warlam-Rodenhuis CC. et al. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicentre randomised trial. PORTEC Study Group. Post Operative Radiation Therapy in Endometrial Carcinoma. Lancet. 2000;355:1404-11
2. Wieser V, Abdel Azim S, Sprung S, Knoll K, Kogl J, Hackl H. et al. TNFalpha signalling predicts poor prognosis of patients with endometrial cancer. Carcinogenesis. 2020;41:1065-73
3. Travaglino A, Raffone A, Stradella C, Esposito R, Moretta P, Gallo C. et al. Impact of endometrial carcinoma histotype on the prognostic value of the TCGA molecular subgroups. Arch Gynecol Obstet. 2020;301(6):1355-63
4. Wang QA, Yang Y, Liang X. LncRNA CTBP1-AS2 sponges miR-216a to upregulate PTEN and suppress endometrial cancer cell invasion and migration. J Ovarian Res. 2020;13:37
5. Jiang SW, Li J, Podratz K, Dowdy S. Application of DNA methylation biomarkers for endometrial cancer management. Expert Rev Mol Diagn. 2008;8:607-16
6. Richardson B. Impact of aging on DNA methylation. Ageing Res Rev. 2003;2:245-61
7. Takai N, Desmond JC, Kumagai T, Gui D, Said JW, Whittaker S. et al. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res. 2004;10:1141-9
8. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613
9. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP. et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169:824-35 e14
10. Wen J, Lv R, Ma H, Shen H, He C, Wang J. et al. Zc3h13 Regulates Nuclear RNA m(6)A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell. 2018;69:1028-38 e6
11. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885-7
12. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18-29
13. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M. et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626
14. Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF. et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Molecular Cell. 2016;61:507-19
15. Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299-308
16. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285-95
17. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369-76
18. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K. et al. m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074-83
19. Sugiyama Y, Gotoh O, Fukui N, Tanaka N, Hasumi K, Takazawa Y. et al. Two Distinct Tumorigenic Processes in Endometrial Endometrioid Adenocarcinoma. Am J Pathol. 2020;190:234-51
20. Wu J, Frazier K, Zhang J, Gan Z, Wang T, Zhong X. Emerging role of m(6) A RNA methylation in nutritional physiology and metabolism. Obes Rev. 2020;21:e12942
21. Zhang QJ, Luan JC, Song LB, Cong R, Ji CJ, Zhou X. et al. m6A RNA methylation regulators correlate with malignant progression and have potential predictive values in clear cell renal cell carcinoma. Exp Cell Res. 2020;392:112015
22. Yang F, Liu D, Deng Y, Wang J, Mei S, Ge S. et al. Frequent promoter methylation of HOXD10 in endometrial carcinoma and its pathological significance. Oncol Lett. 2020;19:3602-8
23. Teixeira SR, Abreu CM, Parkes L, Davies J, Yao S, Sawhney MA. et al. Direct monitoring of breast and endometrial cancer cell epigenetic response to DNA methyltransferase and histone deacetylase inhibitors. Biosens Bioelectron. 2019;141:111386
24. Soslow RA, Tornos C, Park KJ, Malpica A, Matias-Guiu X, Oliva E. et al. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int J Gynecol Pathol. 2019;38(Suppl 1):S64-S74
25. Wilkinson M, Murphy S, Sinclair P, Heneghan H, le Roux CW, Brennan DJ. Patient perceptions and understanding of obesity related endometrial cancer. Gynecol Oncol Rep. 2020;32:100545
26. Mechanick JI, Garber AJ, Handelsman Y, Garvey WT. American Association of Clinical Endocrinologists' position statement on obesity and obesity medicine. Endocr Pract. 2012;18:642-8
27. Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P. et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724-6
28. Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C. et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med. 2015;373:895-907
29. Liu Y, Wang R, Zhang L, Li J, Lou K, Shi B. The lipid metabolism gene FTO influences breast cancer cell energy metabolism via the PI3K/AKT signaling pathway. Oncol Lett. 2017;13:4685-90
30. Boucher J, Mori MA, Lee KY, Smyth G, Liew CW, Macotela Y. et al. Impaired thermogenesis and adipose tissue development in mice with fat-specific disruption of insulin and IGF-1 signalling. Nat Commun. 2012;3:902
31. Hirayama M, Wei FY, Chujo T, Oki S, Yakita M, Kobayashi D. et al. FTO Demethylates Cyclin D1 mRNA and Controls Cell-Cycle Progression. Cell Rep. 2020;31:107464
32. Wen L, Pan X, Yu Y, Yang B. Down-regulation of FTO promotes proliferation and migration, and protects bladder cancer cells from cisplatin-induced cytotoxicity. BMC Urol. 2020;20:39
33. Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373-84
34. George JT, Jolly MK, Xu S, Somarelli JA, Levine H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017;77:6415-28
35. Tanabe S, Kawabata T, Aoyagi K, Yokozaki H, Sasaki H. Gene expression and pathway analysis of CTNNB1 in cancer and stem cells. World J Stem Cells. 2016;8:384-95
36. Zhang Y, Liu X, Liu L, Li J, Hu Q, Sun R. Expression and Prognostic Significance of m6A-Related Genes in Lung Adenocarcinoma. Med Sci Monit. 2020;26:e919644
37. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H. et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388-99
38. Zhu J, Wang M, Hu D. Deciphering N(6)-Methyladenosine-Related Genes Signature to Predict Survival in Lung Adenocarcinoma. Biomed Res Int. 2020;2020:2514230
Author contact
![]() Corresponding author: Yanzhi Du, 845 Lingshan Road, Shanghai 200135, China. Tel: 86-21-2028-4516; Fax: 86-21-2028-4516; E-mail: duyzedu.cn.
Corresponding author: Yanzhi Du, 845 Lingshan Road, Shanghai 200135, China. Tel: 86-21-2028-4516; Fax: 86-21-2028-4516; E-mail: duyzedu.cn.