Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(17):5099-5105. doi:10.7150/jca.55134 This issue Cite
Research Paper
Genomic Profiling of Circulating Tumor DNA from Patients with Extensive-Stage Small Cell Lung Cancer Identifies Potentially Actionable Alterations
Jing Yang1,2*, Xiangyun Wang3*, Jingli Lu1,2, Hui Chen4, Xiaochen Zhao4, Chan Gao4, Yuezong Bai4, Qiwen Zhang1,2, Xiaomin Fu5, Xiaojian Zhang1,2 ![]()
1. Department of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China.
2. Henan Key Laboratory of Precision Clinical Pharmacy, Zhengzhou University, Zhengzhou, China.
3. Department of Respiratory and Critical Care Medicine, Second Affiliated Hospital of Naval Medical University, Shanghai.
4. The Medical Department, 3D Medicines Inc., Shanghai, China.
5. Department of Cancer Immunotherapy, The Affiliated Cancer Hospital of Zhengzhou University and Henan Cancer Hospital, Zhengzhou, China.
*Co-first authors with equal contributions to this work.
Received 2020-10-28; Accepted 2021-5-10; Published 2021-6-22
Abstract

Comprehensive genomic profiling may help uncover potentially actionable alterations in small cell lung cancer (SCLC) patients who have progressed on standard chemotherapy. However, tissue procurement may be extremely challenging for extensive-stage patients. We aimed to investigate the possibility of genomic profiling and detecting actionable alterations from blood in Chinese SCLC patients. Blood samples collected from extensive-stage SCLC pateints were subjected to circulating tumor DNA (ctDNA) extraction and targeted-next generation sequencing (NGS) using a 150-gene panel. A total of 1,300 aberrations were detected in 128 genes and 89.2% (116/130) patients harbored at least one oncogenic alteration. The most frequently mutated genes included TP53 (82.3%), RB1 (56.2%), LRP1B (40.8%) etc. and 54.6% of the patients had concurrent TP53/RB1 mutations. The RTK/RAS/RAF axis was the most frequently mutated oncogenic pathway. Samples harboring alterations in the DNA damaging repair (DDR), Notch, PI3K/mTOR, RTK/RAS/RAF, and Wnt pathways exhibited significantly higher blood tumor mutational burden (bTMB) than their wildtype counterparts. Classification based on OncoKB criteria detected potentially actionable alterations in about 50% of the population, half of which were bTMB-H and bMSI-H, indicating response to immune checkpoint inhibitors. Alterations in the RTK/RAS/RAF, DDR, and PI3K/mTOR also suggested potential sensitivity to matched targeted therapies or emerging investigational agents. Blood-based panel NGS is promising for delineating genomic landscape of SCLC and may also shed some light on treatment selection for Chinese SCLC patients.
Keywords: ctDNA, small cell lung cancer (SCLC), genomic profiling, targetable alterations
Introduction
Small cell lung cancer (SCLC) is a highly malignant neuroendocrine tumor that accounts for up to 15% of all lung cancers [1]. Although the majority of SCLC patients respond well to chemotherapy initially, nearly all patients develop resistance within months and their 5-year survival is only ~6% [2]. In recent years, the advent of immune checkpoint inhibitors (ICIs) has greatly expanded the therapeutic armamentarium of SCLC. In recurrent SCLC patients treated with nivolumab ± ipilimumab or pembrolizumab, the efficacy was demonstrated to be highly correlated with tumor mutational burden levels [3, 4]. In addition to immunotherapy, a number of targeted therapies are also being developed to treat SCLC, and there already exists some evidence showing off-label use of targeted agents benefiting SCLC patients [5]. For instance, human epidermal growth factor receptor 2 (HER2)-positive relapsed patients and those carrying deleterious mutations in BRCA1 responded favorably to trastuzumab- and olaparib-based regimens, respectively [6, 7]. Therefore, next-generation sequencing (NGS)-based comprehensive genotyping is becoming increasingly important for guiding SCLC management.
However, most SCLC patients are diagnosed with extensive-stage disease and tissue availability may pose significant challenges for those seeking genomic characterization. Circulating tumor DNA (ctDNA) sequencing, as a non-invasive alternative, has proven highly concordant with tissue-based sequencing and its utility has been widely explored for guiding treatment, evaluating efficacy, and predicting progression in multiple malignancies [8-11]. Recently, ctDNA sequencing has been applied by multiple studies to unveil potentially targetable mutations in Caucasian SCLC patients, however, little is known about the genomic landscape of Chinese SCLC population in blood [12-15]. In this work, we profiled genomic alterations among 130 Chinese extensive-stage SCLC patients and more importantly, uncovered potentially actionable targets, the value of which to inform treatment decision warrant further validation in larger sample sizes.
Materials and Methods
Patients and sample collection
For genomic landscaping, patients were retrospectively included between January, 2015 and May, 2020 if they were histopathologically or cytologically diagnosed with extensive-stage SCLC and had sufficient peripheral blood for NGS analysis of ctDNA using a 150-gene panel (3D Medicines). Patients were excluded if they had other prior or concomitant tumors. For clinical validation of the potentially targetable alterations, an extensive-stage SCLC patient admitted at Changzheng Hospital, Shanghai was given matched targeted therapy according to NGS analysis of ctDNA. Ten milliliters of peripheral blood were collected from each patient for NGS testing. This study was carried out in accordance with the Declaration of Helsinki and informed consents were obtained from all participants.
Circulating tumor DNA sequencing and variant calling
Circulating tumor DNA sequencing was performed using a panel targeting the whole coding regions of 150 cancer-related genes with a median sequencing depth of 3852 at 3D Medicines Inc., a College of American Pathologists (CAP) accredited and Clinical Laboratory Improvement Amendments (CLIA) certified laboratory (Shanghai, China). DNA isolation, library preparation, targeted hybrid capture and sequencing were performed as previously described [16]. Libraries were prepared with 30-60 ng of cell-free DNA using the Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences) and resulting DNA fragments were tagged with unique molecular identifiers (UMIs) to reduce background noise. Hybrid capture was conducted using the xGen Exome Research Panel v2 (Integrated DNA Technologies), followed by 75bp paired-end sequencing on a NextSeq 500 platform (Illumina).
Sequencing reads were mapped to the reference human genome hg19 using the Burrows-Wheller Aligner (v0.7.12). Duplicate reads were removed using Picard (v1.130). Synonymous mutations were excluded from analyses. Non-synonymous single nucleotide variants (SNVs) and insertions/deletions (indels) were called if their allele frequencies (AFs) were ≥0.3% using MuTect (v1.1.7) and Pindel (v0.2.5a8), respectively. Copy number variations (CNVs) were reported using an in-house developed script with a cut-off of 6 copies. Fusions were called using an in-house developed script with at least 3 supporting reads.
Blood-TMB and blood-MSI determination and identification of actionable targets
Blood TMB (bTMB) was defined as the total number of somatic SNVs and indels in the coding regions examined, including missense, truncating and inframe mutations. Samples in the top quartile of bTMB levels (≥6) were classified as bTMB-H. Blood MSI (bMSI) status was also examined for 100 samples, where a panel of 100 microsatellite markers were evaluated using a proprietary algorithm and any sample with ≥ 20% unstable loci was defined as bMSI-H [17]. Clinically actionable alterations were identified using the OncoKB Precision Oncology Database [18].
Statistical analyses
Between-group differences in bTMB were assessed using a Mann-Whitney U test. The correlation in SNVs/indels frequency between our study and the NATURE_2015 cohort was examined using Pearson correlation test [19].
Results
Patient characteristics and the genomic landscape of Chinese SCLC patients in blood
A total of 130 blood samples were obtained from extensive-stage SCLC patients. The median age of the patients was 65 years (range 42-91) and 87.7% were male (Table 1). The median number of oncogenic alterations was 3 (range 0-15). Two of the 100 samples subjected to bMSI testing were bMSI-H and 36/130 (27.7%) samples were defined as bTMB-H. Overall, 1,300 aberrations were detected in 128 genes and 116 (89.2%) patients harbored at least one oncogenic alteration (Fig. 1A).
Patient characteristics
| Characteristics | |
|---|---|
| Patients | 130 |
| Median age, y (range) | 65 (42-91) |
| Sex | |
| Female | 16 (12.3%) |
| Male | 114 (87.7%) |
| bMSI status | |
| bMSI-H | 2 (1.5%) |
| bMSS | 98 (75.4%) |
| N/A | 30 (23.1%) |
| bTMB status | |
| bTMB-H | 36 (27.7%) |
| bTMB-L | 94 (72.3%) |
| Median oncogenic GAs (range) | 3 (0-15) |
Significantly altered genes (A) and pathways (D) of 130 Chinese extensive-stage SCLC patients. B. The top 30 SNVs/indels in our cohort and the NATURE_2015 cohort. C. The correlation in mutation frequency between our cohort and the NATURE_2015 cohort. E. Correlation between bTMB level and mutated pathways. ns, not significant; *, p<0.05; ***, p<0.001; ****, p<0.0001.
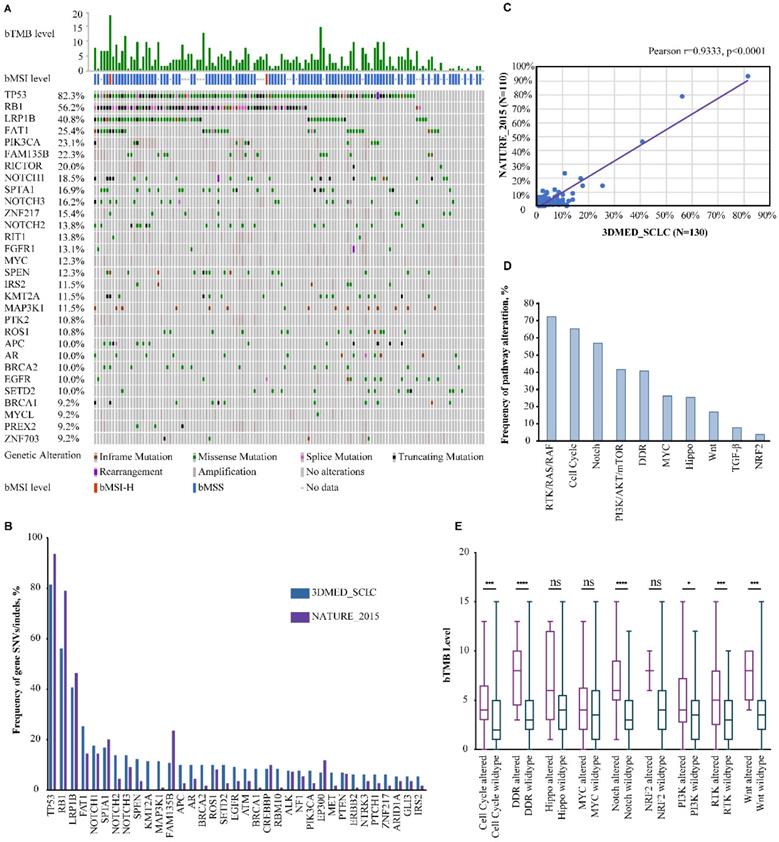
The most frequently mutated genes were TP53 (82.3%), RB1 (56.2%), LRP1B (40.8%), FAT1 (25.4%), PIK3CA (23.1%), FAM135B (22.3%), RICTOR (20.0%), NOTCH1 (18.5%), SPTA1 (16.9%), and NOTCH3 (16.2%). The most frequently amplified genes were PIK3CA in chromosome (chr) 3 (19.2%), RICTOR in chr 5 (18.5%), MYC in chr 8 (12.3%), RIT1 in chr 1 (12.3%), and FAM135B in chr 8 (11.5%). Gene rearrangement was detected in 5 samples: NOTCH1-LCNL1 (chr 9), PTEN-intragenic (chr 10), TNRC6B-EP300 (chr 22), TP53 (chr 17)-DHRS3 (chr 1), and FGFR1-PLEKHA2 (chr 8), of which PTEN-intragenic is likely to cause loss of function while others were all likely-oncogenic. The mutational landscape was highly consistent with that of an external SCLC cohort, NATURE_2015 (Pearson r=0.9333, p<0.0001) (Figure 1B&C) [19].
As inactivating mutations of tumor suppressor genes TP53 and RB1 were both reported to be obligatory in SCLC, we analyzed the co-mutation rate of these two genes [19]. Of the 130 samples, 71 (54.6%) had concurrent TP53/RB1 alterations while 36 and 2 were mutated only in TP53 and RB1, respectively. SNVs/indels in LRP1B, SPEN and ATM and amplifications of PIK3CA, FAM135B, RICTOR, RIT1, PTK2, MYCL, DDR2, NTRK1, CCNE1 and CD274 were more common among TP53/RB1 co-mutated samples.
Alterations in cancer-related signaling pathways and their correlations with bTMB
To gain more insight into the molecular mechanism underlying SCLC pathogenesis, the alterational profiles of 10 critical oncogenic signaling pathways were also investigated [20, 21]. Strikingly, 72.3% of the patients carried at least one alteration in the RTK/RAS/RAF pathway (Fig. 1D). SNVs/indels in ROS1, EGFR, ALK, NF1, and MET occurred in 10.0%, 9.2%, 7.7%, 7.7%, and 6.9% of the study population and amplifications of RIT1, FGFR1, IRS2, NTRK1, and BRAF were observed in 12.3%, 10.0%, 7.7%, 5.4%, and 3.1% of the patients (Table 2). Similar to previous reports, alterations in the cell cycle (65.4%), Notch (56.9%), PI3K/mTOR (41.5%), DNA damaging repair (DDR) (40.8%) and MYC (26.2%) pathways were also among the most mutated pathways [13, 22]. DDR alterations have been reported to be associated with higher TMB loads in several cancers, therefore, the correlation between alterations in DDR as well as other signaling pathways and TMB status was also explored. With bMSI-H samples excluded, samples carrying alterations in the cell cycle, DDR, Notch, PI3K/mTOR, RTK/RAS/RAF, and Wnt pathways had significantly higher bTMB than their wild-type counterparts (Fig. 1E).
Alteration in RTK/RAS/RAF signaling
| Gene | SNV/indel, N (%) | Amplification, N (%) |
|---|---|---|
| ROS1 | 13 (10%) | 1 (0.8%) |
| EGFR | 12 (9.2%) | 2 (1.5%) |
| ALK | 10 (7.7%) | 0 |
| NF1 | 10 (7.7%) | 1 (0.8%) |
| MET | 9 (6.9%) | 2 (1.5%) |
| HER2 | 8 (6.2%) | 3 (2.3%) |
| NTRK3 | 8 (6.2%) | 1 (0.8%) |
| IRS2 | 7 (5.4%) | 10 (7.7%) |
| BRAF | 6 (4.6%) | 4 (3.1%) |
| ERBB4 | 5 (3.8%) | 0 |
| HRAS | 4 (3.1%) | 0 |
| PDGFRA | 4 (3.1%) | 0 |
| PDGFRB | 4 (3.1%) | 0 |
| RAF1 | 4 (3.1%) | 0 |
| CBL | 3 (2.3%) | 0 |
| FGFR1 | 3 (2.3%) | 13 (10%) |
| FGFR2 | 3 (2.3%) | 1 (0.8%) |
| FGFR3 | 3 (2.3%) | 0 |
| KRAS | 3 (2.3%) | 2 (1.5%) |
| NTRK1 | 3 (2.3%) | 7 (5.4%) |
| NTRK2 | 3 (2.3%) | 0 |
| PTPN11 | 3 (2.3%) | 0 |
| JAK2 | 2 (1.5%) | 1 (0.8%) |
| RET | 2 (1.5%) | 3 (2.3%) |
| RIT1 | 2 (1.5%) | 16 (12.3%) |
| ARAF | 1 (0.8%) | 0 |
| ERRFI1 | 1 (0.8%) | 3 (2.3%) |
| KIT | 1 (0.8%) | 0 |
| MAP2K1 | 1 (0.8%) | 1 (0.8%) |
| NRAS | 1 (0.8%) | 1 (0.8%) |
Potentially actionable genomic alterations
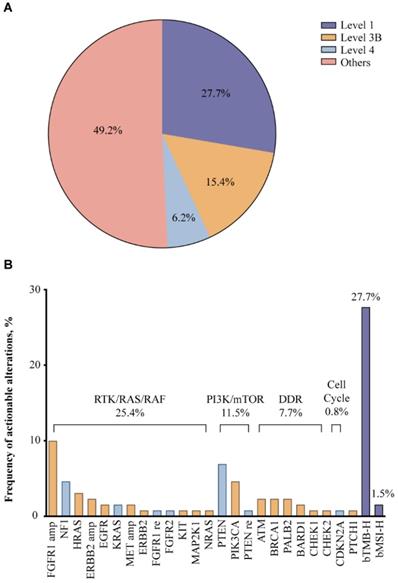
According to OncoKB classification, potentially actionable alterations were detected in about 50% of the patients (Fig. 2A). Level 1 markers included bTMB-H and bMSI-H, accounting for 27.7% of the study population, indicating that these patients may benefit from ICIs (Fig. 2B). Around 15.4% of the patients harbored level 3B alterations such as SNVs/indels of PIK3CA, HRAS, ATM, BRCA1 and PALB2, and amplifications of FGFR1, HER2, and MET, while level 4 alterations were found in 6.2% of the patients, including SNVs/indels of PTEN, NF1 and KRAS. Most of these potentially targetable genes could be categorized into oncogenic pathways: RTK/RAS/RAF pathway (25.4%), PI3K/mTOR pathway (11.5%), and DDR pathways, more specifically the homologous recombination (HR) pathway (7.7%). Given previous success with therapeutic agents targeting the RTK/RAS/RAF and PI3K/mTOR pathways in other malignancies and multiple emerging investigational agents in SCLC, alterations in these pathways may predict sensitivity to targeted therapies [23-25]. In addition, DDR pathway alterations may also predict benefit from ICIs, considering accumulating evidence on improved efficacy of ICIs in DDR-mutated NSCLC [26].
Potentially actionable alterations based on OncoKB criteria (A) and the frequency of potentially actionable alterations (B) in 130 Chinese extensive-stage SCLC patients.
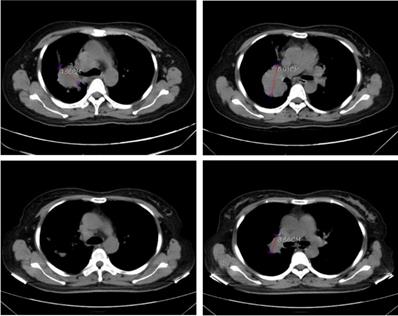
In order to validate the feasibility of leveargeing ctDNA-based testing to guide targeted therapy in SCLC, a 49-year-old female with a confirmed diagnosis of extensive-stage SCLC was subjected to NGS analysis of her peripheral blood using the 150-gene panel described above. The patient presented with a right upper lobe lesion measuring 4.66 cm, a right hilar lesion measuring 5.91 cm and multiple hepatic and bone metastases and had failed two cycles of chemotherapy with etoposide plus cisplatin previously. NGS anlaysis revealed mutations in EGFR (L858R at 56.4% and T790M at 55.14%), ATM (R3008H at 0.52%), PTEN (R130Q at 56.82% and S59* at 8.92%; * denotes a stop codon), and TP53 (P278S at 68.89%). Considering the co-occurrence of L858R and T790M, the patient was started on osimertinib (80 mg/day, po) on June 1st, 2020. A computerized tomography (CT) scan conducted on June 26th, 2020 showed that the patient had achieved partial response (PR) with almost complete regression in the right upper lobe lesion and a 38.1% reduction in the right hilar lesion (Figure 3). Blood tests performed on July 2nd, 2020 also showed a remarkable drop in the levels of tumor biomarkers since the onset of osimertinib treatment (Table 3). The patient continued to receive osimertinib until disease progression (PD) as shown by a CT scan on September 22nd, 2020.
Computerized tomography (CT) scans showing tumor regression in the right upper lobe lesion and the right hilar lesion following treatment with osimeritinib in an extensive-stage SCLC patient harboring EGFR L858R and T790M mutations.
Levels of tumor biomarkers before and after osimertinib treatment
| Biomarker | 06-02-2020 | 07-02-2020 | Reference range |
|---|---|---|---|
| NSE | 166.6 μg/L | 14.2 μg/L | < 16.3 μg/L |
| CYFRA211 | 122.6 μg/L | 1. 7 μg/L | < 3.3 μg/L |
| CEA | 1877 μg/L | 90.09 μg/L | 0-5 μg/L |
| CA19-9 | 2662 U/ml | 111.90 U/ml | 0-39 U/ml |
| CA125 | 562.2 U/ml | 33.32 U/ml | 0-35 U/ml |
| SCC | 1.2 ng/ml | 0.4 ng/ml | 0-1.5 U/ml |
Discussion
Our analyses revealed that genomic profiling using ctDNA may effectively capture the genomic complexity of SCLC by detecting 1,300 mutations in 128 genes. The most frequently altered genes included TP53, RB1, LRP1B etc., and over two thirds of the patients carried aberrations in the RTK/RAS/RAF pathway. Altertions in critical oncogenic pathways were also correlated with a higher bTMB load. More importantly, around 50% of the patients carried potentially actionable alterations, half of which might be sensitive to ICIs.
Blood-based genomic characterization of Chinese SCLC patients has been reported previously, but that study only included 22 patients and primarily focused on delineating clonal evolution [15]. As for studies conducted in Caucasian populations, Carter L et al only included 31 patients, and copy-number aberrations in circulating tumor cells, not ctDNA, were used to differentiate chemosensitive from chemorefractory disease [27]. Although Mohan S et al reported potential therapeutic targets in over 50% of the 69 patients included, only 30 had extensive-stage disease and ICI was not proposed as a treatment option [12]. In contrast, with a cohort of 130, our study represents the largest study interrogating genomic features among Chinese SCLC patients from blood. We also discovered that around 30% (bTMB-H and DDR-mutants) of the patients may respond to ICI. It is true that the study by Devarakonda S. et al shared some similarities with our study, but they used four different panels (54-gene to 73-gene) for genomic profiling while our entire cohort was analyzed using a 150-gene panel, which was previously validated for bTMB determination in lung cancer [16]. Therefore, bTMB-H was identified as an important predictor of ICI efficacy in our cohort.
Loss of TP53 and RB1, often co-occurring, are almost universal in SCLC according to tissue sequencing [19]. Indeed, TP53 mutation was present in 76.3% of the cohort, but RB1 was only mutated in 56.2%, however, when only SNVs/indels were taken into account, our result was only slightly lower than the 64% previously reported for Chinese SCLC patients in blood, although both were lower than the incidence of SNVs/indels based on tissue-testing [15, 22]. This was not completely unprecedented as Devarakonda S. et al also observed a remarkably lower RB1 mutation frequency from the blood of Caucasian SCLC patients compared to tissue testing [13]. This inconsistency could have arised from population differences, a low fraction of ctDNA, challenges of detecting certain variations from ctDNA, etc.
The unusually high prevalence of RTK/RAS/RAF pathway alterations also caught our attention, while alterations in the cell cycle, Wnt and PI3K/mTOR signaling pathways were most common according to tissue analyses in Chinese SCLC patients [22]. Again, Devarakonda S. et al had a similar finding in their ctDNA profiling, indicating the importance of RTK/RAS/RAF pathway in SCLC [13]. This is consistent with previous literature showing the essential role of the RTK/RAS/MAPK signaling in tumor growth, metastasis, and metabolism of SCLC [28]. Amplifications of FGFR1, HER2, and MET of this pathway as detected in our study may suggest therapeutic opportunities, given the emerging investigational agents or approved drugs targeting these aberrations either in SCLC or in other solid tumors [29, 30]. Indeed, this notion was supported by a chemo-resistant SCLC case achieving PR following administration of osimertinib upon detection of T790M in EGFR from blood. Likewise, alterations in HR pathway genes such as BRCA1, ATM, PALB2, etc. may suggest sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors olaparib and talazoparib, both of which are being investigated for treating HR pathway-mutated (NCT03009682) or BRCA1-mutated SCLC [31]. Apart from matched targeted therapies, alterations in the Notch and DDR pathways may also predict benefit from ICIs, as they were both found to be associated with higher bTMB. Notch pathway alterations were reported to be correlated with a higher degree of tumor immune infiltration and improved response to ICI in non-small cell lung cancer [32]. We also for the first time reported two cases of bMSI-H SCLC, both of which were also classified as bTMB-H. Although rarely seen in SCLC, a bMSI-H phenotype may potentially predict response to ICI treatment.
Our study is limited by the retrospective design and the lack of paried tumor tissue. Patients' demographic data and treatment data were also missing, which prevented us from analyzing the correlation between genomic alterations and demographic characteristics/efficacy of matched treatment. Clonal hematopoiesis might have also interefered with alteration calling, which is a common issue with blood-based testing. Additionally, only one case received matched targeted therapy according to testing results, and the value of ctDNA analysis to guide clinical decision needs to be confirmed in larger studies. That being said, our work confirmed the feasibility of ctDNA testing in SCLC and provided us with more insight into pontetial targets for drug development.
Abbreviations
SCLC: small cell lung cancer; NGS: next-generation sequencing; DDR: DNA damaging repair; ctDNA: circulating tumor DNA; ICI: immune checkpoint inhibitor; HER-2: human epidermal growth factor receptor 2; UMI: unique molecular identifier; indels: insertions and deletions; CNV: copy number variation; SNV: single nucleotide variation; AF: allele frequency; bTMB: blood tumor mutational burden; bMSI: blood microsatellite instability; HR: homologous recombination.
Acknowledgements
This study was financially supported by Wu Jieping Medical Foundation (No. 320.6750.19088-73). We thank Dr. Bin Li for assistance with data analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378:1741-55
2. Pietanza MC, Byers LA, Minna JD, Rudin CM. Small cell lung cancer: will recent progress lead to improved outcomes? Clin Cancer Res. 2015;21:2244-55
3. Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A. et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell. 2018;33:853-61 e4
4. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353-65
5. Dawkins JBN, Webster RM. The small-cell lung cancer drug market. Nat Rev Drug Discov. 2020;19:507-8
6. Kinehara Y, Minami T, Kijima T, Hoshino S, Morimura O, Otsuka T. et al. Favorable response to trastuzumab plus irinotecan combination therapy in two patients with HER2-positive relapsed small-cell lung cancer. Lung Cancer. 2015;87:321-5
7. Thomas A, Vilimas R, Trindade C, Erwin-Cohen R, Roper N, Xi L. et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J Thorac Oncol. 2019;14:1447-57
8. Rothwell DG, Ayub M, Cook N, Thistlethwaite F, Carter L, Dean E. et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat Med. 2019;25:738-43
9. Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C. et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223-38
10. Blackhall F, Frese KK, Simpson K, Kilgour E, Brady G, Dive C. Will liquid biopsies improve outcomes for patients with small-cell lung cancer? Lancet Oncol. 2018;19:e470-e81
11. Almodovar K, Iams WT, Meador CB, Zhao Z, York S, Horn L. et al. Longitudinal Cell-Free DNA Analysis in Patients with Small Cell Lung Cancer Reveals Dynamic Insights into Treatment Efficacy and Disease Relapse. J Thorac Oncol. 2018;13:112-23
12. Mohan S, Foy V, Ayub M, Leong HS, Schofield P, Sahoo S. et al. Profiling of Circulating Free DNA Using Targeted and Genome-wide Sequencing in Patients with SCLC. J Thorac Oncol. 2020;15:216-30
13. Devarakonda S, Sankararaman S, Herzog BH, Gold KA, Waqar SN, Ward JP. et al. Circulating Tumor DNA Profiling in Small-Cell Lung Cancer Identifies Potentially Targetable Alterations. Clin Cancer Res. 2019;25:6119-26
14. Du M, Thompson J, Fisher H, Zhang P, Huang CC, Wang L. Genomic alterations of plasma cell-free DNAs in small cell lung cancer and their clinical relevance. Lung Cancer. 2018;120:113-21
15. Nong J, Gong Y, Guan Y, Yi X, Yi Y, Chang L. et al. Circulating tumor DNA analysis depicts subclonal architecture and genomic evolution of small cell lung cancer. Nat Commun. 2018;9:3114
16. Wang Z, Duan J, Cai S, Han M, Dong H, Zhao J. et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients With Non-Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019;5:696-702
17. Wang Z, Zhao X, Gao C, Gong J, Wang X, Gao J. et al. Plasma-based microsatellite instability detection strategy to guide immune checkpoint blockade treatment. J Immunother Cancer. 2020;8(2):e001297
18. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J. et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017. 2017
19. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G. et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47-53
20. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC. et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321-37 e10
21. Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD. et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018;23:239-54 e6
22. Hu J, Wang Y, Zhang Y, Yu Y, Chen H, Liu K. et al. Comprehensive genomic profiling of small cell lung cancer in Chinese patients and the implications for therapeutic potential. Cancer Med. 2019;8:4338-47
23. Sakre N, Wildey G, Behtaj M, Kresak A, Yang M, Fu P. et al. RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR. Oncotarget. 2017;8:5992-6002
24. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM. et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93
25. Rolfo C, Raez L. New targets bring hope in squamous cell lung cancer: neurotrophic tyrosine kinase gene fusions. Lab Invest. 2017;97:1268-70
26. Ricciuti B, Recondo G, Spurr LF, Li YY, Lamberti G, Venkatraman D. et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin Cancer Res. 2020;26:4135-42
27. Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F. et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med. 2017;23:114-9
28. Cristea S, Coles GL, Hornburg D, Gershkovitz M, Arand J, Cao S. et al. The MEK5-ERK5 Kinase Axis Controls Lipid Metabolism in Small-Cell Lung Cancer. Cancer Res. 2020;80:1293-303
29. Tao CW, Chen MY, Tseng CM, Lapke N, Chen SJ, Tan KT. Advanced Lung Adenocarcinoma Patient with ERBB2 Amplification Identified by Comprehensive Genomic Profiling Benefits from Trastuzumab. Case Rep Oncol Med. 2020;2020:9072173
30. Nowak JA. HER2 in Colorectal Carcinoma: Are We There yet? Surg Pathol Clin. 2020;13:485-502
31. de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S. et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017;7:620-9
32. Zhang K, Hong X, Song Z, Xu Y, Li C, Wang G. et al. Identification of Deleterious NOTCH Mutation as Novel Predictor to Efficacious Immunotherapy in NSCLC. Clin Cancer Res. 2020;26:3649-61
Author contact
![]() Corresponding author: Xiaojian Zhang, Department of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China. Henan Key Laboratory of Precision Clinical Pharmacy, Zhengzhou University, Zhengzhou, China. No. 50, JianShe Road, Zhengzhou, Henan, China. Phone: 13803819758, E-mail: fcczhangxjedu.cn.
Corresponding author: Xiaojian Zhang, Department of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China. Henan Key Laboratory of Precision Clinical Pharmacy, Zhengzhou University, Zhengzhou, China. No. 50, JianShe Road, Zhengzhou, Henan, China. Phone: 13803819758, E-mail: fcczhangxjedu.cn.