Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(17):5241-5248. doi:10.7150/jca.48989 This issue Cite
Review
Gasdermin E-mediated programmed cell death: An unpaved path to tumor suppression
Yueyuan Wang1, Jingyu Peng1, Xiao Xie1, Zhihao Zhang1, Mingxi Li2, Ming Yang1 ![]()
1. Department of Breast Surgery, The First Hospital of Jilin University, Changchun, People's Republic of China.
2. Department of Neurology and Neuroscience Center, The First Hospital of Jilin University, Changchun, People's Republic of China.
Received 2020-6-2; Accepted 2021-6-7; Published 2021-6-26
Abstract

Hearing loss-associated protein gasdermin E (GSDME), an effector of secondary necrosis, has been identified in a new pathway of programmed cell death (PCD). GSDME epigenetic silencing and mutations resulting in loss-of-function have been reported in cancer tissues. Additionally, GSDME upregulation inhibits tumor proliferation as well as colony forming ability, and reduces the incidence of lymphatic metastasis, demonstrating that GSDME may act as a tumor suppressor. Here, we have focused on the molecular mechanisms of GSDME-mediated PCD, and tried to reveal the crosstalk between this cell death pathway and apoptosis, autophagy, GSDMD-mediated pyroptosis. Moreover, we concluded the anti-cancer activity of GSDME include forming permeable membranes, and triggering anti-cancer immunity. Thus, GSDME was potential to be a novel target for cancer prevention and treatment.
Keywords: GSDME, programmed cell death, cancer, immunity
1. Introduction
Gasdermin E (GSDME) belongs to the GSDM-family, which also includes GSDMA, GSDMB, GSDMC, GSDMD, and Pejvakin (PJVK) [1-3]. GSDME has long been regarded to be associated with hearing loss (HL), in which its specific mutations form induces the occurrence of HL. The gene encoding GSDME is localized on human chromosome 7p15, and is also known as deafness autosomal dominant 5 (DFNA5) [4]. It consists of 10 exons that encode a 496 amino acid protein with a molecular weight of 55 kDa [4-6]. GSDME can be divided into three distinct domains, including a globular domain A (exon 2-exon 6), a hinge region (exon 6-exon 7), and a globular domain B (exon 7-exon 10) [6]. As a member of gasdermin family which is widely expressed [2], the presence of GSDME gets a lot of attention. Till now, it has been found medium expression in brain, cardiac, and kidney cells, and low expression in cancer cells [7]. The estrogen receptor in breast cancer cell lines is inversely associated with GSDME methylation and expression [8]. Investigations into the relationship between GSDME and tumors have demonstrated that it is negatively related to proliferation, colony formation, invasion, and even the incidence of lymphatic metastasis [9, 10]. Moreover, the level of GSDME expression has been linked to cancer patient outcome; a study showed that tumors deficient in GSDME developed drug resistance [5], and drug susceptibility was restored upon exogenous GSDME transfection in these cells [11].
Programmed cell death (PCD) pathways are crucial for growth, survival, homeostasis, and innate immunity of all multicellular organisms, and GSDME-dependent cell death is a newly discovered PCD. After GSDME was discovered to participate in cell death modulation [12], increasing attention was given to this gene. The GSDME-N domain shows necrotic activity, the toxic region is located in exons 2 and 6, and the GSDME-C domain (exon 8-10) regulates the necrosis-inducing activity, to prevent improper cell death [5, 6, 13-15]. The N-terminal domain of GSDME mediates PCD with distinct morphological characteristics, affecting plasma membrane permeability and water influx, leading to cell swelling with typical bubbles and osmotic lysis [13, 14]. Even though there are different forms of GSDME associated with HL on the genomic level, they all lead to exon 8 skipping with a complete structure of the necrosis-inducing part, which indicates that mutant type (MT) GSDME functions the same way as the GSDME-N-terminal domain [5]. In addition, GSDME is one of the targets of p53, indicating that it may take part in cell cycle arrest and mediate PCD through the mitochondrial pathway [16-19].
In this review, we describe the molecular process of GSDME-mediated PCD and summarize therapeutic advances associated with GSDME and cancer suppression.
2. The crosstalk between GSDME-mediated PCD and conventional PCD
Resisting cell death leads to multiple diseases and is considered a hallmark of cancer [20]. How to induce PCD in tumors is a popular research target in cancer therapy. Here, we listed several known PCD types, including apoptosis, pyroptosis (mediated by GSDMD) and autophagy.
Apoptosis is a process of cell death with cell shrinkage, which leads to a decrease in cell size and a denser cytoplasm. “Apoptotic bodies” consisting of cytoplasm with tight packed organelles are phagocytosed by macrophages or other cells, then degraded within phagolysosomes [21].
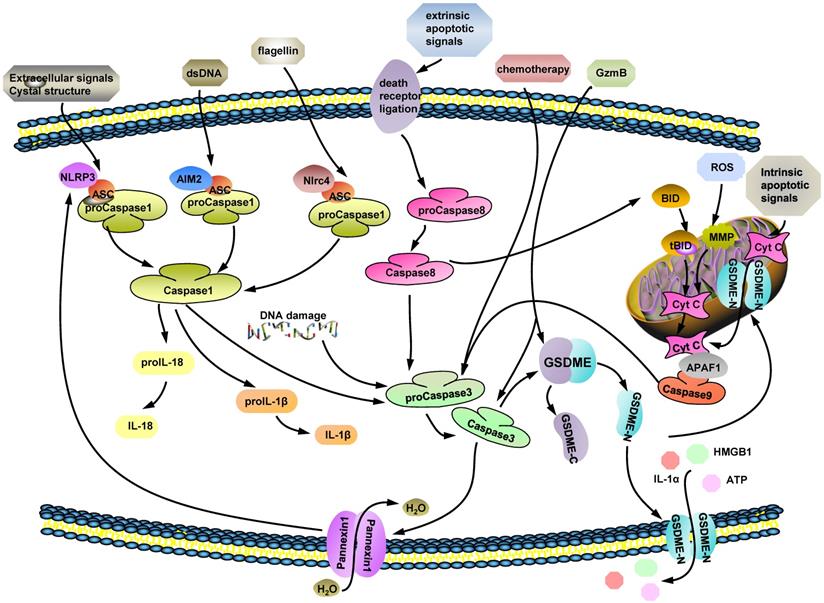
Distinct with apoptosis, GSDMD-mediated pyroptosis is characterized by cells swelling, osmotic lysis and inflammasomes release [20]. The important components of GSDMD-dependent pyroptosis are the inflammasomes, such as nucleotide-binding domain and leucine-rich repeat containing receptors (NLRs) and absent in melanoma (AIM)-like receptors [20, 22]. Unlike AIMs activation, through sensing endogenous or pathogen-derived double-stranded DNA (dsDNA) directly [23-25], NLR (NLRP3, NLRC4) activators includes multiple pathogenic signals [25, 26], including reactive oxygen species (ROS), mitochondrial DAMPs, bacterial pore-forming toxins, extracellular ATP, uric acid crystals, which are sensed by NLRP3 [27, 28], while flagellin and muramyl dipeptide are meant to active NLRC4 [29, 30]. Toll-like receptor 4 (TLR4) signaling triggers full inflammasome activation like NLRP3, by either post-translational modifications or subcellular re-localization of protein, while TLR4 co-engagement may also suppress inflammasome-dependent inflammation at later time-points [26]. Pattern-recognition receptors (PRRs), recognize certain damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). Until then, they could assemble the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), which links procaspase 1/8, to make up inflammasome complexes what are key to pyroptosis. ASC, as a bridge protein, binds to procaspase-1 through CARD-CARD interactions, and to procaspase 8, NLRs, and AIMs by pyrin domain (PYD)-PYD interactions, without interference [22, 31-34]. The ASC assembly recruits procaspase-1, driving the activation of caspase-1 and -3 [20, 35, 36]. Then, GSDMD is cut by caspase 1 and divided into C-domain and N-domain which would bind to plasma membrane (Fig. 1).
The possible molecular mechanisms of GSDME-mediated PCD. (ⅰ) Caspase 3 is a GSDME cleavage executor and facilitates to produce GSDME-N domain by cutting off full-length GSDME at D270. Ectocytic stimulus, such as: extracellular signals crystal structure, certain dsDNA and flagellin, mentioned in figure trigger caspase 1 activation by multifarious pathways. (ⅱ) Some extrinsic apoptotic signals are recognized by death receptor ligation on cytomembrane and active caspase 8. Both caspase 1 and 8 are upstream factors of caspase 3 and their activated forms induce caspase 3 cleavage. (ⅲ) Chemotherapeutic drugs and intracellular DNA damage stimulate procaspase 3 to turn into active caspase 3. (ⅳ) GzmB is a newly found factor which is effectively cut GSDME directly to release its N-terminal domain and takes roles in caspase 3 activation process as well. In conclusion, caspase 3 and GzmB, as the executors of GSDME cleavage, are modulated by various of cytokines.
Autophagy both mediates cell death and, under certain circumstances, helps cells to survive, which is the case during tumorigenesis and resistance [37-39]. It has been illustrated that GSDME-N domain production elevation is related to protective autophagy attenuation, resulting in cell sensitivity to doxorubicin [40].
We collected the connections between these forms of PCD and GSDME-mediated PCD to provide insight into the current understanding of tumor resistance and cancer treatment development.
2.1. GSDME-mediated PCD and apoptosis
Apoptosis, a complex protease cascade process, is initiated by a wide-range of stimuli, including the MAPK-MEK pathway [41], oxidative stress [21], and intrinsic and extrinsic apoptotic signals. The most essential component of apoptosis is caspase-3, which also serves as the unique intrinsic signal that triggers GSDME cleavage. Additionally, apoptotic pathways are repressed in GSDME-knockout mice via gene expression microarray experiments [6]. However, a number of genes are upregulated after MT GSDME transfection, and they modulate different apoptotic pathways, including oxidative stress, mitochondria dysfunction, pentose phosphate pathway activation, ER stress, and MAPK signaling [17-19, 42]
MAPK pathways are continuously activated in yeast and human cell lines within exogenous MT GSDME, under mediation of extracellular signal regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, which is followed by involvement of certain mitochondrial proteins, including Fis1, Por1, Aac1, and Aac3 [18]. Furthermore, intrinsic and extrinsic apoptotic signals induce GSDME-N domain production [14], which in turn enhances apoptosis by permeabilizing the mitochondrial membrane and facilitating Cyt c release [43]. Classic mitochondrial apoptotic pathways, such as the BIM-Bax-Cyt c-APAF1-Smac-caspase-GSDME axis, also play vital roles in GSDME-mediated PCD [11, 14]. Overall, caspase 3-GSDME forms a self-amplifying feedback loop between GSDME-mediated PCD and apoptosis, as well as bridges extrinsic and intrinsic apoptosis pathways.
2.2. GSDME-mediated PCD and GSDMD-mediated pyroptosis
Programmed necrosis mainly refers to necroptosis, pyroptosis, ferroptosis, NETosis and mitochondrial permeability transition (MPT)-dependent necrosis [44]. The cleavage of GSDME is induced by caspase 3 while GSDMD is triggered by caspase 1/4/5/11. Inflammatory caspases 1/4/5/11, initiator caspases 8/9, and the executioner caspase-7, are all unable to produce a pore-forming GSDME-N-terminal domain directly [13, 45]. Therefore, GSDMD and GSDME were once thought to function independently. However, canonical inflammasomes (NLRP3, AIM2, and NLRC4), of which activation is the crucial step of GSDMD-dependent pyroptosis, function as early events leading to caspase-3 cleavage following caspase 1/8 activation [21, 35, 46, 47]. Caspase-8 is not only the apical caspase in DNA-induced apoptosis, but induces caspase-3 activation in AIM2-, NLRP3-, and NLRC4-dependent pyroptotic pathways when caspase-1 is absent, with little or no requirement for either caspase-9 or caspase-2 [46, 47]. Additionally, caspase-1 facilitates caspase-3 activity regardless of whether caspase-8 is present [35]. Therefore, caspase-3 cleavage is driven by caspase-1 or caspase-8. In addition, the channel-forming membrane protein pannexin-1 is a substrate of caspase-3/7 during apoptosis [47-49] and forms pores on plasma membranes, which allow for water influx but not inflammatory cytokines release [20].
ROS accumulation, DNA damage, and extrinsic and intrinsic apoptotic signaling induce caspase-3 activation [21, 47, 50]. GSDME, downstream of caspase-3[13, 14, 45, 51], can be cut into two segments; the GSDME-N-terminal domain that is capable of forming pores on cell membranes [13, 14, 43, 52], and the structural autoinhibition of gasdermin E is destroyed when some sites in GSDME-C terminal domain mutate [53] . The active form of GSDME permeates mitochondrial membranes and promotes cytochrome C (Cyt c) release and caspase-3 activation, contributing to a self-amplifying feed-forward loop, augmenting cytotoxic effects [43]. Granzyme B (GzmB), released by natural killer (NK) cells, not only cleaves GSDME after D270, where the caspase-3 cleavage site was located, but cuts and activates caspase-3 [54, 55]. Overall, both caspase-3 and GzmB cleave GSDME effectively.
Cleaved caspase-3 blocks the GSDMD-mediated pathway specifically by cleaving GSDMD at D88 (D87 in human cells), deactivating the protein [14, 36]. Overall, GSDME-mediated PCD and GSDMD-mediated pyroptotic pathways engage in bidirectional crosstalk in cell death processes (Fig. 1).
3. 3. GSDME and cancer
3.1. GSDME expression and tumorigenesis inhibition
Low level of GSDME expression is a common feature of most cancer cells. From epigenetic studies, GSDME epigenetic silencing occurs in about 52-65% of primary cancers [6], such as gastric cancer [56], colorectal cancer [57, 58] and breast cancer [9, 10]; the incidence of GSDME hypermethylation indicated lymph node metastasis and a worse five-year survival rate in breast cancer [9, 10] as well as strong lymphatic vessel invasion and high TNM stage in colorectal cancer [58]. At present, promoter DNA hypermethylation is the most common feature responsible for GSDME epigenetic inactivation [59]. Moreover, some tumors avoid GSDME-mediated tumor suppression through loss-of-function mutations, which surround the caspase-3 cleavage site to abolish GSDME-mediated tumor cytotoxicity [54].
To identify whether GSDME is a tumor suppressor or not, a series of experiments were performed. Transfection of WT GSDME plasmids into cancer cells, including lung carcinoma, glioblastoma, melanoma, hepatocellular carcinoma, colorectal cancer, and triple-negative breast cancer cells, leads to significant tumor repression [6, 9, 43, 57, 60]. Similarly, GSDME downregulation leads to an obvious increase in growth, colony formation and invasion, while G2/M arrest and caspase 3/8 activation by the Fas/FasL pathway after GSDME gene transfection [60, 61]. When came to a mouse model of melanoma, GSDME-KO tumors behave faster than those expressing GSDME in not only formation but growing to reach sacrificial threshold [43]. A GSDME-/- intestinal cancer mouse model showed a more atypical hyperplasia rather than tumor formation, and less moderate mucosal inflammation, than GSDME positive mice, in both chemically induced and genetically modified groups [62].
3.2. GSDME amplified PCD mechanism
3.2.1. In vitro
Melanoma with GSDME deletion is known to resist etoposide treatment [9]. The small molecular target drugs BRAFi + MEKi (the combination of BRAF inhibitors and MEK inhibitors), have been shown to kill tumors through MEK-ERK1/2 signal attenuation and mitochondrial depolarization, causing caspase-3 cleavage and GSDME-N domain accumulation. Cells resistant to this combination treatment do not undergo GSDME-mediated pyroptosis, unless treated with other drugs that stimulate the GSDME cleavage process [63]. Additionally, iron synergizes with CCCP, which induces ROS signals, results in the mitochondrial outer membrane protein Tom20 oxidizing and oligomerizing. Bax, downstream of oxidized Tom20, is recruited to mitochondria to facilitate Cyt c release and activate the caspase cascade, ultimately leading to GSDME-N domain production [64]. Additionally, protective autophagy, which usually occurs during chemotherapy, is a new challenge in melanoma treatment. Doxorubicin induces GSDME-mediated PCD, as well as eEF-2K activation, a kinase related to autophagy. Silencing eEF-2K amplifies GSDME cleavage triggered by doxorubicin, and decreases inappropriate autophagy, which leads to stronger tumor inhibition [40].
Polo-like kinase 1 (PLK1) inhibitor augments the chemosensitivity of esophageal squamous cell carcinoma cells through the BAX-caspase-3-GSDME pathway and inhibits DNA repair [65]. In gastric cancer, intracytoplasmic contents flow from the pores formed by GSDME-N domain assembly upon 5-FU treatment [45]. As2O3 exerts HCC suppression with GSDME cleavage and downregulation of DNMT-related proteins containing Dnmt3a, Dnmt3b, and Dnmt1, which take part in elevation of DNA methyltransferase expression [66, 67]. In colon cancer cells, lobaplatin triggers ROS accumulation and JNK phosphorylation, resulting in intrinsic apoptotic signal initiation and GSDME-mediated PCD [14].
For lung cancer cells, whether traditional chemotherapeutic drugs or novel small molecular inhibitors, extrinsic and intrinsic apoptotic pathways are vital for induction of GSDME-N domain accumulation; the anti-tumor effectiveness of these drugs is weakened in GSDME deficient cells [11, 51]. The mitochondrial pathway is prominent in the activation of GSDME [14, 18, 68]. Dioscin induces a Bax/Bcl ratio decrease and GSDME-mediated PCD through the JNK/p38 pathway in human osteosarcoma [69]. Osthole disequilibrates the mitochondrial membrane potential to raise ROS levels in ovarian carcinoma cells, resulting in GSDME-dependent pyroptosis rather than apoptosis [70]. In vitro, galangin decreases glioblastoma multiforme cell, but not non-cancer cell, viability. It also triggers autophagy and GSDME-mediated PCD, where the Bax/caspase-3 axis plays an important role [71].
GSDME mRNA production in the human acute lymphoblastic leukemia cell line CEM increases after exposure to steroids. According to Webb et al., forskolin alone reduced cell growth but not death, however, it contributed to the apoptotic response when synergized with dexamethasone via the PKA cAMP pathway [72, 73]. GSDME epigenetic inactivation is induced by promoter DNA hypermethylation commonly[59], thus, decitabine, a common anti-cancer medicine, elevates tumor cell chemosensitivity through reversing GSDME silencing [56, 57]
3.2.2. In vivo
As the previous experiment mentioned, decitabine reversed GSDME silencing in human tumors [56, 57]. This function was also confirmed in mice that decitabine significantly elevated the DFNA5 gene expression of macrophages, colon cancer cells, and breast carcinoma cells [74]. It has been proved that treating specific tumor-bearing mice with combination of decitabine and cisplatin (DDP) in corresponding order, demethylating DFNA5 gene with decitabine primarily which followed by DDP, mediated a stronger tumor-killing effect than treating with DDP or decitabine alone. Moreover, a surprisingly increased presence of cytotoxic T lymphocytes (CTLs) in the tumor microenvironment was shown in mice treated with liposome cis-platinum + decitabine. This combination led to a shift of native CD8+T cells to the central memory CD8+T cells in the spleen, which considered to be efficient for mediating protective immunity [74]. Experiment has been conducted recently to testify that ectopic mGSDME expression in 4T1E and B16 cells markedly inhibited tumor growth, whereas 4T1E tumors overexpressing lose-of-function (LOF) mutants of GSDME did not have more functional tumor infiltrating lymphocytes (TILs) [54]. Moreover, regrowth of residual disease after removing BRAFi + MEKi was sooner and lager in GSDME-KO tumors than control group [63]. Anyhow, the evidence to certify that GSDME serves as a cancer fighter by amplifying PCD is still far away from enough and fulfilled, lots of work called for been done.
3.3. GSDME-mediated PCD activates the immune response
In 2011, Douglas Hanahan and Robert A. Weinberg extended the hallmarks of cancer from six to ten; avoiding immune destruction is one of the hallmarks [75]. The pore-forming GSDME-N domain helps the release of pro-inflammatory factors (e.g. IL-1α, IL-1β, IL-18, high-mobility group box 1 (HMGB1) and adenosine 5′triphosphate (ATP)) in a lysis-dependent manner [13, 14, 43, 55, 63, 76], leading to a strong inflammatory reaction even though it has nothing to do with IL-1β/18 maturation. Immunocytes receive these stimuli, like HMGB1 and ATP, and progress to cell death processes. Additionally, its expression relates to the tumor microenvironment, which stimulates the recruitment of tumor infiltrating lymphocytes (TILs), tumor-associated macrophages, and inflammatory signal production, like GzmB and perforin (PFN) [54]. Due to immunity participation, GSDME-mediated anti-cancer pathways involve systemic responses and does not kill tumors alone.
3.3.1. Phagocytosis amplification and immunocytes activation
Pro-necrotic molecules promote intra-tumoral immunity through immunocytes, thus, GSDME, a pro-necrotic molecule, is significantly positively associated with myeloid cells, especially macrophages [77]. Consistently, the markers of macrophages increase significantly in tumors with ectopic GSDME expression [54], indicating that GSDME enhances the phagocytosis of tumors. The activation and proliferation of TILs is beneficial in sustained tumor suppression [78]. NK cells in ectopic GSDME expression tumors express more GzmB and PFN, similar to CD8+ T cells, although with no increase in quantity [54]. Of note, there is more secretion of IFNγ and TNF from CD8+T cells, suggesting the elevation of immunocyte activation. Furthermore, BRAFi + MEKi induces GSDME-mediated PCD, following by intra-tumoral immune reactions involving the release of inflammatory factors, intra-tumor dendritic cell (DC) activation, and T cell abundance extension. Less immunocyte activation and shorter tumor regression under BRAFi+MEKi therapy are detected in GSDME-knockout tumors than in control counterparts [63]. This gene is well-correlated to the anti-tumor immunocyte response.
3.3.2. GzmB from NK cells
GzmB, an exogenous serine endopeptidase, is found in a variety of cancers. It is generally released from TILs with PFN, and regulates caspase-independent cell death upon intracellular delivery of PFN [79, 80]. When GSDME-overexpressing tumors are incubated with NK cells, GzmB, from NK cells, cleaves GSDME effectively and initiates GSDME-mediated pyroptotic pathway [54]. Similarly, during chimeric antigen receptor (CAR) T cell therapy, cytolytic T cells release GzmB and drive GSDME cleavage. This reaction triggers cytokine release syndrome (CRS) in patients, neutralizing the effectiveness of CAR T treatment. Furthermore, the effect of GzmB is amplified by caspase, for it cuts caspase-3/7 to produce their active forms [55, 79, 80]. Taken together, GzmB mediates extrinsic and caspase-independent pathways of GSDME-mediated PCD.
4. Conclusion
While GSDME has been of interest for decades, associated studies remain limited. GSDME has been shown to play a crucial role in various molecular processes, including HL [4, 81], tumor suppression [57], PCD-mediation [6, 18] and acting as an anti-tumor immunity promoter [54, 63]. However, recent research found out that treating GSDME-/- mice with cisplatin, they showed less tissue damages and weight loss [13]. Therefore, GSDME was benefit for anti-cancer therapy outcomes or not was still unclear and more experiments combining GSDME and anti-cancer therapy was urged to conducted.
Despite adverse reactions, such as CRS [55], severe weight loss upon chemotherapy [13], and induction of renal tubulointerstitial fibrosis progression [82], the significant anti-tumor potential of GSDME should not be ignored. Some researchers propose that GSDME may be a promising biomarker for cancer detection, for predicting the 5-year patient survival rate [83], and for use in GSDME-mediated PCD initiated immunogenic cell death (ICD) in GSDME-overexpressing cells [54]. The standard of ICD is that secondary-tumor formation could be prevented after injection with tumor cells experiencing ICD [84], meaning that GSDME-dependent cell death results in sustainable tumor regression. P53, an acknowledged tumor suppressor, commands various of downstream genes transcriptional activation. DFNA5, one of those genes, has been performed to be strongly induced by exogenous and endogenous p53. Concertedly, the ability of promoting cell death of DFNA5 can only be exerted in the presence of p53 while its effect reversed in the absence of p53 to entice the resistance to etoposide. A possible explanation of this relation is that a potential p53-binding sequence locates in DFNA5 intron 1 and DFNA5 is a mediator in p53-inducing cell death process, thus DFNA5's function might be disordered when p53 is negative or LOF [16].
More than acting as a tumor suppressor, GSDME functions as a potential therapeutic target in the battle against cancer. It has been proved that chemotherapeutic drugs like cisplatin, paclitaxel or doxorubicin successfully induced GSDME-N domain formation, which was benefit to cancer cells killing, including lung cancer [51] and colon cancer [85]. Small molecular inhibitors, effecting on KRAS, EGFR, ALK in lung cancer [11], BRAF and MEK in melanoma cells [63], elicit concurrent GSDME-mediated PCD. Additionally, PLK1 kinase inhibitor can be utilized as adjuvant in cisplatin-driven oesophageal squamous cell carcinoma therapy since it enhances the chemosensitivity by eliciting GSDME cleavage [65]. Excessive levels of ROS increase cellular oxidative stress which is harmful for cell surviving and disruption of ferritin lead to elevation of ROS [64]. Iron combining with ROS-inducing drugs facilitates ROS level to be sensed by Tom20-Bax-caspase-GSDME axis with mitochondrial intrinsic apoptotic dependency, what has been detected by Zhou's group. Interestingly, knockdown GSDME could attenuate pyroptosis and LDH release while this inhibitory effect is abolished when GSDMD replaced GSDME [64]. Hence, GSDME might serve as an executor in cancer cells death. But, the detail of how GSDME mediates cancer therapy has not been completely revealed and whether GSDME cleavage boosts cancer treatment in vivo has not been ultimately confirmed.
Currently, the clinical application of GSDME remains a future prospect, and the molecular mechanisms underlying its activity remain to be precisely characterized. Further studies that unveil GSDME biological functions will improve the current understanding of its role in cancer proliferation and invasion, which will expedite the development of novel anti-cancer therapies.
Abbreviations
GSDME: gasdermin E; PCD: programmed cell death; PJVK: Pejvakin; DFNA5: deafness autosomal dominant 5; ICERE-1: inversely correlated to estrogen receptor expression; MT: mutant type; PRR: pattern-recognition receptor; DAMPs: damaged-associated molecular patterns; PAMPs: pathogen-associated molecular patterns; NLRs: nucleotide-binding domain and leucine-rich repeat containing receptors; AIM: absent in melanoma; dsDNA: double-stranded DNA; ROS: reactive oxygen species; ASC: apoptosis-associated speck-like protein containing a caspase recruitment domain; CARD: N-terminal caspase recruitment; PYD: pyrin domain; Cyt c: cytochrome C; GzmB: Granzyme B; NK: natural killer; ERK: extracellular signal regulated kinase; JNK: c-Jun N-terminal kinase; eEF-2k: Eukaryotic elongation factor-2 kinase; MAPK: mitogen-activated protein kinase; BRAFi + MEKi: the combination of BRAF inhibitors and MEK inhibitors; CCCP: carbonyl cyanide m-chlorophenyl hydrazine; PLK1: Polo-like kinase 1; DNMT: DNA methyltransferase; PKA: protein kinase A; cAMP: cyclic adenosine moriophosphate; TILs: tumor infiltrating lymphocytes; IL-18: Interleukin 18; IL-1β: Interleukin1β; PFN: perforin; IFNγ: interferon γ; TNF: tumor necrosis factor; DC: dendritic cell; CAR: chimeric antigen receptor; CRS: cytokine release syndrome; ICD: immunogenic cell death; TNM: tumor, lymph node, metastasis; HMGB1: high-mobility group box 1; ATP: adenosine 5′triphosphate; CTLs: cytotoxic T lymphocytes; DDP: cisplatin; TLR: toll-like receptor; LOF: lose of function.
Acknowledgements
Y Wang, J Peng, Z Zhang and X Xie screened the reference. Y Wang drafted the manuscript. Y Wang, J Peng and M Li discussed and revised the manuscript. All authors read and approved final manuscript. Thanks to all authors for their time and effort and department of breast surgery from the first hospital of Jilin University for technical support.
Funding
This work was supported by the First Hospital of Jilin University and Jilin University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Feng S, Fox D, Man SM. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J Mol Biol. 2018;430:3068-3080
2. Tamura M, Tanaka S, Fujii T. et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618-629
3. Zheng Z, Deng W, Lou X. et al. Gasdermins: pore-forming activities and beyond. Acta Biochim Biophys Sin (Shanghai). 2020;52:467-474
4. Van Laer L, Huizing E H, Verstreken M. et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194-197
5. Li YQ, Peng JJ, Peng J. et al. The deafness gene GSDME: its involvement in cell apoptosis, secondary necrosis, and cancers. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:1043-1048
6. Op de Beeck K, Van Camp G, Thys S. et al. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet. 2011;19:965-973
7. Thompson DA, Weigel RJ. Characterization of a gene that is inversely correlated with estrogen receptor expression (ICERE-1) in breast carcinomas. Eur J Biochem. 1998;252:169-177
8. Croes L, Beyens M, Fransen E. et al. Large-scale analysis of DFNA5 methylation reveals its potential as biomarker for breast cancer. Clin Epigenetics. 2018;10:51
9. Kim MS, Lebron C, Nagpal JK. et al. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem Biophys Res Commun. 2008;370:38-43
10. Fujikane T, Nishikawa N, Toyota M. et al. Genomic screening for genes upregulated by demethylation revealed novel targets of epigenetic silencing in breast cancer. Breast Cancer Res Treat. 2010;122:699-710
11. Lu H, Zhang S, Wu J. et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin Cancer Res. 2018;24:6066-6077
12. Lage H, Helmbach H, Grottke C. et al. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001;494:54-59
13. Wang Y, Gao W, Shi X. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99-103
14. Rogers C, Fernandes-Alnemri T, Mayes L. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128
15. Op de Beeck K, Schacht J, Van Camp G. Apoptosis in acquired and genetic hearing impairment: the programmed death of the hair cell. Hear Res. 2011;281:18-27
16. Masuda Y, Futamura M, Kamino H. et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J Hum Genet. 2006;51:652-664
17. Gregan J, Van Laer L, Lieto L D. et al. A yeast model for the study of human DFNA5, a gene mutated in nonsyndromic hearing impairment. Biochim Biophys Acta. 2003;1638:179-186
18. Van Rossom S, Op de Beeck K, Hristovska V. et al. The deafness gene DFNA5 induces programmed cell death through mitochondria and MAPK-related pathways. Front Cell Neurosci. 2015;9:231
19. Van Rossom S, Op de Beeck K, Franssens V. et al. The splicing mutant of the human tumor suppressor protein DFNA5 induces programmed cell death when expressed in the yeast Saccharomyces cerevisiae. Front Oncol. 2012;2:77
20. Fang Y, Tian S, Pan Y. et al. Pyroptosis: A new frontier in cancer. Biomed Pharmacother. 2020;121:109595
21. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
22. Aachoui Y, Sagulenko V, Miao E A. et al. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. 2013;16:319-326
23. Wang B, Tian Y, Yin Q. AIM2 Inflammasome Assembly and Signaling. Adv Exp Med Biol. 2019;1172:143-155
24. Jin T, Perry A, Jiang J. et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561-571
25. Roberts TL, Idris A, Dunn JA. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057-1060
26. Schroder K, Sagulenko V, Zamoshnikova A. et al. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology. 2012;217:1325-1329
27. Sun Q, Scott M J. Caspase-1 as a multifunctional inflammatory mediator: noncytokine maturation roles. J Leukoc Biol. 2016;100:961-967
28. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325-332
29. Zhao Y, Yang J, Shi J. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596-600
30. Gong YN, Shao F. Sensing bacterial infections by NAIP receptors in NLRC4 inflammasome activation. Protein Cell. 2012;3:98-105
31. Ferrao R, Wu H. Helical assembly in the death domain (DD) superfamily. Curr Opin Struct Biol. 2012;22:241-247
32. Lu A, Magupalli VG, Ruan J. et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193-1206
33. Vajjhala PR, Lu A, Brown DL. et al. The Inflammasome Adaptor ASC Induces Procaspase-8 Death Effector Domain Filaments. J Biol Chem. 2015;290:29217-29230
34. Fu TM, Li Y, Lu A. et al. Cryo-EM Structure of Caspase-8 Tandem DED Filament Reveals Assembly and Regulation Mechanisms of the Death-Inducing Signaling Complex. Mol Cell. 2016;64:236-250
35. Sagulenko V, Vitak N, Vajjhala PR. et al. Caspase-1 Is an Apical Caspase Leading to Caspase-3 Cleavage in the AIM2 Inflammasome Response, Independent of Caspase-8. J Mol Biol. 2018;430:238-247
36. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol. 2017;24:507-514.e504
37. Schwartz LM, Smith SW, Jones ME. et al. Do all programmed cell deaths occur via apoptosis? Proc Natl Acad Sci USA. 1993;90:980-984
38. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891-2906
39. Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66-74
40. Yu P, Wang HY, Tian M. et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol Sin. 2019;40:1237-1244
41. Sun Y, Liu WZ, Liu T. et al. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35:600-604
42. Van Laer L, Vrijens K, Thys S. et al. DFNA5: hearing impairment exon instead of hearing impairment gene? J Med Genet. 2004;41:401-406
43. Rogers C, Erkes DA, Nardone A. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689
44. Wallach D, Kang TB, Dillon CP. et al. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154
45. Wang Y, Yin B, Li D. et al. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem Biophys Res Commun. 2018;495:1418-1425
46. Sagulenko V, Thygesen SJ, Sester DP. et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149-1160
47. Chen KW, Demarco B, Heilig R. et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. Embo J. 2019 38
48. Chekeni FB, Elliott MR, Sandilos JK. et al. Pannexin 1 channels mediate 'find-me' signal release and membrane permeability during apoptosis. Nature. 2010;467:863-867
49. Sandilos JK, Chiu YH, Chekeni FB. et al. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303-11311
50. Henry RE, Andrysik Z, Paris R. et al. A DR4:tBID axis drives the p53 apoptotic response by promoting oligomerization of poised BAX. Embo J. 2012;31:1266-1278
51. Zhang CC, Li CG, Wang YF. et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24:312-325
52. Rogers C, Alnemri ES. Gasdermins: novel mitochondrial pore-forming proteins. Mol Cell Oncol. 2019;6:e1621501
53. Ding J, Wang K, Liu W. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111-116
54. Zhang Z, Zhang Y, Xia S. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415-420
55. Liu Y, Fang Y, Chen X. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol. 2020 5
56. Akino K, Toyota M, Suzuki H. et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007;98:88-95
57. Kim M S, Chang X, Yamashita K. et al. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene. 2008;27:3624-3634
58. Yokomizo K, Harada Y, Kijima K. et al. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res. 2012;32:1319-1322
59. Xia X, Wang X, Cheng Z. et al. The role of pyroptosis in cancer: pro-cancer or pro-"host"? Cell Death Dis. 2019;10:650
60. Wang CJ, Tang L, Shen DW. et al. The expression and regulation of DFNA5 in human hepatocellular carcinoma DFNA5 in hepatocellular carcinoma. Mol Biol Rep. 2013;40:6525-6531
61. Stennicke HR, Jurgensmeier JM, Shin H. et al. Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem. 1998;273:27084-27090
62. Croes L, Fransen E, Hylebos M. et al. Determination of the Potential Tumor-Suppressive Effects of Gsdme in a Chemically Induced and in a Genetically Modified Intestinal Cancer Mouse Model. Cancers (Basel). 2019 11
63. Erkes DA, Cai W, Sanchez IM. et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020;10:254-269
64. Zhou B, Zhang JY, Liu XS. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171-1185
65. Wu M, Wang Y, Yang D. et al. A PLK1 kinase inhibitor enhances the chemosensitivity of cisplatin by inducing pyroptosis in oesophageal squamous cell carcinoma. EBioMedicine. 2019;41:244-255
66. Miremadi A, Oestergaard MZ, Pharoah P D. et al. Cancer genetics of epigenetic genes. Hum Mol Genet. 2007 16 Spec No 1:R28-49
67. Hu J, Dong Y, Ding L. et al. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct Target Ther. 2019;4:28
68. Rogers C, Alnemri E S. Gasdermins in Apoptosis: New players in an Old Game. Yale J Biol Med. 2019;92:603-617
69. Ding Q, Zhang W, Cheng C. et al. Dioscin inhibits the growth of human osteosarcoma by inducing G2/M-phase arrest, apoptosis, and GSDME-dependent cell death in vitro and in vivo. J Cell Physiol. 2020;235:2911-2924
70. Liang J, Zhou J, Xu Y. et al. Osthole inhibits ovarian carcinoma cells through LC3-mediated autophagy and GSDME-dependent pyroptosis except for apoptosis. Eur J Pharmacol. 2020;874:172990
71. Kong Y, Feng Z, Chen A. et al. The Natural Flavonoid Galangin Elicits Apoptosis, Pyroptosis, and Autophagy in Glioblastoma. Front Oncol. 2019;9:942
72. Medh RD, Webb MS, Miller A L. et al. Gene expression profile of human lymphoid CEM cells sensitive and resistant to glucocorticoid-evoked apoptosis. Genomics. 2003;81:543-555
73. Medh RD, Saeed MF, Johnson BH. et al. Resistance of human leukemic CEM-C1 cells is overcome by synergism between glucocorticoid and protein kinase A pathways: correlation with c-Myc suppression. Cancer Res. 1998;58:3684-3693
74. Fan JX, Deng RH, Wang H. et al. Epigenetics-Based Tumor Cells Pyroptosis for Enhancing the Immunological Effect of Chemotherapeutic Nanocarriers. Nano Lett. 2019;19:8049-8058
75. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674
76. Schneider KS, Groß CJ, Dreier RF. et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep. 2017;21:3846-3859
77. Stoll G, Ma Y, Yang H. et al. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. Oncoimmunology. 2017;6:e1299302
78. Pages F, Galon J, Dieu-Nosjean M C. et al. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29:1093-1102
79. Yang X, Stennicke HR, Wang B. et al. Granzyme B mimics apical caspases. Description of a unified pathway for trans-activation of executioner caspase-3 and -7. J Biol Chem. 1998;273:34278-34283
80. Metkar SS, Wang B, Ebbs M L. et al. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol. 2003;160:875-885
81. de Beeck KO, Van Laer L, Van Camp G. DFNA5, a gene involved in hearing loss and cancer: a review. Ann Otol Rhinol Laryngol. 2012;121:197-207
82. Wen S, Wang ZH, Zhang CX. et al. Caspase-3 Promotes Diabetic Kidney Disease Through Gasdermin E-Mediated Progression to Secondary Necrosis During Apoptosis. Diabetes Metab Syndr Obes. 2020;13:313-323
83. Ibrahim J, Op de Beeck K, Fransen E. et al. Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med. 2019;8:2133-2145
84. Galluzzi L, Buque A, Kepp O. et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97-111
85. Yu J, Li S, Qi J. et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193
Author contact
![]() Corresponding author: Ming Yang, Department of Breast Surgery, The First Hospital of Jilin University, Changchun 130021, People's Republic of China (e-mail: yangmingedu.cn).
Corresponding author: Ming Yang, Department of Breast Surgery, The First Hospital of Jilin University, Changchun 130021, People's Republic of China (e-mail: yangmingedu.cn).