Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(18):5543-5561. doi:10.7150/jca.54699 This issue Cite
Review
Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy
Run Huang#, Huan Chen#, Jiayu Liang, Yi Li, Jiali Yang, Chuang Luo, Youshan Tang, Yu Ding, Xing Liu, Qing Yuan, Hong Yu, Yingchun Ye, Wenfeng Xu, Xiang Xie ![]()
Public Center of Experimental Technology, The school of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan Province, 646000, China.
#These authors contributed equally to this work.
Received 2020-10-19; Accepted 2021-6-30; Published 2021-7-25
Abstract

Reactive oxygen species (ROS) play a dual role in the initiation, development, suppression, and treatment of cancer. Excess ROS can induce nuclear DNA, leading to cancer initiation. Not only that, but ROS also inhibit T cells and natural killer cells and promote the recruitment and M2 polarization of macrophages; consequently, cancer cells escape immune surveillance and immune defense. Furthermore, ROS promote tumor invasion and metastasis by triggering epithelial-mesenchymal transition in tumor cells. Interestingly, massive accumulation of ROS inhibits tumor growth in two ways: (1) by blocking cancer cell proliferation by suppressing the proliferation signaling pathway, cell cycle, and the biosynthesis of nucleotides and ATP and (2) by inducing cancer cell death via activating endoplasmic reticulum stress-, mitochondrial-, and P53- apoptotic pathways and the ferroptosis pathway. Unfortunately, cancer cells can adapt to ROS via a self-adaption system. This review highlighted the bidirectional regulation of ROS in cancer. The study further discussed the application of massively accumulated ROS in cancer treatment. Of note, the dual role of ROS in cancer and the self-adaptive ability of cancer cells should be taken into consideration for cancer prevention.
Keywords: reactive oxygen species, cancer, pro-tumor, anti-cancer, self-adaption system, therapy
Introduction
The Global Burden of Disease 2017 Study showed that deaths from noncommunicable diseases represented 73.4% of deaths in 2017 [1]. Cancer causes the second largest number of fatalities following cardiovascular disease [1]. Between 2007 and 2017, the number of cancer cases increased by 33%, and the average annual age-standardized incidence rates for all cancers increased [2]. Global cancer statistics 2018 revealed 18.1 million new cancer cases and 9.6 million cancer-related deaths in 2018 [3]. Malignant tumors have become the main killer among humans. The treatment of cancer has gained the attention of the medical community because of the rapid increase in the global cancer burden. An increasing number of studies have emerged demonstrating that reactive oxygen species (ROS) are highly intertwined with various cancers [4], such as lung cancer [5], colorectal cancer [6], breast cancer [7], hepatocellular cancer [8], and cervical cancer [9], which are among the top 10 cancers ranked by the highest number of incident cases globally [2]. The mechanism of ROS in carcinogenesis needs to be considered to determine more efficient therapeutics against cancer. This review highlighted the 'double-edged sword' role of ROS in cancer formation, development, and suppression. In addition, the self-adaption mechanism of cancer cells was discussed. The study also focused on the therapeutic approaches targeting ROS to treat cancers, thus providing an insight into how pro- and anti-tumorigenic ROS signaling pathways are involved in the treatment of cancer.
Physical properties of ROS
ROS exist in two forms. One form comprises free oxygen radicals including superoxide, hydroxyl radicals, nitric oxide, alkoxyl radicals, and peroxyl radicals [10]. The other form comprises nonradical ROS, including hydrogen peroxide (H2O2), organic hydroperoxides, and hypochlorite [10]. ROS are mainly generated by the mitochondrial electron transport chain and tricarboxylic acid cycle [11]. Besides, the peroxisome and the endoplasmic reticulum (ER) can produce oxidants [12]. Many enzymes also promote the production of ROS, such as nicotinamide adenine dinucleotide phosphate oxidase (NOX), xanthine oxidase, nitric oxide synthase, cyclooxygenases, cytochrome P450 enzymes, and lipoxygenases [12]. Additionally, exogenous stress, such as that from ionizing radiation, chemotherapeutic drugs and environmental insult, is also the main cause of ROS generation [13]. ROS can oxidize intracellular lipids, proteins and deoxyribonucleic acid (DNA), thus leading to the accumulation of damaged biological molecules. ROS induced DNA damage can include single- or double-strand breakage, base modifications, deoxyribose modification, and DNA cross-linking [13]. Moreover, lipid peroxidation results in the formation of reactive aldehydes, including malondialdehyde and 4-hydroxy-2-nonenal, which have high reactivity toward protein and DNA [14]. Although this damaging aspect of ROS biology persists, oxidant species are increasingly being understood to have beneficial effects. ROS can regulate cellular proliferation, differentiation, survival, and even apoptosis processes [15]. ROS thus play a dual role in cell development.
Potential mechanisms involved in the cancer promoting effects of ROS
Promoting effects of ROS in cancer
The physiological levels of ROS are important to regulate crucial cellular processes such as cellular proliferation, differentiation, survival, and even apoptosis in normal cells [15]. Once ROS exceed normal levels, the high levels of ROS can initiate tumorigenesis and promote tumor progression [16]. Excess ROS cause gene mutations, and subsequently activate oncogenes and inhibit tumor suppressor genes, such as Kirsten rat sarcoma viral oncogene (KRAS) and tumor suppressor P53, thus leading to cancer initiation [17]. Not only that, but increased ROS also promote tumor development by reducing the function of T cells and natural killer (NK) cells and promoting the recruitment and M2 polarization of macrophages, thereby promoting tumor progression [18-21]. Infiltration of T cells, NK cells, and M1 macrophages in breast cancer has been reported to prevent tumor development [22]. Furthermore, high levels of ROS induce tumor invasion and metastasis via epithelial-mesenchymal transition (EMT) [23]. Below, we further discuss the potential mechanisms of ROS in promoting cancer.
Potential mechanisms of promoting effects of ROS in cancer
ROS promote cancer formation via initiating DNA mutation
DNA is the carrier of genetic information essential for the survival and reproduction of organisms. The evolution of normal cells into cancer cells involves genetic instability [24]. The overexpression of exogenous and endogenous ROS often leads to DNA mutations, affecting the interpretation and transmission of genetic information [25]. The DNA mutations lead to the inactivation of tumor suppressor genes and/or the activation of oncogenes, allowing cells to proliferate outside their normal growth restraints, and promote tumorigenesis by favoring tumor cell proliferation, migration, and resistance to apoptosis [26]. ROS can cause hotspot codon mutagenesis in P53, and the mutations of P53 have been reported in liver cancer and breast cancer [25, 27]. Besides, ROS also cause mutagenesis in the oncogene rat sarcoma (Ras) gene, and the mutation of Ras is closely related to skin cancer and colorectal cancer [28-30]. In another study, excessive H2O2 produced by myeloid cells has been shown to trigger genome-wide DNA mutations in intestinal epithelial cells to stimulate invasive growth during intestinal tumorigenesis [16].
ROS can lead to DNA damage and cause DNA mutations (Fig. 1). ROS can oxidize nucleobases and form different types of oxidative damage products that have mutational properties [31]. Guanine (G), with a lower oxidation potential compared with other bases [32], is most easily oxidized by ROS, leading to the formation of an oxidized product 7,8-dihydro-8-oxo-2´-deoxyguanosine (8-oxo-dG), which is the most extensively studied oxidative damage product [33]. 8-oxo-dG acts as a mutagenic lesion due to the loss of base-pairing specificity and mispairing with adenine (A) [34]. When ROS oxidizes guanine in DNA, 8-oxo-dG acts as a template, leading to G→T (thymine) transversion mutations [35]. When ROS oxidize deoxyguanosine triphosphate in the nucleotide pool, 8-oxo-dG acts as a substrate, resulting in A→C (cytosine) mutations [35]. ROS also oxidize A with two main products: 2-hydroxy-2'-deoxyadenosine (2-OH-dA) [36] and 7,8-dihydro-8-oxo-2'-deoxyadenosine (8-oxo-dA) [32]. Mutagenic replication of 2-OH-dA as a template causes A→C, A→G, A→T substitutions, and misincorporation of 2-OH-dA as a substrate causes GC→AT transition [36, 37]. Besides, 8-oxo-dA can lead to A→G and A→C mutations [38]. Other oxidative DNA lesions, such as 5-hydroxy-2'-deoxycytidine, 5,6-dihydro-5,6-dihydroxy-2'-deoxyuridine, and 5-hydroxy-2'-deoxyuridine oxidized from cytidine, as well as 5,6-dihydro-5,6-dihydroxy-2'-deoxythymidine oxidized from thymine, have also been shown to be mutagenic [31]. In addition, ROS can cause hydrogen bond breakage, unfolding, and double- and single-strand breakage of the DNA double-helix structure, leading to the exposure of more purine and pyrimidine residues to ROS that further facilitate the oxidation of nucleobases [39], which promotes DNA mutations.
Elevated-ROS cause nuclear DNA, thus leading to tumorigenesis. High levels of ROS oxidize adenine in DNA, thus leading to the formation of 8-oxo-dA and 2-OH-dA; in turn, 8-oxo-dA can lead to A→G and A→C mutations, and 2-OH-dA causes A→C, A→G, and A→T substitutions. ROS also oxidize guanine in DNA, resulting in the formation of 8-oxo-dG, which leads G→T transversion mutations. dGTP and dATP in the nucleotide pool are also oxidized by ROS, yielding 8-oxo-dGTP and 2-OH-dATP. The misincorporation of 8-oxo-dGTP and 2-OH-dATP as a substrate causes A→C and GC→AT mutation, respectively. Moreover, ROS impair the DNA damage repair system by downregulating the function of hOGG1 and BRCA2, thus leading to nuclear DNA mutations and favoring tumorigenesis. dATP, deoxyadenosine triphosphate; BRCA2, breast cancer susceptibility gene 2.
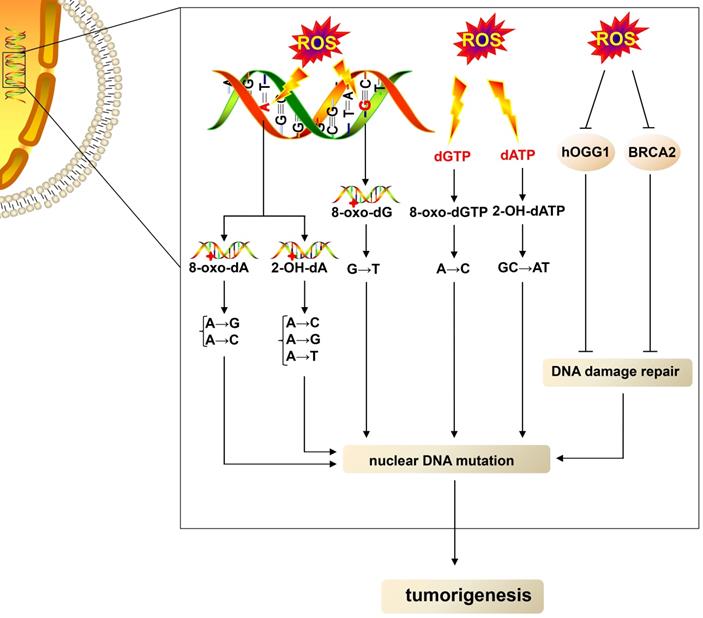
The DNA damage repair system is vital in dealing with DNA damage to avoid DNA mutations. DNA damage repair methods include base excision repair (BER) for base modifications, mismatch repair for mismatched bases, nucleotide excision repair (NER) for intra-strand cross-links and thymidine dimers, and nonhomologous end joining and homologous recombination (HR) for double-strand DNA breaks [40]. ROS can not only damage DNA but also repress the part DNA repair process (Fig. 1). ROS can oxidize human 8-oxoguanine DNA N-glycosylase 1 (hOGG1) and inhibit its activity, resulting in the impairment of BER [41]. Besides, ROS block HR by inducing proteasomal degradation of breast cancer susceptibility gene 2, one of the key factors for HR [42]. In summary, ROS not only cause direct DNA damage but also impair the DNA damage repair system, leading to DNA mutations and thus favoring tumor growth.
ROS promote cancer development via regulating immune cells
T cells
High levels of ROS can block the differentiation, maturation, and activation of T cells and induce T-cell death to suppress the antitumor function of T cells (Fig. 2). The upregulation of ROS enhances the production of vascular endothelial growth factor (VEGF) via stabilizing hypoxia-inducible factor 1 α (HIF-1α) [43, 44]. The overproduction of VEGF then blocks the differentiation and/or migration of thymus-committed progenitors, thus interfering with T-cell development and contributing to tumor-associated immune deficiencies [45]. The production of ROS can also reduce the expression of the CD3ζ chain, thus blocking the differentiation and activation of T cells [46, 47]. The CD3ζ chain, a component of the T-cell antigen receptor complex, is important in T-cell differentiation and activation, which mediates signal transduction of T cells [47]. With the decrease in the expression of the CD3ζ chain, the functions of T cells, such as proliferative ability, cytotoxic activity, and cytokine production are inhibited, finally suppressing the immune response [46-48]. In line with this, the reduced expression of CD3ζ has been reported in several cancers, such as gastric adenocarcinoma, breast cancer, and head and neck cancer [49-51]. In the tumor microenvironment, granulocytic or polymorphonuclear myeloid-derived suppressor cells, which are immunosuppressive cells, have the ability to overexpress ROS to inhibit T-cell activation and function, thus promoting tumor metastasis [18]. A reduction of ROS in myeloid-derived suppressor cells significantly increases the number of active CD8+ and CD4+ T lymphocytes, which efficiently suppresses the proliferation of tumor cells [52]. Besides influencing the development of T cells, elevated levels of ROS also induce T-cell death. ROS trigger extracellular signal-regulated kinase (ERK) phosphorylation and subsequently lead to the activation of poly(ADP-ribose) polymerase (PARP) 1, which causes the release of apoptosis-inducing factor, leading to T-cell death [53]. Consistent with these findings, the in vivo blockade of ROS reversed the selective loss of selective CD4+ T lymphocytes and delayed nonalcoholic fatty liver disease-promoted hepatocellular carcinoma (HCC) [54].
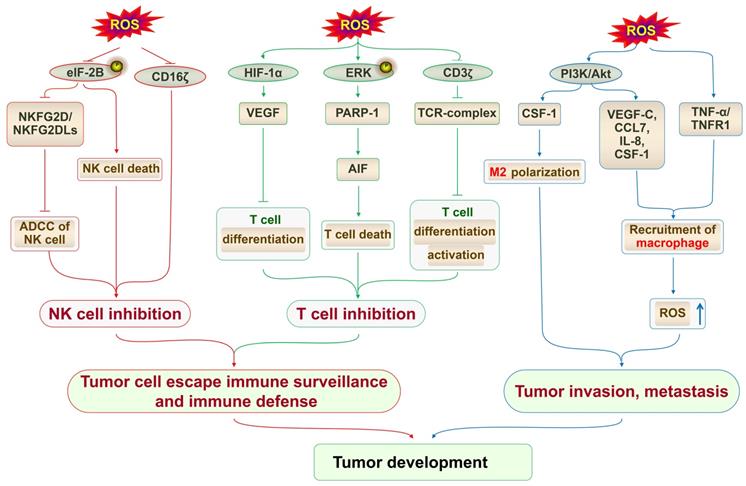
The upregulation of ROS inhibits T cells and NK cells, recruits macrophages, and induces M2 macrophage polarization. The overproduction of ROS attenuates the phosphorylation of eIF2B, thereby downregulating NKG2D/NKG2Dls and suppressing NK cell-mediated ADCC activity. Elevated ROS downregulate CD16ζ expression in NK cells, thus inhibiting the cytotoxicity of NK cells. The upregulation of ROS enhances the production of VEGF via stabilizing HIF-1α, thus interfering with T-cell differentiation. ROS trigger ERK activation of PARP-1, thereby causing the release of AIF and finally leading to T-cell death. The production of ROS also decreases CD3ζ chain expression, thereby impairing the formation of TCR, and blocking the differentiation and activation of T cells. The inhibition of T cells and NK cells favors tumor development. Increased ROS activate the PI3K/Akt signaling pathway and promote the secretion of VEGF-C, CCL7, IL-8, and CSF-1, thus resulting in macrophage recruitment. ROS also activate TNF-α/TNFR1 signaling, which results in macrophage recruitment. Macrophages in turn elevate the secretion of ROS and finally promote tumor invasion and metastasis. ROS/PI3K/Akt signaling-dependent CSF-1 production induces M2 polarization of macrophages, thus contributing to tumor cell growth. NKG2DLs, NKG2D ligands; ADCC, antibody-dependent cell-mediated cytotoxicity; AIF, apoptosis-inducing factor; CCL7, cytokines chemokine (C-C motif) ligand 7.
NK cells
Besides controlling T cells, high levels of ROS facilitate cancer cell distant metastasis by downregulating NK cell function (Fig. 2). Elevated levels of ROS downregulate CD16ζ expression in NK cells, which is an important signal transduction molecule for NK cell functions, thus inhibiting the cytotoxicity of NK cells [46]. Besides, the overproduction of ROS can attenuate the phosphorylation of eukaryotic initiation factor 2B, downregulate the expression of natural killer group 2 member D (NKG2D) and NKG2D ligands, and subsequently reduce the release of cytotoxic granules of NK cells, thus suppressing NK cell-mediated antibody-dependent cell-mediated cytotoxicity activity and finally promoting breast cancer growth and metastasis [7, 55]. Furthermore, ROS reduce interferon-γ (IFN-γ) production in NK cells, consequently downregulating NK cell-mediated clearance of malignant cells and finally resulting in melanoma metastasis [56]. Elevated levels of ROS also can lead to the death of NK cells by inducing apoptosis [57]. Primary human chronic myelomonocytic leukemia (CMML) cells release ROS, leading to the death of NK cells, which work as an immune escape mechanism in CMML [58]. Additionally, liver-resident NK cells undergo apoptosis because of the increased production of mitochondrial ROS, which allows tumors to evade liver NK cell surveillance, ultimately leading to colorectal liver metastatic tumors [19].
Macrophages
Macrophages are of two types: cancer-inhibiting M1 type and cancer-promoting M2 type [59]. Tumor-associated macrophages (TAMs) show an M2 phenotype, the infiltration of which contributes to cancer malignant progression [60]. ROS can regulate both recruitment and M2 polarization of macrophages to promote cancer development [20] (Fig. 2). ROS have been reported to inactivate phosphatase and tensin homolog [61], a well-known negative regulator of phosphatidylinositide 3-kinase (PI3K), the activated form of which phosphorylates protein kinase B (Akt) [62]. The mechanisms reported so far suggest that NOX4-induced ROS activate the PI3K/Akt signaling pathway and lead to the secretion of VEGF-C, cytokines chemokine (C-C motif) ligand 7, interleukin-8, and colony-stimulating factor-1 (CSF-1), resulting in macrophage recruitment [20]. Besides, myeloid-derived H2O2 activates tumor necrosis factor (TNF-α)/TNFR1 signaling in intestinal epithelial cells (IEC), increasing the recruitment of macrophages and in turn elevating the secretion of H2O2, which can trigger IEC mutation and promote tumor invasion [16]. In addition, ROS are essential to polarize macrophages toward the M2 status [21]. A previous study revealed that long-term arsenic exposure could trigger M2 macrophage differentiation via ROS production [63]. Studies have found that the reduction of ROS production specifically blocks the differentiation of M2 macrophages [21]. ROS/PI3K/Akt signaling-dependent CSF-1 production induces M2 polarization of macrophages, thus contributing to tumor cell growth [20]. Of note, the high levels of ROS promote TAMs to generate TNF-α in the primary melanoma microenvironment, thus promoting tumor invasion and metastasis [64].
ROS promote cancer invasion and metastasis via EMT
EMT is a process in which epithelial cells transform into mesenchymal cells. Upon the initiation of EMT, epithelial cells lose cell-cell junctions and cell polarity. Then, the cells experience both cytoskeleton remodeling and extracellular matrix (ECM) protein degradation [65]. These processes of EMT have endowed cancer cells with invasive and metastatic capacities. Finally, the cancer cells undergoing EMT can evade from their original epithelial layer. Recently, an increasing number of studies have described ROS as the main cause of EMT in cancer cells, finally promoting cancer cell invasion and metastasis (Fig. 3) [66].
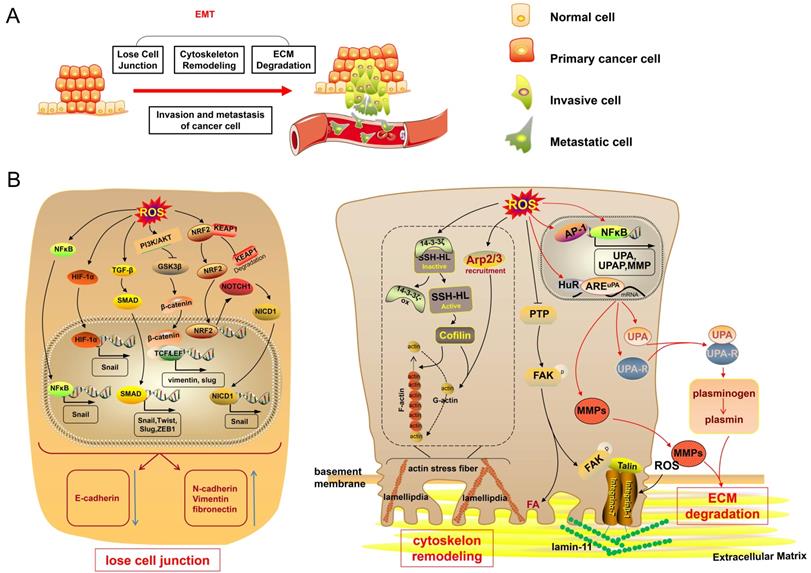
Excess ROS trigger EMT, promoting cancer cell invasion and metastasis. (A) When undergoing EMT, cancer cells experience cell-cell junction dissociation, cytoskeleton remodeling and ECM degradation. These processes of EMT endow cancer cells with invasive and metastatic capacities. (B) High levels of ROS promote the expression of the transcription factors Snail, Slug, Twist and ZEB1 via the NF-κB, HIF-1α, TGF-β/SMAD, PI3K/Akt/GSK-3β and NRF2/NOTHC1/NICD1 signaling pathways, thereby repressing the expression of E-cadherin and promoting N-cadherin, Vimentin, and fibronectin, disrupting cell-cell junctions and initiating the EMT process. Elevated-ROS oxidize 14-3-3ζ and subsequently activate SSH-1L, thus resulting in the activation of cellular cofilin depolymerizing F-actin. Increased-ROS also recruit Arp2/3. Cofilin and Arp2/3, thus resulting in maintenance of actin treadmilling, formation of lamellipodia and cytoskeletal extension. Increased ROS inhibit PTP, thereby sustaining the phosphorylation of FAK, and recruiting talin, which reacts with the extracellular domain of integrin; integrin α7β1 modified by ROS links to laminin-111 in the ECM. ROS promote AP-1 and NF-κB activation of the transcription of MMPs, uPA, and uPAR; ROS also enhance the binding of HuR with AREuPA and consequently promote uPA and uPAR expression. Activated uPA leads to the transformation of plasminogen into plasmin. MMPs and plasmin result in ECM degradation. NICD1, NOTHC1 intracellular domain; N-cadherin, neural cadherin; PTP, protein tyrosine phosphatase.
Oxidative stress is an essential factor causing cell-cell junction dissociation [67]. The elevated levels of ROS can activate nuclear factor-κB (NF-κB) and promote the expression of transcription factor Snail, which downregulates the expression of epithelial cadherin (E-cadherin) and promotes the expression of neural cadherin and vimentin [68, 69], thus resulting in the disruption of cell-cell junctions and triggering the EMT process [23]. Moreover, ROS can promote the expression of transforming growth factor-β (TGF-β), and TGF-β favors the formation of trimeric drosophila mothers against decapentaplegic protein (SMAD) complexes [23, 70]. The SMAD complexes can translocate to the nucleus and activate mesenchymal genes as well as Snail, Twist, Slug, and ZEB1 genes, in turn enhancing the expression of vimentin and fibronectin and repressing E-cadherin expression [70]. Besides, the upregulation of ROS also promotes HIF-1α-induced transcription of Snail, thus leading to the repression of E-cadherin [71]. Furthermore, ROS also stabilize nuclear factor erythroid 2-related factor 2 (NRF2) via the degradation of Kelch-like ECH-associated protein 1 (KEAP1) to enhance the transactivation of Notch homolog 1, translocation-associated (Drosophila) (NOTHC1), promoting the NOTHC1 intracellular domain translocation to the nucleus and leading to Snail transcription, thereby promoting the EMT of cancer cells and favoring cancer cell invasion and metastasis [72]. In addition to the aforementioned signaling, the ROS-activated PI3K/Akt signaling pathway can inhibit glycogen synthase kinase 3β (GSK-3β), the activation of which is involved in the proteasomal degradation of Snail [23, 73, 74]. The inhibition of GSK-3β can promote the nuclear-translocation of β-catenin, following which nuclear β-catenin binds with TCF/LEF and enhances the transcription of vimentin and slug [75], finally inducing EMT. ROS can lead to the loss of cell-cell junctions via NF-κB, TGF-β, HIF-1α, NRF2, and PI3K/Akt signaling, inducing transition to the mesenchymal phenotype.
ROS also cause cytoskeleton remodeling, favoring cancer cell migration, and thus promoting the EMT process. The cytoskeleton consists of actin filaments, intermediate filaments, and microtubules, and it is important to maintain the cell shape [76]. Actin filaments are twisted by two strands of actin polymer fiber actin (F-actin), and F-actin is polymerized by actin monomer spherical actin (G-actin). The actin filament is a dynamic structure responsible for cell migration capacity. ROS can regulate polymerization and depolymerization between G-actin and F-actin to achieve dynamic actin reorganization. ROS can oxidize 14-3-3ζ, which inactivates slingshot-1L (SSH-1L), thus alleviating the inhibition of SSH-1L [77]. SSH-1L ultimately results in the dephosphorylation and activation of cellular cofilin; the activation of which leads to the depolymerization of F-actin, thus providing actin monomers to further polymerize [77, 78]. Besides, ROS induce Arp2/3 recruitment, which can nucleate actin monomers to assemble a new actin filament meshwork [78, 79]. ROS-regulated cofilin and Arp2/3 maintain actin treadmilling and drive cells to form actin-rich membrane projections called lamellipodia, which promote cytoskeletal extension [78, 79]. Subsequently, ROS increase the formation of actin stress fibers [80], which are microfilament bundles formed by actin filaments [81]. Additionally, ROS also inhibit protein tyrosine phosphatase to sustain the phosphorylation of focal adhesion kinase (FAK) [23]. On the one hand, FAK can lead to focal adhesion, and actin stress fibers located on the inside of the cell membrane adhere to the focal adhesion spot [82]. On the other hand, FAK can recruit talin, an actin-binding protein, which reacts with the extracellular domains of integrins [83]. Integrins are cell surface adhesion molecules, and integrin α7β1 modified by ROS is a link to laminin-111 in the ECM [84]. Thus, a connection exists between the ECM and intracellular actin cytoskeleton, which promotes cell migration.
During the EMT process, cancer cells acquire an invasive phenotype by recognizing and attaching to the ECM to degrade the basement membrane and ECM proper. Several studies have indicated that ROS can cause ECM degradation by regulating proteolytic enzymes, such as matrix metalloproteinases (MMPs) and serine proteases, both of which are thought to be involved in tumor malignancy [85, 86]. ROS mainly promote activator protein-1 (AP-1) and NF-κB-induced transcription of MMPs, causing protein matrix degradation [87, 88]. Besides, ROS activate mitogen-activated protein kinase (MAPK), AP-1, and NF-κB signaling pathways to promote the expression of urokinase plasminogen activator (uPA) and uPA receptor (uPAR) [85, 86, 89, 90]. ROS also enhance the binding of mRNA-stabilizing factor Hu antigen R (HuR) with AU-rich element (ARE)uPA to promote the expression of uPA and uPAR [91]. Activated uPA, a serine protease, leads to the transformation of plasminogen into plasmin, resulting in MMP activation, ECM degradation, and eventual cancer cell invasion [23, 86].
Potential mechanisms underlying the anti-cancer effects of ROS
Anti-cancer effects of ROS
In the last few years, several studies have demonstrated a dual role of ROS (“pro-tumor” and “anti-cancer”) [92]. In the early stage, high levels of ROS are crucial in promoting tumor initiation and tumor progression, as discussed earlier. However, the massive accumulation of ROS plays an antioncogenic role in cancer. Extensive accumulation of ROS can inhibit tumor cell proliferation [93] and lead to cancer cell death, which can be explained by the activation of ER stress-, mitochondrial-, P53- apoptotic pathways and the ferroptosis pathway in cancer [94-96] (Fig. 4, Fig. 5). The anti-cancer effects of ROS can provide an additional avenue for cancer treatment. Thus, it is of great interest to discuss the potential mechanisms of the anti-cancer effects of ROS.
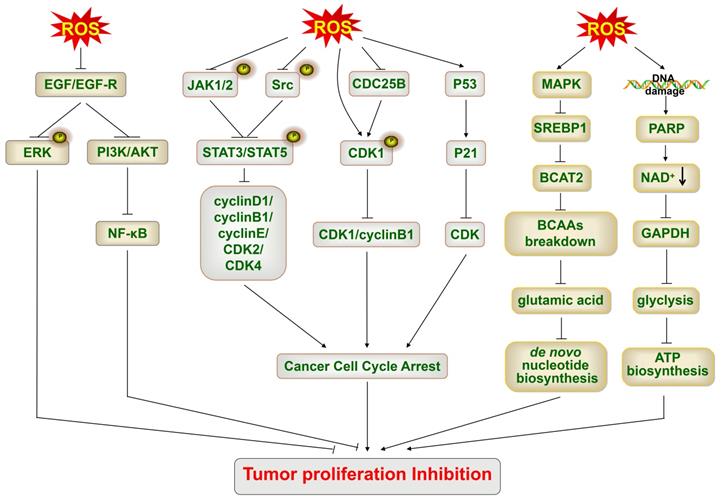
The accumulation of ROS leads to cancer cell growth inhibition. The accumulation of ROS inhibits EGF/EGF-R, then blocks ERK and PI3K/Akt/NF-κB signaling, and inhibits cancer cell proliferation. The accumulation of ROS decreases the phosphorylation of JAK1/2 and Src, thus blocking the phosphorylation of STAT3/STAT5 and downregulating the expression of cyclin D1, cyclin B1, cyclin E, CDK2 and CDK4, and finally leading to cell cycle arrest. ROS phosphorylate CDK1 (inactive) directly or indirectly by inactivating cdc25B, thus suppressing the CDK1/cyclin B complex and resulting in cell cycle arrest. ROS activate p53 and then trigger p21, an inhibitor of most CDKs, thus finally leading to cell cycle arrest. ROS accumulation activates MAPK, suppresses SREBP1 and inhibits BCAT2, thus decreasing the breakdown of BCAAs and the synthesis of glutamic acid, and finally leading to a decrease in de novo nucleotide biosynthesis. ROS accumulation induces DNA damage and activates PARP, thereby leading to a decrease in NAD+, causing the inactivation of GAPDH and inhibition of glycolysis, and ultimately leading to the depletion of ATP and inducing an energetic crisis. EGF-R, EGF receptor; SREBP1, sterol regulatory element-binding protein 1; BCAT2, branched-chain amino acid transaminase 2; BCAAs, branched-chain amino acids; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NAD+, nicotinamide adenine dinucleotide oxidized form.
The accumulation of ROS causes cancer cell apoptosis. Accumulated-ROS induce DNA damage, thus causing the activation of ATM and ATR, and activating P53. Abundant ROS reactivate mutant P53 protein, and P53 subsequently promotes the expression of pro-apoptotic proteins, BAX, MDM2 and Puma, thus subsequently leading to cancer cell apoptosis. ROS oxidize cardiolipin, thereby resulting in Cyt c detachment from the membrane. Oxidized cardiolipin translocates to the outer membrane and recruits the pro-apoptotic protein BAX to the mitochondrial membrane, thus enhancing MOMP and generating Cyt c-traversable pores. ROS also activate ASK/JNK and ASK/P38 axis and promote the release of Cyt c in a manner dependent on BAX. Cyt c interacts with Apaf-1, thus resulting in apoptosome formation, leading to caspase-9 and caspase-3/7 activation, and causing cancer cell apoptosis. The accumulation ROS also causes cancer cell apoptosis via the ER stress pathway through the PERK/eIF2α/ATF-4/CHOP axis and IRE1α/JNK/CHOP axis. MDM 2, murine double minute 2; Puma, p53 upregulated modulator of apoptosis; Apaf1, apoptotic protease activating factor 1; PERK, protein kinase-like ER kinase; IRE1α, inositol-requiring enzyme-1α; ATF4, activating transcription factor-4.
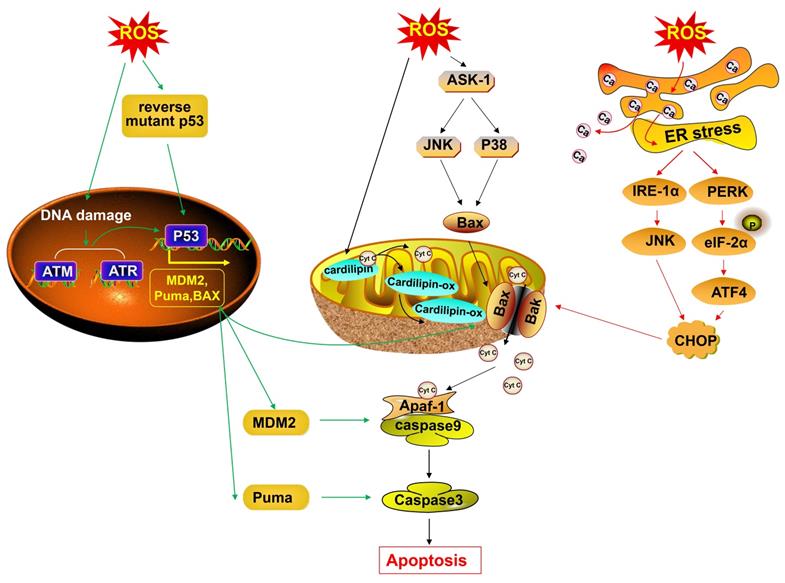
Potential mechanisms of anti-cancer effects of ROS
ROS inhibit cancer cell proliferation
ROS inhibit the proliferation signaling pathway
Cancer cells are characteristic of uncontrolled cell proliferation and cell growth. ROS can inhibit cancer cell growth by inhibiting cell proliferation. Epidermal growth factor (EGF) and EGF receptor (EGFR) signaling play critical roles in tumor growth. When ROS levels exceed the self-regulating ability of cancer cells, ROS directly decrease the expression of EGF and EGFR and markedly inhibit the phosphorylation of EGFR; as a result, downstream cell proliferation signaling molecules are inhibited, such as ERK and PI3K/Akt [97-99]. Vitamin C has been reported to promote the production of ROS and inhibit the phosphorylation of ERK by decreasing EGF release and EGF receptor phosphorylation, thus suppressing the proliferation of thyroid cancer cells [99]. Similarly, Koumine also promotes the production of ROS, which inhibits the phosphorylation of ERK, consequently suppressing the proliferation of hepatocellular carcinoma cells [93]. In line with this, the administration of H2O2 can impair the phosphorylation of ERK1/2 in breast cancer cells in a dose-dependent manner, thus attenuating cancer cell proliferation [100]. Additionally, excessive ROS inhibit EGF-induced EGFR mediated PI3K/Akt signaling and ultimately block androgen-independent prostate cancer cell proliferation [97]. In addition, ROS block the PI3K/Akt/NF-κB signaling pathway, which subsequently leads to the inhibition of the proliferation of human nonsmall cell lung cancer, A549 cells; the same mechanism associated with oenothein B-mediated suppression of cancer cell growth [98].
ROS inhibit the cell cycle
Somatic cell proliferation mainly depends on mitosis. Somatic cell mitosis is periodic and referred to as the cell cycle. The cell cycle is composed of four phases: the gap 1 (G1) phase, synthesis (S) phase, G2 phase, and mitosis (M) phase [101]. A specific cyclin can bind to a specific cyclin-dependent kinase (CDK) to form a complex, regulating different phases of the cell cycle. CDK4 and CDK6 can bind to cyclin D to regulate the early G1 phase [102]. CDK2 binds to cyclin E to cause S-phase initiation, and later CDK2/cyclin A complex formation promotes DNA replication in the S phase [101]. CDK1/cyclin B complex can regulate G2 to M phase transition [101]. The accumulation of ROS in multiple myeloma cells reduces the phosphorylation of Janus kinase (JAK)1, JAK2, and Src, thus blocking the phosphorylation of signal transducers and activators of transcription3 (STAT)/STAT5 and downregulating the expression of cyclin D1, cyclin B1, cyclin E, CDK2, and CDK4, finally leading to cell cycle arrest and attenuating tumor growth [103]. The overexpression of ROS in human nonsmall cell lung cancer cells also promotes CDK1 phosphorylation and suppression of cyclin B1 and CDK1 expression, subsequently blocking CDK1/cyclin B1 complex formation and leading to G2/M phase arrest [104]. The accumulation of ROS also controls cell cycle regulatory molecules, such as cell division cycle (cdc)25 and CDK inhibitors, to suppress tumor growth [101]. LGH00031 accelerates cellular ROS production to inactivate cdc25B, which dephosphorylates tyrosine15 of CDK1 to activate the CDK1/cyclin B complex, resulting in cell cycle arrest and inhibition of cancer cell growth [105]. Physalin A increases the levels of intracellular ROS to activate p53 and then trigger p21, an inhibitor of most CDKs, finally leading to cell cycle arrest of human nonsmall cell lung cancer cells [104].
ROS suppress the biosynthesis of nucleotides and ATP
Sufficient nucleotides and ATP are required to meet the needs of highly mitotic cells. The accumulation of ROS can impair the ability of cancer cells to synthesize nucleotides and ATP, thus failing to meet their demand for cell proliferation [106, 107]. ROS accumulation mediates the activation of the MAPK pathway in pancreatic cancer cells, suppresses sterol regulatory element-binding protein 1, and inhibits its target gene branched-chain amino acid transaminase 2, causing a decrease in the breakdown of branched-chain amino acids and thus reducing the synthesis of glutamic acid, finally leading to a decrease in de novo nucleotide biosynthesis [106]. Moreover, ROS accumulation inhibits glycolysis by the inactivation of glyceraldehyde 3-phosphate dehydrogenase via both direct post-translational modifications and depletion of the nicotinamide adenine dinucleotide oxidized form, which is consumed by PARP activated by ROS-induced DNA damage, ultimately leading to the depletion of ATP, inducing an energetic crisis, and finally killing KRAS and BRAF mutant colorectal cancer cells [107].
ROS induce tumor cell apoptosis
ER stress-mediated apoptosis
The accumulation of ROS in cancer cells can cause apoptosis via the ER stress-mediated pathway. ROS can cause the release of Ca2+ ions from the ER lumen, subsequently resulting in defects in ER chaperones and other proteins, thus leading to ER stress with the accumulation of unfolded/misfolded proteins [108]. ER stress caused by the accumulation of ROS induces the phosphorylation of eIF2α via RNA-dependent protein kinase-like ER kinase, allowing the preferential translation of activating transcription factor-4, which in turn mediates the induction of pro-apoptotic C/EBP homologous protein (CHOP), leading to cancer cell apoptosis [94, 109]. Moreover, ROS-induced ER stress can mediate cancer cell apoptosis via the inositol-requiring enzyme-1α/ c-Jun NH(2)-terminal kinase (JNK)/CHOP pathway [110]. Persistent ER stress initiates apoptotic cascades that play fundamental roles in the mechanism underlying the anticancer properties of ROS [109, 111-113]. The alantolactone-mediated accumulation of ROS breaks the imbalance of intracellular redox status in triple-negative breast cancer cell lines, increasing the levels of unfolded proteins in the ER and resulting in ER stress, subsequently leading to cancer cell apoptosis [113]. Additionally, β-phenethyl isothiocyanate and curcumin also trigger ER stress-mediated apoptosis via ROS accumulation to treat cancers [111, 112].
Mitochondrial apoptotic pathway
In addition to ER stress-mediated apoptosis, the mitochondrial apoptotic pathway regulated by ROS accumulation has anticancer effects. The mechanisms reported so far suggest that the accumulation of ROS can drive the release of pro-apoptotic factor cytochrome c (Cyt c) from the mitochondria to the cytosol, which is a key step in the mitochondrial pathway, finally inducing cancer cell apoptosis [114]. Cyt c is anchored to the outer surface of the mitochondrial inner membrane via combination with cardiolipin, a mitochondria-specific phospholipid [115]. The release of Cyt c relies on disrupting the interaction of Cyt c with cardiolipin and enhancing mitochondrial outer membrane permeabilization (MOMP), which is regulated by pro-apoptotic proteins Bcl-2-associated X protein (BAX) and Bcl-2-antagonist killer (BAK) [116]. ROS can oxidize cardiolipin, which results in Cyt c detachment from the membrane [116]. The oxidized cardiolipin translocates to the outer membrane and helps to recruit pro-apoptotic protein BAX to the mitochondrial membrane, thus enhancing MOMP and generating Cyt c-traversable pores [117]. Besides, ROS can activate apoptosis signal-regulating kinase 1, a member of the mitogen-activated protein kinase kinase kinase superfamily, to activate JNK and P38 MAPK, the activation of which promotes the release of Cyt c on the mitochondrial inner membrane in a manner dependent on BAX [118-120]. Moreover, the accumulation of ROS can mediate the ER stress pathway and then activate the expression of pro-apoptotic protein Noxa [136, 137]. Activated Noxa translocates into the mitochondria and subsequently inhibits anti-apoptotic proteins, thereby reversing the inhibition of BAX and BAK, forming MOMP, and finally promoting the release of Cyt c [121, 122]. Then, Cyt c interacts with apoptotic protease activating factor 1 to form an apoptosome, leading to caspase-9 and caspase-3 activation and subsequent cell apoptosis [123]. The administration of α-hederin can induce the overexpression of ROS and activate the mitochondrial apoptotic pathway, finally triggering the apoptosis of gastric cancer cells [95]. Similarly, in vitro studies have found that metal thiourea complexes can work as a new antitumor metallodrug to kill HeLa cells via a burst of the ROS-regulated mitochondria apoptotic pathway [114].
The P53 apoptotic pathway
Most cancers have abrogation of P53 function with a wide range of mutation frequencies [124]. The hot mutations of P53 are DNA contact mutation (P53R273H), which impairs P53 contact with DNA, and conformational mutation (P53R175H), which affects the stability of the DNA-binding domain, resulting in the loss of function [124]. Notably, ROS can act as an intermediate medium to reverse the abrogated P53 function. Piperlongumine reverses the R273H mutant P53 protein by overproducing ROS and leads to a restoration of the functional status of mutant P53 in human colon cancer cells, inducing the transcription of the P53 target gene, murine double minute 2, and BAX and finally resulting in cancer cell apoptosis [125]. R-goniothalamin-induced abundant ROS reactivate the R175H mutant P53 protein in human breast cancer cells, and then P53 promotes the expression of pro-apoptotic proteins: p21cip1, BAX, and p53 upregulated modulator of apoptosis, causing tumor cell apoptosis [96]. ROS not only enhance the transcription effect of P53 but also translocate P53 to mitochondria, contributing to BAX mitochondrial recruitment and mitochondrial Cyt c release, thus leading to cancer cell apoptosis [126]. In addition, the accumulation of ROS can trigger cancer cell DNA damage, and ROS-induced DNA damage causes the activation of DNA damage sensors and regulators such as ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR), thus activating P53 and subsequently leading to cancer cell apoptosis [127, 128].
ROS-induced ferroptosis
Ferroptosis is a form of iron-dependent cell death characterized by the lethal accumulation of lipid-based ROS and excessive accumulation of lipid peroxidation [129]. Excessive iron from aberrant iron metabolism and the loss of cellular antioxidant capacity can cause the accumulation of ROS, thereby inducing catastrophic alterations in cellular redox metabolism and leading to massive lipid peroxidation in the plasma membrane and loss of cell viability, and eventually ferroptosis [130, 131]. Cancer cells are more vulnerable to ferroptosis than noncancerous cells, owing to the powerful iron dependency feature [131]. ROS-induced ferroptosis is one of the most important means of inhibiting cancer. Several ferroptosis inducing drugs have been used to treat cancer. Erastin, artesunate, and sorafenib have been reported to inactivate glutathione peroxidase 4, an important antioxidant, via inhibiting the synthesis of glutathione (GSH), thereby inducing oxidative stress in cancer cells and ultimately leading to ferroptosis [129, 132, 133]. Ferroptosis inducers have led to high expectations regarding the potential of ferroptosis to be a new promising method of killing cancer cells.
Self-adaption system in cancer
As mentioned earlier, the massive accumulation of ROS is generally detrimental to cancer cells. ROS can inhibit cancer cell growth by suppressing the synthesis of ATP and nucleotides, leading to cell cycle arrest and blocking cancer cell proliferation. ROS also cause cancer cell death through several apoptotic pathways. However, cancer cells use the self-adaption system, including the antioxidant system, DNA damage repair pathway, and metabolism reprogramming, to inhibit the accumulation of ROS and ameliorate ROS-induced damage, so as to maintain their development and survival (Fig. 6).
ROS utilize the antioxidant system, DNA damage repair pathway, and metabolic reprogramming to inhibit the accumulation of ROS and repair ROS-induced damage. ROS activate NRF2, thereby promoting the expression of NQO1, CAT, SOD1/2 and PRDX1/3, and decreasing ROS levels. ROS activate the expression of KLF14 via the PI3K/Akt, p42/p44 MAPK, AMPK, and PKC pathways; KLF14 then couples with p300 and CBP, and promotes the expression of HO-1, which restores cellular redox balance. ROS accumulation inhibits PKM2, thus leading to G-6-P flux into the PPP and generating sufficient NADPH to produce GSH, thereby maintaining ROS homeostasis. ROS trigger LIPG upregulation and induce the lipolysis of extracellular lipoproteins to compensate for the ROS-impeded fatty acid synthesis pathway. The upregulated LIPG also protects against oxidative damage via the accumulation of intracellular lipid droplets. ROS activate KRAS/AMPK signaling, thus inducing metabolic reprogramming toward aerobic glycolysis. ROS activate ATM, which in turn phosphorylates BRCA1 and triggers HR for DNA repair and activation of P53, which in turn triggers the NER pathway. ROS activate the ATM-chk2 and ATR-chk1 axis, thereby causing cell cycle arrest of cancer cells, preventing the cells from undergoing mitosis and favoring DNA repair. NQO1, NADH dehydrogenase, quinone 1; CAT, catalase; PKC, protein kinase C; G-6-P, glucose-6-phosphate; PPP, pentose phosphate pathway.
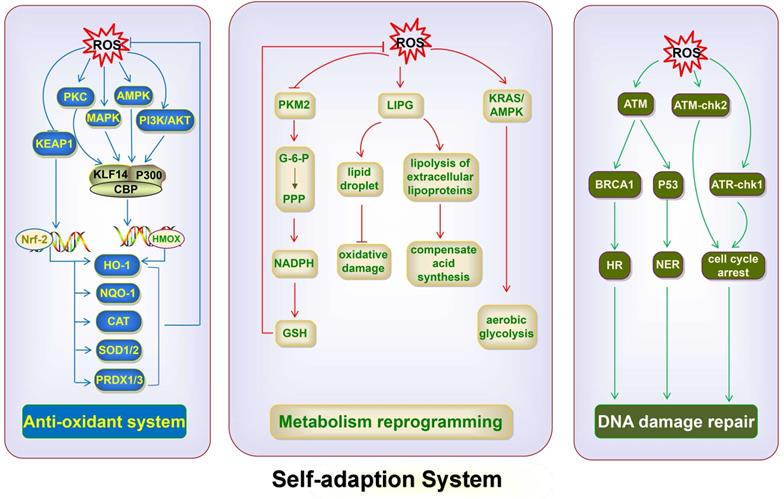
Antioxidant system
The antioxidant system is essential to maintain cellular redox balance. Accumulating evidence shows that NRF2 is pivotal in antioxidant responses. Importantly, it is a key gene for cancer cells to maintain oxidative balance. The target antioxidant genes of NRF2 include reduced nicotinamide adenine dinucleotide dehydrogenase, quinone 1, SOD1/2, catalase, heme oxygenase-1 (HO-1), thioredoxin reductase 1, peroxiredoxin (PRDX)1, and PRDX3, all of which are antioxidant enzymes that reduce ROS levels [134]. Under oxidative stress, ROS can cause the binding of 5′-adenosine monophosphate-activated protein kinase (AMPK) to HCC-specific fructokinase A (KHK-A), induce KHK-A phosphorylation of serine at position 80, and subsequently phosphorylate p62 at S28 [135]. Consequently, p62 oligomerization occurs and the aggregation of p62 with KEAP1 that inhibits NRF2 is enhanced, thereby increasing nuclear translocation and activation of NRF2 to alleviate ROS, and finally promoting the development of HCC [135]. Besides, accumulated ROS lead to lactate dehydrogenase A nuclear translocation to promote the production of α-hydroxybutyrate (α-HB) in HPV-induced cervical tumors [9]. Then, α-HB triggers the disruptor of telomeric silencing 1-like-mediated histone H3K79 hypermethylation, inducing the activation of NRF2 to control cellular redox balance [9]. Cancer cells also use other factors, besides NRF2, to control antioxidant activity. Oxidative stress activates the expression of Krüppel-like factor 14 (KLF14) via multiple pathways, including the PI3K/Akt, p42/p44 MAPK, AMPK, and protein kinase C pathways, in castration-resistant prostate cancer. Then, KLF14 couples with p300 and CREB-binding protein (CBP) to enhance the transcriptional activation of heme oxygenase 1, which encodes the antioxidant enzyme HO-1 that helps cancer cells restore cellular redox balance under androgen-depleted conditions, resulting in cancer cell survival [136].
DNA damage repair pathway
Cancer cells use the DNA damage repair pathway to adapt to DNA damage caused by chemotherapy and radiotherapy (RT), finally leading to therapeutic resistance. The key regulators of the DNA damage repair process are ATM and ATR [137]. ATM and ATR can recognize DNA lesions and phosphorylate downstream molecules to transmit DNA damage signals, leading to cell cycle arrest and finally completing DNA repair [138]. Under oxidative stress, ROS regulate the activation of ATM, which recognizes DNA damage [138]. ATM triggers signal transduction of checkpoint kinase 2 (Chk2), and the ATM-Chk2 axis causes cell cycle arrest of cancer cells, preventing the cells from undergoing mitosis and favoring DNA repair [139]. Besides, ATM can both phosphorylate BRCA1, which is required for HR [140], and activate P53, which can trigger the NER pathway [141, 142], finally leading to DNA repair. Additionally, ROS can induce the phosphorylation of ATR, which in turns phosphorylate Chk1, resulting in the cell cycle arrest of cancer cells and better repair of DNA damage [143, 144]. The overproduction of ROS activates the ATR-Chk1 axis, resulting in resistance to cisplatin in ovarian cancer cells, and the inhibition of ATR and Chk1 reverses chemotherapeutic resistance [145]. The findings provided strong evidence that accumulated ROS triggered the DNA damage repair pathway to resist the ROS-caused damage in cancer cells.
Metabolism reprogramming
Metabolism reprogramming is also a critical way to resist ROS-induced inhibition of cancer cell development, besides antioxidant system and DNA damage repair pathway. Cancer cells can change their metabolic pathway to maintain the oxidative balance and their nutrient needs and cause therapeutic resistance. Accumulated ROS can trigger metabolism reprogramming to reduce the levels of ROS in cancer cells. The accumulation of ROS inhibits glycolytic enzyme pyruvate kinase M2 (PKM2) via the oxidation of Cys358 in human lung cancer cells [146]. The inhibition of PKM2 leads to glucose-6-phosphate flux into the pentose phosphate pathway, which generates sufficient nicotinamide adenine dinucleotide phosphate oxidase to produce GSH, thus maintaining ROS homeostasis and favoring cancer cell survival [146]. The change in the abundance of selected lipids can promote tumor growth and progression by eliminating ROS in colorectal cancer development [147]. The accumulation of ROS can also trigger metabolism reprogramming to maintain the nutritional needs of cancer cells. A study has shown that severe oxidative stress triggers endothelial lipase (LIPG) upregulation, which induces the lipolysis of extracellular lipoproteins, to compensate the ROS-impeded fatty acid synthesis pathway in breast cancer cells, thus supporting tumor progression [148]. The upregulated LIPG also results in the accumulation of intracellular lipid droplets in breast cancer cells, which protects against oxidative damage, finally supporting survival [148]. Additionally, metabolic reprogramming leads to tumor treatment tolerance. Gemcitabine-induced ROS activate KRAS/AMPK signaling, which induces metabolic reprogramming toward aerobic glycolysis, enhancing stem-like cell properties in pancreatic cancer [149]. Oxidative stress-induced metabolic reprogramming is one of the most important reasons for the therapeutic resistance of pancreatic cancer because cancer stem cells are insensitive to external physical and chemical factors that kill tumor cells [149].
Scientific significance of the dual role of ROS in cancer
The incidence of cancer has increased gradually, and cancer acts as the leading cause of death among humans. Fortunately, a large number of therapeutics have been developed. Increased levels of ROS can suppress tumor cell growth and lead to apoptosis (Table 1). Recent studies show that a large number of therapeutic regimens to treat cancer depend on the elevation of ROS levels. However, ROS have a dual role in cancer, and cancer cells own a self-adaption system to resist the harmful effects of ROS. In normal cells or during early stages of cancer, the upregulation of ROS can cause gene mutations and tumor initiation that favor tumor invasion and metastasis (Table 2). If the ROS levels increase significantly, the therapeutic effect cannot be achieved; instead, ROS promote the development of cancer. Thus, different methods should be used to treat cancers in different stages. In the early carcinogenesis stage, attention should be paid to the therapeutic regimens depending on the downregulation of ROS. Nanoparticles (NPs) containing ROS-scavenging nitroxide radicals are synthesized to abrogate ROS-mediated cell proliferation and metastasis to attenuate cancers [150]. However, in advanced cancer stages, enhancing the production of ROS to kill tumor cells and inhibiting the antioxidant system, DNA damage repair pathway, and metabolism reprogramming should be considered, as inhibition can effectively resolve therapeutic resistance. During the course of cancer treatment, considering the different roles of ROS in different stages of cancer can effectively increase the preventive and anticancer effects.
The anti-cancer effects of ROS
| Function | Result | Effect |
|---|---|---|
| Decrease EGF and EGFR expression | Inhibit the proliferation signaling pathway | Inhibit cancer cell proliferation |
| Inhibit EGFR phosphorylation | ||
| Reduce JAK1, JAK2, and Src phosphorylation | Cell cycle arrest | |
| Promote CDK1 phosphorylation | ||
| Suppress cyclin B1 and CDK1 expression | ||
| Inactivate cdc25B, | ||
| Activate p53 | ||
| Activate MAPK | Decrease de novo nucleotide biosynthesis | |
| Activate PARP | Cause ATP depletion | |
| Cause Ca2+ ions release from the ER lumen | Induce ER stress-mediated apoptosis | Lead to cancer cell death |
| Oxidize cardiolipin | Trigger mitochondria-mediated apoptosis | |
| Activate ASK-1 | ||
| Trigger cancer cell DNA damage | Activate P53-medicated apoptosis | |
| Reverses the R273H mutant P53 protein | ||
| Reactivate the R175H mutant P53 protein |
The promoting effects of ROS in cancer
| Function | Result | Effect |
|---|---|---|
| Oxidize nucleobases | Form oxidative damage products | DNA mutation, subsequently promoting cancer formation |
| BRCA2 degradation | Inhibit BRCA2 | |
| Oxidize hOGG1 | Inhibit hOGG1 activity | |
| Stabilize HIF-1α | Interfere with T-cell development | Inhibit T cell function, finally promoting cancer development and metastasis. |
| Reduce CD3ζ chain expression | Block T cells differentiation and activation | |
| Phosphorylate ERK | T cells apoptosis | |
| Downregulate CD16ζ expression | Inhibit NK cells cytotoxicity | Inhibit NK cell function, subsequently promoting tumor growth and metastasis |
| Attenuate eIF-2B phosphorylation | Suppress ADCC of NK cell | |
| Inactivate PTEN | Macrophage recruitment, M2 polarization of macrophage | Promote tumor growth, invasion and metastasis |
| Activate TNF-α/TNFR1 | ||
| Activate NF-κB | Lose cell-cell junction | Trigger EMT process in cancer cells, finally promoting cancer cell invasion and metastasis |
| Promote TGF-β expression | ||
| Activate HIF-1α | ||
| Cause KEAP1 degradation | ||
| Activate PI3K/Akt | ||
| Oxidize 14-3-3ζ | Cytoskeleton remodeling | |
| Induce Arp2/3 recruitment | ||
| Inhibit protein tyrosine phosphatase | ||
| Activate MAPK, AP-1 and NF-κB | ECM degradation |
Clinical application of the dual role of ROS in cancer
Many traditional chemotherapeutic agents used in clinical settings, such as doxorubicin (DOX) and pirarubicin, can induce intracellular ROS accumulation and trigger the mitochondrial apoptotic pathway, finally leading to cancer cell death [151]. However, some chemotherapeutic agents, such as anthracyclines, have cytotoxic effects on normal cells [152]. Many laboratories are committed to finding natural drug ingredients with lower cytotoxic effects on normal cells to treat malignant tumors [153]. Galangin (GG), a naturally active flavonoid extracted primarily from the root of Alpinia officinarum Hance, can kill cancer cells without causing cytotoxicity in normal cells [154]. Experiments have demonstrated that the natural drug GG, combined with tumor necrosis factor-related apoptosis-inducing ligand, enhances ROS production, leading to the apoptosis of human breast cancer cells [155]. Other natural drugs, such as curcumin and parthenolide, also increase the levels of ROS to cause cancer cell apoptosis [112, 156]. Besides chemotherapy, RT also becomes an important means to treat malignant tumors in clinic. RT uses ionizing radiation to promote the production of ROS to treat cancer [157]. Ionizing radiation not only leads to the radiolysis of water to produce large amounts of ROS [158] but also promotes the expression of NOX1 to overexpress ROS [159]. The ROS produced react with DNA, finally leading to DNA strand breaks [160]. Failure to perform normal functions after DNA strand breaks causes cancer cell death [161].
However, the emergence of treatment resistance hinders the efficacy of cancer treatment. The antioxidant system of cancer cells is important in tumor treatment resistance. High mucin 1 C-terminal subunit expression in urothelial carcinoma cells stabilizes the expression of x-cystine/glutamate transporter and increases intracellular GSH levels, thus causing cisplatin resistance by urothelial carcinoma [162]. Also, after radiation treatment, free radical species can stabilize HIF1, the master regulator of oxygen homeostasis, finally buffering ROS generated by radiation, and hence mitigating the effect of RT [163]. It is important to find drugs that inhibit the function of the antioxidant system to solve the problem. Fortunately, a study has shown that piperlongumine can inhibit glutathione and thioredoxin systems, thus reducing ROS clearance and enhancing the radioresponse of colorectal cancer cells [164]. Besides, the activation of the DNA damage repair pathway can counteract DNA damage caused by chemotherapy and RT, which is one of the main causes of treatment resistance. Radiation can upregulate the expression of the ATM gene, which triggers DNA damage repair, finally mitigating the ROS-induced DNA lesions [165, 166]. Obviously, inhibiting the RT-induced DNA damage repair pathway is a useful way to solve the problems. Another study has reported that the combination treatment of resveratrol and capsaicin, besides RT, can inhibit ATM activation and thus radiosensitize pancreatic tumor cells toward cell death [167].
New therapies have partly resolved the treatment resistance to RT and chemotherapy. However, the biggest problem at present is the inability to deliver targeted therapies. Radiotherapy and chemotherapy can induce ROS production in nontargeted tissues, and the high concentrations of ROS can damage normal cells and even cause carcinogenesis. Therefore, targeted treatments have become a hot research spot, and new therapies have emerged based on the requirement. The advent of photodynamic therapy (PDT) has enabled the precise and effective treatment of tumors. PDT is an outstandingly selective, effective, and noninvasive cancer therapeutic methodology comprising a light source, photosensitizer, and oxygen [168]. A specific wavelength of laser light excites the photosensitizer absorbed by the tissue, and the excited-state photosensitizer transfers energy to the surrounding oxygen molecules, generating a highly active singlet oxygen (an ROS), which can kill cancer cells precisely. However, hypoxia often occurs in solid tumors with very low oxygen concentrations [169]. Under hypoxic conditions, the generation of singlet oxygen is reduced because of the low oxygen level, and hence the therapeutic efficacy of PDT in clinical treatment is greatly suppressed [170]. Initiator-loaded gold nanocages, as the free radical generator, can generate ROS under different oxygen tensions on exposure to near-infrared light irradiation [171], effectively solving the problem of PDT resistance caused by hypoxia.
The application of nanoparticles has opened new avenues to develop cancer therapies, with the advancement in nanotechnological techniques and further investigations on nanoparticles in biomedical sciences. NPs can be divided into inorganic nanomaterials such as iron oxide NPs (IO NPs) and cerium oxide NPs (CONPs), and organic nanomaterials such as polymeric micelles [172-174]. It is possible to further reduce the side effects and improve the efficacy of tumor treatment due to the selective targeting capabilities and superior efficacy of NPs [175]. IO NPs, serving as a drug vehicle, conjugate with anticancer drug DOX, which can promote the production of ROS, forming magneto-sensitive NPs [173]. These NPs can selectively target using magnetic fields in cancer treatment [173]. IO NPs can also link with linoleic acid hydroperoxide (LAHP). Under the acidic-pH condition, Fe2+ ions are released from IO NPs and react with LAHP, triggering the generation of singlet oxygen (1O2), which can kill cancer cells [176]. IO-LAHP NPs can produce tumor-specific 1O2 to treat cancer due to the overall acidic environment of solid tumors [177], which contribute to targeted treatment [176]. Besides selectively killing cancer cells, NPs can combine with other therapies, such as RT and PDT, to enhance the efficacy of cancer treatment. Experiments have demonstrated that CONP pretreatment facilitates the production of RT-induced ROS in human pancreatic cancer cells [174], indicating that CONPs can serve as an RT sensitizer to improve pancreatic cancer treatment. Likewise, polymeric micelles are used in PDT for cancer therapy. Polymeric micelles, hemoglobin (Hb), and zinc phthalocyanine form an Hb-conjugated photosensitizer carrier, which has an oxygen self-compensating ability in PDT [172]. Under light irradiation, the Hb-conjugated photosensitizer carrier can induce the production of more ROS, thus causing cancer cell death [172]. NPs can target and control the release of ROS to reduce side effects. They can also combine with other therapies to improve the efficacy of cancer treatment. Hence, NPs have broad application prospects and development space.
Summary and perspectives
Anticancer treatment and early cancer prevention have become hot topics in medical research due to the increased morbidity and mortality of cancer. Targeting ROS has become an efficient way to treat cancer. Gaining a deeper understanding of the various mechanisms of ROS can provide better insights into cancer treatment. This review illustrated the dual role of ROS in promoting cancer occurrence and development and inhibiting cancer growth. In the early stages, elevated levels of ROS act as pro-tumorigenesis factors, which can induce DNA mutation and EMT, thus favoring cancer initiation, development, invasion, and metastasis. Besides, a high level of ROS inhibits T cells and NK cells and promotes the recruitment and M2 polarization of macrophages; consequently, cancer cells escape immune surveillance and immune defense. Interestingly, excessively accumulated-ROS repress cancer cell growth by inhibiting the production of essential substances needed for cell life activities, such as nucleotides and ATP, causing cell cycle arrest and blocking cancer cell proliferation. ROS also induce cancer cell death by activating ER stress-, mitochondrial-, and P53-apoptotic pathways and the ferroptosis pathway. Of note, tumor cells can establish a self-adaption system (antioxidant response, DNA damage repair pathway, and metabolism reprogramming) to deal with massively accumulated ROS and the damage they cause. Furthermore, the review discussed the methods and development of cancer treatment in recent years and also focused on therapeutics using ROS to treat cancer. The characterization of the dual role of ROS in cancer and the self-adaptive ability of cancer cells may provide a new idea for the prevention of carcinogenesis and the development of new therapeutic regimens for cancers.
An increasing number of studies have shown that various cancer cells always have high levels of ROS [4]. ROS can promote the development of cancer by inducing gene mutation, mitochondrial dysfunction, and EMT, and activating anti-apoptotic pathways. In particular, ROS can inhibit the function of immune cells. Eventually, cancer cells escape from immune surveillance and immune defense. Interestingly, age-related thymic involution is closely connected with elevated levels of ROS [178]. A previous study showed that a murine oxidative stress model via ozone inhalation exhibited accelerated thymic involution [179]. Consistent with this finding, the administration of antioxidant compounds can restore the size of the thymus [180]. The thymus is a site for T-cell differentiation and development. The thymic involution leads to the decline of T-cell output with decreased immune function [181]. A mathematical model of cancer incidence has shown that the aging immune system leads to an increased risk of cancer [182]. Therefore, it is speculated that ROS-mediated thymic involution is closely associated with carcinogenesis, thus providing a new basis for immune remodeling to treat cancer. However, the specific mechanism of ROS causing thymus involution to regulate cancer development is not clear.
Acknowledgements
This study was partly supported by grants from the National Natural Science Foundation of China (81800670), Luzhou City Bureau of Science and Technology (2020LZXNYDZ05), National Undergraduate Innovation and Entrepreneurship Training Program (S201910703072, S202010632005, 2020360), and Joint Project of the People's Government of Luxian County and Southwest Medical University (2019LXXNYKD06).
Abbreviations
ROS: reactive oxygen species; H2O2: hydrogen peroxide; ER: endoplasmic reticulum; NOX: nicotinamide adenine dinucleotide phosphate oxidase; DNA: deoxyribonucleic acid; KRAS: Kirsten rat sarcoma viral oncogene; NK: natural killer; EMT: epithelial-mesenchymal transition; Ras: rat sarcoma; G: guanine; 8-oxo-dG: 7,8-dihydro-8-oxo-2´-deoxyguanosine; A: adenine; T: thymine; C: cytosine; 2-OH-dA: 2-hydroxy-2'-deoxyadenosine; 8-oxo-dA: 7,8-dihydro-8-oxo-2'-deoxyadenosine; BER: base excision repair; NER: nucleotide excision repair; HR: homologous recombination; hOGG 1: human 8-oxoguanine DNA N-glycosylase 1; VEGF: vascular endothelial growth factor; HIF-1 α: hypoxia-inducible factor 1 α; ERK: extracellular signal-regulated kinase; PARP: poly(ADP-ribose) polymerase 1; HCC: hepatocellular carcinoma; NKG2D: natural killer group 2 member D; IFN-γ: interferon-γ; CMML: chronic myelomonocytic leukemia; TAMs: tumor-associated macrophages; PI3K: phosphatidylinositide 3-kinase; Akt: protein kinase B; CSF-1: colony-stimulating factor-1; TNF-α: tumor necrosis factor; IEC: intestinal epithelial cells; E-cadherin: epithelial cadherin; ECM: extracellular matrix; NF-κB: nuclear factor-κB; TGF-β: transforming growth factor-β; SMAD: trimeric drosophila mothers against decapentaplegic protein; NRF2: nuclear factor erythroid 2-related factor 2; KEAP1: Kelch-like ECH-associated protein 1; NOTHC1: translocation-associated (Drosophila); GSK-3β: glycogen synthase kinase 3β; F-actin: polymer fiber actin; G-actin: monomer spherical actin; SSH-1L: slingshot-1L; FAK: focal adhesion kinase; MMPs: matrix metalloproteinases; AP-1: activator protein-1; MAPK: mitogen-activated protein kinase; uPA: urokinase plasminogen activator; uPAR: uPA receptor; HuR: Hu antigen R; ARE: AU-rich element; EGF: epidermal growth factor; EGFR: EGF receptor; G1: gap 1; S: DNA synthesis; M: mitosis; CDK: cyclin-dependent kinase; JAK: Janus kinase; STAT: signal transducers and activators of transcription; cdc25: cell division cycle 25; CHOP: C/EBP homologous protein; JNK: c-Jun NH(2)-terminal kinase; Cyt c: cytochrome c; MOMP: mitochondrial outer membrane permeabilization; BAX: Bcl-2-associated X protein; BAK: Bcl-2-antagonist killer; ATM: ataxia telangiectasia mutated; ATR: ataxia telangiectasia and Rad3-related; HO-1: heme oxygenase-1; PRDX1: peroxiredoxin 1; AMPK: 5′-adenosine monophosphate-activated protein kinase; KHK-A: fructokinase A; α-HB: α-hydroxybutyrate; KLF14: Krüppel-like factor 14; CBP: CREB-binding protein; Chk2: checkpoint kinase 2; PKM2: pyruvate kinase M2; GSH: glutathione; LIPG: endothelial lipase; RT: radiotherapy; PDT: photodynamic therapy; NPs: nanoparticles; DOX: doxorubicin; GG: galangin; IO NPs: iron oxide NPs; CONPs: cerium oxide NPs; LAHP: linoleic acid hydroperoxide; 1O2: singlet oxygen; Hb: hemoglobin.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Global regional, national age-sex-specific mortality for 282 causes of death in 195 countries territories, 1980-2017. a systematic analysis for the Global Burden of Disease Study 2017. Lancet (London, England). 2018;392:1736-88
2. Global Burden of Disease Cancer C, Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F. et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019;5:1749-68
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
4. Kruk J, Aboul-Enein HY. Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types. Mini reviews in medicinal chemistry. 2017;17:904-19
5. Zhu M, Jiang Y, Wu H, Shi W, Lu G, Cong D. et al. Gambogic Acid Shows Anti-Proliferative Effects on Non-Small Cell Lung Cancer (NSCLC) Cells by Activating Reactive Oxygen Species (ROS)-Induced Endoplasmic Reticulum (ER) Stress-Mediated Apoptosis. Med Sci Monit. 2019;25:3983-8
6. Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB. et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761-73
7. Jin F, Wu Z, Hu X, Zhang J, Gao Z, Han X. et al. The PI3K/Akt/GSK-3β/ROS/eIF2B pathway promotes breast cancer growth and metastasis via suppression of NK cell cytotoxicity and tumor cell susceptibility. Cancer biology & medicine. 2019;16:38-54
8. Shao S, Duan W, Xu Q, Li X, Han L, Li W. et al. Curcumin Suppresses Hepatic Stellate Cell-Induced Hepatocarcinoma Angiogenesis and Invasion through Downregulating CTGF. Oxid Med Cell Longev. 2019;2019:8148510
9. Liu Y, Guo JZ, Liu Y, Wang K, Ding W, Wang H. et al. Nuclear lactate dehydrogenase A senses ROS to produce alpha-hydroxybutyrate for HPV-induced cervical tumor growth. Nat Commun. 2018;9:4429
10. Sun Y, Lu Y, Saredy J, Wang X, Drummer Iv C, Shao Y. et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020;37:101696
11. Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer. 2014;14:709-21
12. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411-21
13. Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. 2010;38:96-109
14. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65-87
15. Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981-90
16. Canli O, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M. et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell. 2017;32:869-83 e5
17. Jackson JH. Potential molecular mechanisms of oxidant-induced carcinogenesis. Environmental health perspectives. 1994;102(Suppl 10):155-7
18. Zhong LM, Liu ZG, Zhou X, Song SH, Weng GY, Wen Y. et al. Expansion of PMN-myeloid derived suppressor cells and their clinical relevance in patients with oral squamous cell carcinoma. Oral Oncol. 2019;95:157-63
19. Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD. et al. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol Res. 2019;7:335-46
20. Zhang J, Li H, Wu Q, Chen Y, Deng Y, Yang Z. et al. Tumoral NOX4 recruits M2 tumor-associated macrophages via ROS/PI3K signaling-dependent various cytokine production to promote NSCLC growth. Redox Biol. 2019;22:101116
21. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013;23:898-914
22. Tower H, Ruppert M, Britt K. The Immune Microenvironment of Breast Cancer Progression. Cancers (Basel). 2019;11:1375
23. Jiang J, Wang K, Chen Y, Chen H, Nice EC, Huang C. Redox regulation in tumor cell epithelial-mesenchymal transition: molecular basis and therapeutic strategy. Signal Transduct Target Ther. 2017;2:17036
24. Reis AH, Vargas FR, Lemos B. Biomarkers of genome instability and cancer epigenetics. Tumour Biol. 2016;37:13029-38
25. Jiang D, Rusling JF. Oxidation Chemistry of DNA and p53 Tumor Suppressor Gene. ChemistryOpen. 2019;8:252-65
26. Kontomanolis EN, Koutras A, Syllaios A, Schizas D, Mastoraki A, Garmpis N. et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020;40:6009-15
27. Brancato B, Munnia A, Cellai F, Ceni E, Mello T, Bianchi S. et al. 8-Oxo-7,8-dihydro-2'-deoxyguanosine and other lesions along the coding strand of the exon 5 of the tumour suppressor gene P53 in a breast cancer case-control study. DNA Res. 2016;23:395-402
28. Assi M. The differential role of reactive oxygen species in early and late stages of cancer. Am J Physiol Regul Integr Comp Physiol. 2017;313:R646-R53
29. Zhao Y, Chaiswing L, Bakthavatchalu V, Oberley TD, St Clair DK. Ras mutation promotes p53 activation and apoptosis of skin keratinocytes. Carcinogenesis. 2006;27:1692-8
30. Margetis N, Kouloukoussa M, Pavlou K, Vrakas S, Mariolis-Sapsakos T. K-ras Mutations as the Earliest Driving Force in a Subset of Colorectal Carcinomas. In Vivo. 2017;31:527-42
31. Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutation research. 1998;400:99-115
32. Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013;5:a012559
33. Qing X, Shi D, Lv X, Wang B, Chen S, Shao Z. Prognostic significance of 8-hydroxy-2'-deoxyguanosine in solid tumors: a meta-analysis. BMC Cancer. 2019;19:997
34. Cilli P, Ventura I, Minoprio A, Meccia E, Martire A, Wilson SH. et al. Oxidized dNTPs and the OGG1 and MUTYH DNA glycosylases combine to induce CAG/CTG repeat instability. Nucleic Acids Res. 2016;44:5190-203
35. Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G--T and A--C substitutions. The Journal of biological chemistry. 1992;267:166-72
36. Fotouhi A, Hagos WW, Ilic M, Wojcik A, Harms-Ringdahl M, de Gruijl F. et al. Analysis of mutant frequencies and mutation spectra in hMTH1 knockdown TK6 cells exposed to UV radiation. Mutation research. 2013;751-752:8-14
37. Satou K, Harashima H, Kamiya H. Mutagenic effects of 2-hydroxy-dATP on replication in a HeLa extract: induction of substitution and deletion mutations. Nucleic Acids Res. 2003;31:2570-5
38. Kamiya H, Miura H, Murata-Kamiya N, Ishikawa H, Sakaguchi T, Inoue H. et al. 8-Hydroxyadenine (7,8-dihydro-8-oxoadenine) induces misincorporation in in vitro DNA synthesis and mutations in NIH 3T3 cells. Nucleic Acids Res. 1995;23:2893-9
39. Oliveira SC, Oliveira-Brett AM. In situ DNA oxidative damage by electrochemically generated hydroxyl free radicals on a boron-doped diamond electrode. Langmuir. 2012;28:4896-901
40. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129-43
41. Bravard A, Vacher M, Gouget B, Coutant A, de Boisferon FH, Marsin Sp. et al. Redox Regulation of Human OGG1 Activity in Response to Cellular Oxidative Stress. Molecular and Cellular Biology. 2006;26:7430-6
42. van den Tempel N, Zelensky AN, Odijk H, Laffeber C, Schmidt CK, Brandsma I. et al. On the Mechanism of Hyperthermia-Induced BRCA2 Protein Degradation. Cancers (Basel). 2019;11:97
43. Song KH, Kim JH, Lee YH, Bae HC, Lee HJ, Woo SR. et al. Mitochondrial reprogramming via ATP5H loss promotes multimodal cancer therapy resistance. J Clin Invest. 2018;128:4098-114
44. Lee YH, Bae HC, Noh KH, Song KH, Ye SK, Mao CP. et al. Gain of HIF-1alpha under normoxia in cancer mediates immune adaptation through the AKT/ERK and VEGFA axes. Clin Cancer Res. 2015;21:1438-46
45. Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S. et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878-86
46. Kono K, Salazar-Onfray F, Petersson M, Hansson J, Masucci G, Wasserman K. et al. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. European journal of immunology. 1996;26:1308-13
47. Ohno H, Aoe T, Taki S, Kitamura D, Ishida Y, Rajewsky K. et al. Developmental and functional impairment of T cells in mice lacking CD3 zeta chains. The EMBO journal. 1993;12:4357-66
48. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer research. 2001;61:4756-60
49. Lopez-Santalla M, Krishnan S, Valeri AP, Aguilera-Montilla N, Fisher CU, Perez-Blas M. et al. Defective CD3zeta chain expression in Herpesvirus saimiri (HVS)-derived T-cell lines in gastric adenocarcinoma. Cell Immunol. 2005;238:113-22
50. Yu B, Zhang W. Down-regulation of CD3zeta is a breast cancer biomarker associated with immune suppression. Cell Biol Int. 2011;35:165-9
51. Upreti D, Zhang ML, Bykova E, Kung SK, Pathak KA. Change in CD3zeta-chain expression is an independent predictor of disease status in head and neck cancer patients. Int J Cancer. 2016;139:122-9
52. Yin T, Zhao ZB, Guo J, Wang T, Yang JB, Wang C. et al. Aurora A Inhibition Eliminates Myeloid Cell-Mediated Immunosuppression and Enhances the Efficacy of Anti-PD-L1 Therapy in Breast Cancer. Cancer research. 2019;79:3431-44
53. Akhiani AA, Werlenius O, Aurelius J, Movitz C, Martner A, Hellstrand K. et al. Role of the ERK pathway for oxidant-induced parthanatos in human lymphocytes. PLoS One. 2014;9:e89646
54. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P. et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253-7
55. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer research. 2013;73:1777-86
56. Aydin E, Johansson J, Nazir FH, Hellstrand K, Martner A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol Res. 2017;5:804-11
57. Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH. et al. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol. 2011;11:932-8
58. Aurelius J, Hallner A, Werlenius O, Riise R, Möllgård L, Brune M. et al. NOX2-dependent immunosuppression in chronic myelomonocytic leukemia. Journal of leukocyte biology. 2017;102:459-66
59. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P. et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev. 2017;36:105-16
60. Hensler M, Kasikova L, Fiser K, Rakova J, Skapa P, Laco J. et al. M2-like macrophages dictate clinically relevant immunosuppression in metastatic ovarian cancer. J Immunother Cancer. 2020;8:e000979
61. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. The Journal of biological chemistry. 2002;277:20336-42
62. Zhang C, Lan T, Hou J, Li J, Fang R, Yang Z. et al. NOX4 promotes non-small cell lung cancer cell proliferation and metastasis through positive feedback regulation of PI3K/Akt signaling. Oncotarget. 2014;5:4392-405
63. Cui J, Xu W, Chen J, Li H, Dai L, Frank JA. et al. M2 polarization of macrophages facilitates arsenic-induced cell transformation of lung epithelial cells. Oncotarget. 2017;8:21398-409
64. Lin X, Zheng W, Liu J, Zhang Y, Qin H, Wu H. et al. Oxidative stress in malignant melanoma enhances tumor necrosis factor-alpha secretion of tumor-associated macrophages that promote cancer cell invasion. Antioxid Redox Signal. 2013;19:1337-55
65. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
66. Kim YM, Muthuramalingam K, Cho M. Redox Regulation of NOX Isoforms on FAK((Y397))/SRC((Y416)) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells. Cells. 2020;9:1555
67. Inumaru J, Nagano O, Takahashi E, Ishimoto T, Nakamura S, Suzuki Y. et al. Molecular mechanisms regulating dissociation of cell-cell junction of epithelial cells by oxidative stress. Genes Cells. 2009;14:703-16
68. Li W, Cao L, Han L, Xu Q, Ma Q. Superoxide dismutase promotes the epithelial-mesenchymal transition of pancreatic cancer cells via activation of the H2O2/ERK/NF-kappaB axis. Int J Oncol. 2015;46:2613-20
69. Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE. et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123-7
70. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69-84
71. Yun Y, Gao R, Yue H, Guo L, Li G, Sang N. Sulfate Aerosols Promote Lung Cancer Metastasis by Epigenetically Regulating the Epithelial-to-Mesenchymal Transition (EMT). Environmental Science & Technology. 2017;51:11401-11
72. Jin M, Wang J, Ji X, Cao H, Zhu J, Chen Y. et al. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:136
73. Nakanishi A, Wada Y, Kitagishi Y, Matsuda S. Link between PI3K/AKT/PTEN Pathway and NOX Proteinin Diseases. Aging Dis. 2014;5:203-11
74. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29-33
75. Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E-cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer. 2009;9:325
76. Valdivia A, Duran C, San Martin A. The role of Nox-mediated oxidation in the regulation of cytoskeletal dynamics. Current pharmaceutical design. 2015;21:6009-22
77. Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol Biol Cell. 2009;20:2650-60
78. Taulet N, Delorme-Walker VD, DerMardirossian C. Reactive oxygen species regulate protrusion efficiency by controlling actin dynamics. PLoS One. 2012;7:e41342
79. Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009-26
80. Buyukhatipoglu K, Clyne AM. Superparamagnetic iron oxide nanoparticles change endothelial cell morphology and mechanics via reactive oxygen species formation. J Biomed Mater Res A. 2011;96:186-95
81. Martiel JL, Michelot A, Boujemaa-Paterski R, Blanchoin L, Berro J. Force Production by a Bundle of Growing Actin Filaments Is Limited by Its Mechanical Properties. Biophys J. 2020;118:182-92
82. Tapial Martinez P, Lopez Navajas P, Lietha D. FAK Structure and Regulation by Membrane Interactions and Force in Focal Adhesions. Biomolecules. 2020;10:179
83. Pinon P, Parssinen J, Vazquez P, Bachmann M, Rahikainen R, Jacquier MC. et al. Talin-bound NPLY motif recruits integrin-signaling adapters to regulate cell spreading and mechanosensing. J Cell Biol. 2014;205:265-81
84. de Rezende FF, Martins Lima A, Niland S, Wittig I, Heide H, Schroder K. et al. Integrin alpha7beta1 is a redox-regulated target of hydrogen peroxide in vascular smooth muscle cell adhesion. Free Radic Biol Med. 2012;53:521-31
85. Nikitovic D, Corsini E, Kouretas D, Tsatsakis A, Tzanakakis G. ROS-major mediators of extracellular matrix remodeling during tumor progression. Food Chem Toxicol. 2013;61:178-86
86. Lee KH, Kim SW, Kim JR. Reactive oxygen species regulate urokinase plasminogen activator expression and cell invasion via mitogen-activated protein kinase pathways after treatment with hepatocyte growth factor in stomach cancer cells. J Exp Clin Cancer Res. 2009;28:73
87. Huang M, Xin W. Matrine inhibiting pancreatic cells epithelial-mesenchymal transition and invasion through ROS/NF-kappaB/MMPs pathway. Life Sci. 2018;192:55-61
88. Ho BY, Wu YM, Chang KJ, Pan TM. Dimerumic acid inhibits SW620 cell invasion by attenuating H₂O₂-mediated MMP-7 expression via JNK/C-Jun and ERK/C-Fos activation in an AP-1-dependent manner. International journal of biological sciences. 2011;7:869-80
89. Kim MH, Cho HS, Jung M, Hong MH, Lee SK, Shin BA. et al. Extracellular signal-regulated kinase and AP-1 pathways are involved in reactive oxygen species-induced urokinase plasminogen activator receptor expression in human gastric cancer cells. Int J Oncol. 2005;26:1669-74
90. Kim MH, Yoo HS, Kim MY, Jang HJ, Baek MK, Kim HR. et al. Helicobacter pylori stimulates urokinase plasminogen activator receptor expression and cell invasiveness through reactive oxygen species and NF-kappaB signaling in human gastric carcinoma cells. International journal of molecular medicine. 2007;19:689-97
91. Tran H, Maurer F, Nagamine Y. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol Cell Biol. 2003;23:7177-88
92. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Seminars in cell & developmental biology. 2018;80:50-64
93. Yuan Z, Liang Z, Yi J, Chen X, Li R, Wu J. et al. Koumine Promotes ROS Production to Suppress Hepatocellular Carcinoma Cell Proliferation Via NF-κB and ERK/p38 MAPK Signaling. Biomolecules. 2019;9:559
94. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP. et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012;19:1880-91
95. Wang J, Deng H, Zhang J, Wu D, Li J, Ma J. et al. α-Hederin induces the apoptosis of gastric cancer cells accompanied by glutathione decrement and reactive oxygen species generation via activating mitochondrial dependent pathway. Phytotherapy research: PTR. 2020;34:601-11
96. Punganuru SR, Madala HR, Arutla V, Srivenugopal KS. Selective killing of human breast cancer cells by the styryl lactone (R)-goniothalamin is mediated by glutathione conjugation, induction of oxidative stress and marked reactivation of the R175H mutant p53 protein. Carcinogenesis. 2018;39:1399-410
97. Thamilselvan V, Menon M, Stein GS, Valeriote F, Thamilselvan S. Combination of Carmustine and Selenite Inhibits EGFR Mediated Growth Signaling in Androgen-Independent Prostate Cancer Cells. J Cell Biochem. 2017;118:4331-40
98. Pei X, Xiao J, Wei G, Zhang Y, Lin F, Xiong Z. et al. Oenothein B inhibits human non-small cell lung cancer A549 cell proliferation by ROS-mediated PI3K/Akt/NF-κB signaling pathway. Chemico-biological interactions. 2019;298:112-20
99. Su X, Shen Z, Yang Q, Sui F, Pu J, Ma J. et al. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics. 2019;9:4461-73
100. Li Y, Liang R, Zhang X, Wang J, Shan C, Liu S. et al. Copper Chaperone for Superoxide Dismutase Promotes Breast Cancer Cell Proliferation and Migration via ROS-Mediated MAPK/ERK Signaling. Frontiers in pharmacology. 2019;10:356
101. Verbon EH, Post JA, Boonstra J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene. 2012;511:1-6
102. Gao X, Leone GW, Wang H. Cyclin D-CDK4/6 functions in cancer. Adv Cancer Res. 2020;148:147-69
103. Kim C, Lee SG, Yang WM, Arfuso F, Um JY, Kumar AP. et al. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer letters. 2018;431:123-41
104. Kang N, Jian JF, Cao SJ, Zhang Q, Mao YW, Huang YY. et al. Physalin A induces G2/M phase cell cycle arrest in human non-small cell lung cancer cells: involvement of the p38 MAPK/ROS pathway. Molecular and cellular biochemistry. 2016;415:145-55
105. Zhou YB, Feng X, Wang LN, Du JQ, Zhou YY, Yu HP. et al. LGH00031, a novel ortho-quinonoid inhibitor of cell division cycle 25B, inhibits human cancer cells via ROS generation. Acta pharmacologica Sinica. 2009;30:1359-68
106. Dey P, Baddour J, Muller F, Wu CC, Wang H, Liao WT. et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature. 2017;542:119-23
107. Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K. et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science (New York, NY). 2015;350:1391-6
108. Farrukh MR, Nissar UA, Afnan Q, Rafiq RA, Sharma L, Amin S. et al. Oxidative stress mediated Ca(2+) release manifests endoplasmic reticulum stress leading to unfolded protein response in UV-B irradiated human skin cells. Journal of dermatological science. 2014;75:24-35
109. He H, Zhuo R, Dai J, Wang X, Huang X, Wang H. et al. Chelerythrine induces apoptosis via ROS-mediated endoplasmic reticulum stress and STAT3 pathways in human renal cell carcinoma. Journal of cellular and molecular medicine. 2020;24:50-60
110. Kim BR, Park SH, Jeong YA, Na YJ, Kim JL, Jo MJ. et al. RUNX3 enhances TRAIL-induced apoptosis by upregulating DR5 in colorectal cancer. Oncogene. 2019;38:3903-18
111. Hong YH, Uddin MH, Jo U, Kim B, Song J, Suh DH. et al. ROS Accumulation by PEITC Selectively Kills Ovarian Cancer Cells via UPR-Mediated Apoptosis. Frontiers in oncology. 2015;5:167
112. Kim B, Kim HS, Jung EJ, Lee JY, B KT, Lim JM. et al. Curcumin induces ER stress-mediated apoptosis through selective generation of reactive oxygen species in cervical cancer cells. Molecular carcinogenesis. 2016;55:918-28
113. Yin C, Dai X, Huang X, Zhu W, Chen X, Zhou Q. et al. Alantolactone promotes ER stress-mediated apoptosis by inhibition of TrxR1 in triple-negative breast cancer cell lines and in a mouse model. Journal of cellular and molecular medicine. 2019;23:2194-206
114. Yu B, Liu Y, Peng X, Hua S, Zhou G, Yan K. et al. Synthesis, characterization, and antitumor properties of Au(i)-thiourea complexes. Metallomics: integrated biometal science. 2020;12:104-13
115. Orrenius S, Zhivotovsky B. Cardiolipin oxidation sets cytochrome c free. Nature chemical biology. 2005;1:188-9
116. Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1259-63
117. Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. The Journal of biological chemistry. 2011;286:26334-43
118. Gotoh Y, Cooper JA. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. The Journal of biological chemistry. 1998;273:17477-82
119. Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245-51
120. Wang H, Jiang D, Liu J, Ye S, Xiao S, Wang W. et al. Compound K induces apoptosis of bladder cancer T24 cells via reactive oxygen species-mediated p38 MAPK pathway. Cancer biotherapy & radiopharmaceuticals. 2013;28:607-14
121. Jeong S, Yun HK, Jeong YA, Jo MJ, Kang SH, Kim JL. et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer letters. 2019;447:12-23
122. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47-59
123. Yang Y, Yu Y, Wang J, Li Y, Li Y, Wei J. et al. Silica nanoparticles induced intrinsic apoptosis in neuroblastoma SH-SY5Y cells via CytC/Apaf-1 pathway. Environ Toxicol Pharmacol. 2017;52:161-9
124. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008
125. Basak D, Punganuru SR, Srivenugopal KS. Piperlongumine exerts cytotoxic effects against cancer cells with mutant p53 proteins at least in part by restoring the biological functions of the tumor suppressor. Int J Oncol. 2016;48:1426-36
126. Shao J, Li M, Guo Z, Jin C, Zhang F, Ou C. et al. TPP-related mitochondrial targeting copper (II) complex induces p53-dependent apoptosis in hepatoma cells through ROS-mediated activation of Drp1. Cell communication and signaling: CCS. 2019;17:149
127. Jang JY, Kang YJ, Sung B, Kim MJ, Park C, Kang D. et al. MHY440, a Novel Topoisomerase Ι Inhibitor, Induces Cell Cycle Arrest and Apoptosis via a ROS-Dependent DNA Damage Signaling Pathway in AGS Human Gastric Cancer Cells. Molecules (Basel, Switzerland). 2018;24:96
128. Hong M, Li J, Li S, M MA. Acetylshikonin Sensitizes Hepatocellular Carcinoma Cells to Apoptosis through ROS-Mediated Caspase Activation. Cells. 2019;8:1466
129. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
130. Jiang M, Qiao M, Zhao C, Deng J, Li X, Zhou C. Targeting ferroptosis for cancer therapy: exploring novel strategies from its mechanisms and role in cancers. Transl Lung Cancer Res. 2020;9:1569-84
131. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35:830-49
132. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B. et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732-42
133. Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517-32
134. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annual review of pharmacology and toxicology. 2013;53:401-26
135. Xu D, Li X, Shao F, Lv G, Lv H, Lee JH. et al. The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Science advances. 2019;5:eaav4570
136. Luo XH, Liu JZ, Wang B, Men QL, Ju YQ, Yin FY. et al. KLF14 potentiates oxidative adaptation via modulating HO-1 signaling in castrate-resistant prostate cancer. Endocrine-related cancer. 2019;26:181-95
137. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Molecular cell. 2017;66:801-17
138. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084
139. Li X, Wang K, Ren Y, Zhang L, Tang XJ, Zhang HM. et al. MAPK signaling mediates sinomenine hydrochloride-induced human breast cancer cell death via both reactive oxygen species-dependent and -independent pathways: an in vitro and in vivo study. Cell death & disease. 2014;5:e1356
140. Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science (New York, NY). 1999;286:1162-6
141. Batchelor E, Loewer A, Lahav G. The ups and downs of p53: understanding protein dynamics in single cells. Nat Rev Cancer. 2009;9:371-7
142. Ford JM. Regulation of DNA damage recognition and nucleotide excision repair: another role for p53. Mutation research. 2005;577:195-202
143. Willis J, Patel Y, Lentz BL, Yan S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:10592-7
144. Macip S, Kosoy A, Lee SW, O'Connell MJ, Aaronson SA. Oxidative stress induces a prolonged but reversible arrest in p53-null cancer cells, involving a Chk1-dependent G2 checkpoint. Oncogene. 2006;25:6037-47
145. Meng Y, Chen CW, Yung MMH, Sun W, Sun J, Li Z. et al. DUOXA1-mediated ROS production promotes cisplatin resistance by activating ATR-Chk1 pathway in ovarian cancer. Cancer letters. 2018;428:104-16
146. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M. et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science (New York, NY). 2011;334:1278-83
147. Wang Y, Hinz S, Uckermann O, Hönscheid P, von Schönfels W, Burmeister G. et al. Shotgun lipidomics-based characterization of the landscape of lipid metabolism in colorectal cancer. Biochimica et biophysica acta Molecular and cell biology of lipids. 2020;1865:158579
148. Cadenas C, Vosbeck S, Edlund K, Grgas K, Madjar K, Hellwig B. et al. LIPG-promoted lipid storage mediates adaptation to oxidative stress in breast cancer. Int J Cancer. 2019;145:901-15
149. Zhao H, Wu S, Li H, Duan Q, Zhang Z, Shen Q. et al. ROS/KRAS/AMPK Signaling Contributes to Gemcitabine-Induced Stem-like Cell Properties in Pancreatic Cancer. Molecular therapy oncolytics. 2019;14:299-312
150. Shashni B, Nagasaki Y. Nitroxide radical-containing nanoparticles attenuate tumorigenic potential of triple negative breast cancer. Biomaterials. 2018;178:48-62
151. Mizutani H, Hotta S, Nishimoto A, Ikemura K, Miyazawa D, Ikeda Y. et al. Pirarubicin, an Anthracycline Anticancer Agent, Induces Apoptosis Through Generation of Hydrogen Peroxide. Anticancer Res. 2017;37:6063-9
152. Chen Y, Jungsuwadee P, Vore M, Butterfield DA, St Clair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Molecular interventions. 2007;7:147-56
153. Jin X, Zhou J, Zhang Z, Lv H. The combined administration of parthenolide and ginsenoside CK in long circulation liposomes with targeted tLyp-1 ligand induce mitochondria-mediated lung cancer apoptosis. Artificial cells, nanomedicine, and biotechnology. 2018;46:S931-s42
154. Yu S, Gong LS, Li NF, Pan YF, Zhang L. Galangin (GG) combined with cisplatin (DDP) to suppress human lung cancer by inhibition of STAT3-regulated NF-κB and Bcl-2/Bax signaling pathways. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2018;97:213-24
155. Song W, Yan CY, Zhou QQ, Zhen LL. Galangin potentiates human breast cancer to apoptosis induced by TRAIL through activating AMPK. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;89:845-56
156. Guzman ML, Rossi RM, Karnischky L, Li X, Peterson DR, Howard DS. et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163-9
157. Zou Z, Chang H, Li H, Wang S. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis. 2017;22:1321-35
158. Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer letters. 2012;327:48-60
159. Tateishi Y, Sasabe E, Ueta E, Yamamoto T. Ionizing irradiation induces apoptotic damage of salivary gland acinar cells via NADPH oxidase 1-dependent superoxide generation. Biochemical and biophysical research communications. 2008;366:301-7
160. Petragnano F, Pietrantoni I, Di Nisio V, Fasciani I, Del Fattore A, Capalbo C. et al. Modulating the dose-rate differently affects the responsiveness of human epithelial prostate- and mesenchymal rhabdomyosarcoma-cancer cell line to radiation. International journal of radiation biology. 2020;96:823-35
161. Meng DF, Guo LL, Peng LX, Zheng LS, Xie P, Mei Y. et al. Antioxidants suppress radiation-induced apoptosis via inhibiting MAPK pathway in nasopharyngeal carcinoma cells. Biochemical and biophysical research communications. 2020;527:770-7
162. Shigeta K, Hasegawa M, Kikuchi E, Yasumizu Y, Kosaka T, Mizuno R. et al. Role of the MUC1-C oncoprotein in the acquisition of cisplatin resistance by urothelial carcinoma. Cancer Sci. 2020;111:3639-52
163. Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425-37
164. Wang H, Jiang H, Corbet C, de Mey S, Law K, Gevaert T. et al. Piperlongumine increases sensitivity of colorectal cancer cells to radiation: Involvement of ROS production via dual inhibition of glutathione and thioredoxin systems. Cancer letters. 2019;450:42-52
165. Lynam-Lennon N, Reynolds JV, Pidgeon GP, Lysaght J, Marignol L, Maher SG. Alterations in DNA repair efficiency are involved in the radioresistance of esophageal adenocarcinoma. Radiat Res. 2010;174:703-11
166. Gomez-Casal R, Epperly MW, Wang H, Proia DA, Greenberger JS, Levina V. Radioresistant human lung adenocarcinoma cells that survived multiple fractions of ionizing radiation are sensitive to HSP90 inhibition. Oncotarget. 2015;6:44306-22
167. Vendrely V, Amintas S, Noel C, Moranvillier I, Lamrissi I, Rousseau B. et al. Combination treatment of resveratrol and capsaicin radiosensitizes pancreatic tumor cells by unbalancing DNA repair response to radiotherapy towards cell death. Cancer letters. 2019;451:1-10
168. Lan M, Zhao S, Liu W, Lee CS, Zhang W, Wang P. Photosensitizers for Photodynamic Therapy. Advanced healthcare materials. 2019;8:e1900132
169. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-47
170. Larue L, Myrzakhmetov B, Ben-Mihoub A, Moussaron A, Thomas N, Arnoux P. et al. Fighting Hypoxia to Improve PDT. Pharmaceuticals (Basel, Switzerland). 2019;12:163
171. Wang XQ, Gao F, Zhang XZ. Initiator-Loaded Gold Nanocages as a Light-Induced Free-Radical Generator for Cancer Therapy. Angewandte Chemie (International ed in English). 2017;56:9029-33
172. Wang S, Yuan F, Chen K, Chen G, Tu K, Wang H. et al. Synthesis of Hemoglobin Conjugated Polymeric Micelle: A ZnPc Carrier with Oxygen Self-Compensating Ability for Photodynamic Therapy. Biomacromolecules. 2015;16:2693-700
173. Orel V, Shevchenko A, Romanov A, Tselepi M, Mitrelias T, Barnes CH. et al. Magnetic properties and antitumor effect of nanocomplexes of iron oxide and doxorubicin. Nanomedicine: nanotechnology, biology, and medicine. 2015;11:47-55
174. Wason MS, Colon J, Das S, Seal S, Turkson J, Zhao J. et al. Sensitization of pancreatic cancer cells to radiation by cerium oxide nanoparticle-induced ROS production. Nanomedicine: nanotechnology, biology, and medicine. 2013;9:558-69
175. Bisht G, Rayamajhi S. ZnO Nanoparticles: A Promising Anticancer Agent. Nanobiomedicine. 2016;3:9
176. Zhou Z, Song J, Tian R, Yang Z, Yu G, Lin L. et al. Activatable Singlet Oxygen Generation from Lipid Hydroperoxide Nanoparticles for Cancer Therapy. Angewandte Chemie (International ed in English). 2017;56:6492-6
177. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891-9
178. Nacka-Aleksić M, Pilipović I, Kotur-Stevuljević J, Petrović R, Sopta J, Leposavić G. Sexual dimorphism in rat thymic involution: a correlation with thymic oxidative status and inflammation. Biogerontology. 2019;20:545-69
179. Feng R, He W, Ochi H. A new murine oxidative stress model associated with senescence. Mechanisms of ageing and development. 2001;122:547-59
180. Barbouti A, Vasileiou PVS, Evangelou K, Vlasis KG, Papoudou-Bai A, Gorgoulis VG. et al. Implications of Oxidative Stress and Cellular Senescence in Age-Related Thymus Involution. Oxid Med Cell Longev. 2020;2020:7986071
181. Lynch HE, Goldberg GL, Chidgey A, Van den Brink MR, Boyd R, Sempowski GD. Thymic involution and immune reconstitution. Trends in immunology. 2009;30:366-73
182. Palmer S, Albergante L, Blackburn CC, Newman TJ. Thymic involution and rising disease incidence with age. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:1883-8
Author contact
![]() Corresponding author: Public Center of Experimental Technology, The school of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan Province, 646000, China. Tel.:17361102561; Email address: xiangxieedu.cn.
Corresponding author: Public Center of Experimental Technology, The school of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan Province, 646000, China. Tel.:17361102561; Email address: xiangxieedu.cn.