Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(19):5817-5824. doi:10.7150/jca.56307 This issue Cite
Research Paper
OxLDL as an Inducer of a Metabolic Shift in Cancer Cells
Albert V. Bitorina1*, Yvonne Oligschlaeger1*, Lingling Ding1, Tulasi Yadati1, Annemarie Westheim3, Tom Houben1, Rianne D.W. Vaes2, Steven W.M. Olde Damink2, Jan Theys3 ![]() , Ronit Shiri-Sverdlov1
, Ronit Shiri-Sverdlov1 ![]()
1. Department of Molecular Genetics, School for Nutrition and Translational Research in Metabolism (NUTRIM), Maastricht University, Maastricht, The Netherlands.
2. Department of Surgery, School for Nutrition and Translational Research in Metabolism (NUTRIM), Maastricht University, Maastricht, The Netherlands.
3. Department of Precision Medicine, School for Oncology & Developmental Biology (GROW), Maastricht University Medical Centre, Maastricht, Netherlands.
*These authors contributed equally to this work; #Shared last authors.
Received 2020-11-23; Accepted 2021-6-24; Published 2021-8-3
Abstract

Recent evidence established a link between disturbed lipid metabolism and increased risk for cancer. One of the most prominent features related to disturbed lipid metabolism is an increased production of oxidized low-density-lipoproteins (oxLDL), which results from elevated oxidative stress. OxLDL is known to have detrimental effects on healthy cells and plays a primary role in diseases related to the metabolic syndrome. Nevertheless, so far, the exact role of oxLDL in cancer cell metabolism is not yet known. To examine changes in metabolic profile induced by oxLDL, pancreatic KLM-1 cells were treated with oxLDL in a concentration- (25 or 50 µg/ml) and/or time-dependent (4 hr or 8 hr) manner and the impact of oxLDL on oxygen consumption rates (OCR) as well as extracellular acidification rates (ECAR) was analyzed using Seahorse technology. Subsequently, to establish the link between oxLDL and glycolysis, stabilization of the master regulator hypoxia-inducible factor 1-alpha (HIF-1α) was measured by means of Western blot. Furthermore, autophagic responses were assessed by measuring protein levels of the autophagosomal marker LC3B-II. Finally, the therapeutic potential of natural anti-oxLDL IgM antibodies in reversing these effects was tested. Incubation of KLM-1 cells with oxLDL shifted the energy balance towards a more glycolytic phenotype, which is an important hallmark of cancer cells. These data were supported by measurement of increased oxLDL-mediated HIF-1α stabilization. In line, oxLDL incubation also increased the levels of LC3B-II, suggesting an elevated autophagic response. Importantly, antibodies against oxLDL were able to reverse these oxLDL-mediated metabolic effects. Our data provides a novel proof-of-concept that oxLDL induces a shift in energy balance. These data not only support a role for oxLDL in the progression of cancer but also suggest the possibility of targeting oxLDL as a therapeutic option in cancer.
Keywords: Pancreatic cancer cells, oxLDL, metabolic switch, HIF-1α, autophagy
Introduction
Cancer is the second leading cause of deaths worldwide, which currently accounts for nearly ten million deaths [1]. In particular, overweight and obesity, due to excess energy intake and decreased physical activity, have reached pandemic levels worldwide [2] and recent clinical data suggests that individuals with obesity are at an increased risk of developing several types of cancer [3-5].
Mechanisms that are known to link obesity with increased cancer burden, i.e., cancer development, progression and decreased treatment response [6], include adipose tissue dysfunction, increased circulating levels of hormones, perturbed lipid metabolism and low-grade chronic inflammation [7]. Chronic inflammation is strongly associated with increased secretion of cytokines in the circulation, which can lead to cancer-related cellular processes such as cell adhesion, chemotaxis, migration and angiogenesis [8]. Moreover, excess nutrient intake has been shown to increase the expression of multiple growth factors and hormones [9, 10] and is known to induce changes in gut microbiota [11], thereby influencing oncogenesis and tumor progression.
One of the most prominent features related to excess nutrient intake and dysregulated lipid metabolism is the increased production of oxidized cholesterol-rich low-density lipoprotein (oxLDL), which results from elevated oxidative stress. Increased levels of oxLDL is known to have detrimental effects on healthy cells and plays a primary responsible role in metabolic syndrome (MetS)-related diseases such as atherosclerosis [12] and non-alcoholic steatohepatitis (NASH) [13]. Strikingly, increasing evidence also points towards a correlation between circulating levels of oxLDL and the development of cancer [14], suggesting that oxLDL is a potent pro-oncogenic factor. Nevertheless, the exact metabolic role of oxLDL in cancer is not well-understood.
In the present study, we hypothesized that oxLDL induces a metabolic shift in cancer cells. We demonstrate for the first time that incubating KLM-1 pancreatic cancer cells with oxLDL shifts the energy balance towards a more glycolytic phenotype, which is supported by oxLDL-induced stabilization of hypoxia-inducible factor 1-alpha (HIF-1α), a master regulator of glycolysis. In line with these data, we also show that oxLDL, likely due to increased autophagic activity, increases the expression of the autophagosomal marker LC3B-II. Essentially, the addition of EO6 IgM antibodies against oxLDL were able to reverse the oxLDL-mediated metabolic effects.
Altogether, our data provide a proof-of-concept that oxLDL induces a metabolic shift, which is involved in cancer progression. In addition, our data also supports the opportunity of targeting oxLDL as a novel therapeutic option in cancer.
Materials and methods
Cell culture
Human pancreatic cancer cell line KLM-1 (obtained from RIKEN BioResource Center) was cultured in high glucose (4.5 g/L) Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% (v/v) fetal bovine serum (USDA Approved, PAA, Pasching, Austria). Cells were maintained at 37 °C, 5% CO2 in a humidified incubator.
Energy metabolism assay
KLM-1 cells were grown in DMEM + 10% FBS and seeded into 96XF plates @ 24000 cells/well and left to attach overnight at 37 °C. Metabolic profiles were generated by replacing the growth medium with assay medium 1 hr before measurement according to the manufacturer guidelines [15]. Minimally oxidized LDL (Alfa Aesar) concentration was increased to 25 µg/ml over 60 min in steps of 5 µg/ml. Oxygen consumption and extracellular acidification rates were monitored using a Seahorse Bioscience XF96 Extracellular Flux Analyzer. Values were corrected versus baseline as well as for total protein content in each well. Total protein concentration was measured using a Pierce® BCA protein assay kit (Waltham, MA, USA). Data were obtained and depicted using Wave 2.6.0.31 software (Agilent).
Western blot
In 3 independent experiments, KLM-1 cells were grown in DMEM + 10% FBS and seeded into 6 well plates at 500,000 cells/well and left to attach overnight at 37 ºC. Subsequently, cells were treated with oxLDL (25 or 50 µg/ml; minimally oxidized; Tebu-Bio) for 4 hr or 8 hr. Alternatively, cells were either treated with oxLDL alone (50 µg/ml), oxLDL (50 µg/ml) + chloroquine (20 µM), oxLDL (50 µg/ml) + EO6 (14 µg/ml; Avanti) or oxLDL (50 µg/ml) + chloroquine (20 µM) + EO6 (14 µg/ml). Subsequently, cells were washed with cold PBS and lysed with cold RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% Sodium deoxycholate, 1% Triton X-100, 0.1% SDS) supplemented with a mixture of protease and phosphatase inhibitors (Complete and PhosStop; Roche). Lysates were centrifuged at 16,000 xg for 15 min, after which the supernatant was transferred to a new Eppendorf and stored at -20 °C. The total protein concentration was measured using a Pierce® (Waltham, MA, USA) BCA protein assay. Equal amounts of protein were loaded on the gel. After SDS/PAGE, proteins were transferred to nitrocellulose membrane (Bio-Rad), which was blocked with 5% non-fat dry milk for 1h at room temperature. For detection, the membrane was incubated with an antibody against HIF-1α (1:2000 dilution; BD Transduction Laboratories; Catalogue #610959), β-Actin (1:2000 dilution; Cell Signaling, #4967) and LC3B (1:1000 dilution; Cell Signaling, #2775) overnight at 4°C followed by 1h of incubation with donkey-anti-rabbit or rat-anti-mouse detection antibody, respectively, at room temperature (1:2000 Jackson Laboratories, Bar Harbor, ME, USA). Signal was detected with the Biorad ChemiDoc™ XRS+System by enhanced chemiluminescence. Blots were subsequently quantified using ImageJ software (Ver 1.52R). HIF-1α, LC3B-I and LC3B-II levels were first corrected for loading by β-Actin, after which these levels were normalized to medium control or OxLDL respectively. Fold-changes are represented in bar graphs that were generated using GraphPad 6 software.
Statistical analysis
Data were statistically analyzed by performing two-tailed nonpaired t tests using GraphPad Prism (version 6 for Windows; GraphPad Software, San Diego, CA) for comparing differences in ECAR% between control and oxLDL-treated KLM-1 cells. One-way ANOVA with multiple comparisons Tukey post-hoc test was used to compare differences of HIF-1α or LC3B-II between control and oxLDL treated KLM-1 cells pooled from 3 independent experiments. The results were visualized and expressed as the mean ±SEM and considered significant at p ≤ 0.05, * and ** indicate p ≤ 0.01 respectively.
Results
OxLDL increases glycolysis in KLM-1 cells
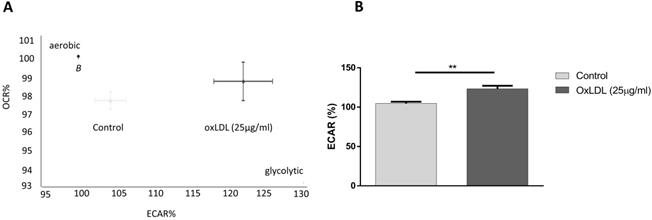
Oxygen consumption rate (OCR) is routinely used as an indicator of mitochondrial oxidative phosphorylation, whereas extracellular acidification rates (ECAR) reflect the state of lactate secretion into the medium, which is strongly linked to the levels of glycolysis in the cell. In order to investigate the effects of oxLDL on cellular energy balance in cancer cells, we titrated 25 µg/ml of oxLDL, under normoxic conditions, to the pancreatic tumor cell line KLM-1 and measured real-time basal changes in OCR and ECAR over the course of 60 min. Whereas no effect was observed on OCR rates (supplemental Fig. 1), our data showed that incubation with oxLDL shifted the energy balance towards a more glycolytic phenotype, as shown by a 20% increase in ECAR compared to control cells (Fig. 1A,B).
OxLDL shifts energy balance to a glycolytic profile. KLM-1 pancreatic cancer cells were titrated with oxLDL (25 µg/ml). (A) Representative energy metabolism diagram of basal OCR vs. ECAR. (B) Difference in ECAR% between control and oxLDL-treated cells. **p<0.01.
OxLDL stabilizes HIF-1α protein
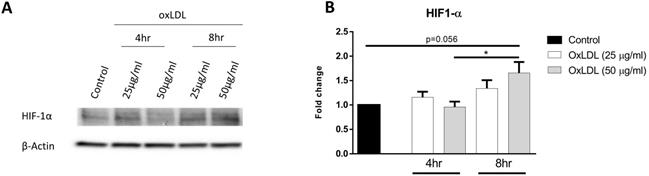
HIF-1α is a master regulator of glycolysis and high levels of the stabilized protein is maintained in cells with high glycolytic rates. In order to determine the effect of oxLDL on HIF-1α protein stabilization, KLM-1 cells were incubated in the absence or presence of different concentrations of oxLDL (25 or 50 µg/ml, respectively) for 4 hr or 8 hr. Our results demonstrated that protein levels of HIF-1α increased in response to 50 but not 25 µg/ml oxLDL treatment compared to control (p=0.056) (Fig. 2A, B; Supplemental Fig. 2). Furthermore, significant differences in HIF-1α protein were observed after incubation of 8 hr compared to 4hr, suggesting that the oxLDL-mediated increase in ECAR resulted from HIF-1α protein stabilization in a dose- and time-dependent manner.
OxLDL stabilizes HIF-1α protein levels. KLM-1 cells were treated with oxLDL (25 or 50 µg/ml) for 4 hr or 8 hr. (A) Representative Western blot of HIF-1α protein levels, (B) Quantification of HIF-1α protein adjusted for β-Actin.
OxLDL increases autophagosomal marker LC3B-II
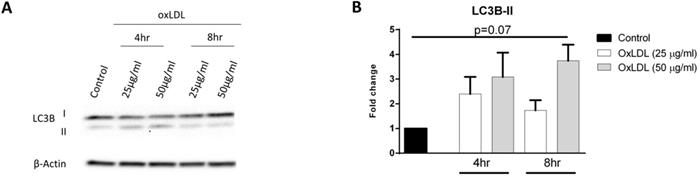
In addition to its involvement in glycolysis, HIF-1α can also modulate autophagy in order to secure additional energy during metabolic stress responses such as hypoxia. It has been shown that the cytosolic protein LC3B-I is converted to autophagosomal marker LC3B-II during the fusion of endosomes with lysosomes, a process known as autophagosome formation [16]. In order to assess whether autophagic activity is induced in response to oxLDL, we treated KLM-1 cells with or without oxLDL (25 and 50 µg/ml, respectively) over a time course of 4 hr and 8 hr. Subsequently, protein levels of LC3B-II were determined with Western blot (Fig. 3A; Supplemental Fig. 2) and quantitative data analysis showed that incubation with oxLDL resulted in a 4-fold increase of LC3B-II protein expression after 8 hr (p=0.07) (Fig. 3B; Supplemental Fig. 2).
OxLDL increases LC3B-II proteins levels. KLM-1 cells were treated with oxLDL (25 or 50 µg/ml) for 4 hr or 8 hr. (A) Representative Western blot of LC3B-I and LC3B-II protein levels, (B) Quantification of LC3B-II protein adjusted for β-Actin.
EO6 reverses oxLDL-induced metabolic changes
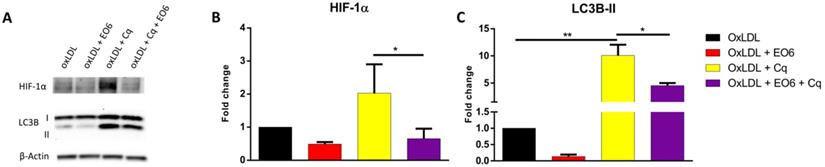
Given the fact that natural IgM antibodies against oxLDL have been shown to ameliorate the adverse effects of oxLDL in several metabolic diseases, including NASH [17] and atherosclerosis [18], we assessed whether EO6 monoclonal antibodies against oxLDL were able to counteract the metabolic effects of oxLDL on HIF-1α and LC3B by treating KLM-1 cells for 8hr with oxLDL (50 µg/ml) in the presence or absence of EO6 (14 µg/ml). Additionally, cells were also treated with chloroquine (Cq; 20 µM) in order to ensure that any observed differences in LC3B-II were due to increased conversion of LC3B-I rather than differences in breakdown of LC3B-II. Chloroquine is a lysosomotropic weak base that diffuses into the lysosome, where it gets trapped, thereby changing the lysosomal pH and subsequently inhibiting autophagic degradation of proteins including LC3B-II [19]. Our results showed that treatment with EO6 significantly reversed oxLDL-mediated increases in HIF-1α protein, in the presence of chloroquine (Fig. 4A, B). Similarly, treatment with EO6 also significantly reversed oxLDL-mediated increases in LC3B-II protein, in the presence of chloroquine (Fig. 4A, C; Supplemental Fig. 2).
EO6 reverses oxLDL-mediated effects on HIF-1α and LC3B-II. KLM-1 cells were treated with oxLDL (50µg/ml) in the presence or absence of chloroquine (Cq; 20 µM) and EO6 antibodies against oxLDL (14 µg/ml) for 8 hr. (A) Representative Western blot of HIF-1α and LC3B-I and LC3B-II protein levels, (B) Quantification of HIF-1α and (C) LC3B-II protein adjusted for β-Actin.
Discussion
Metabolic reprogramming, referred to as an important cellular adaptation mechanism, is unequivocally considered a hallmark of cancer [20]. Many studies have focused on gaining better insight into the complex nature of cancer cell metabolism both in-vitro [21, 22] and in-vivo [23], and recently, the impact of abnormal lipid metabolism in modulating cancer pathophysiology has received increasing interest [24]. In the current study, we provide novel insights into the role of oxLDL in regulating cancer cell metabolism.
Due to their high proliferation rate, cancer cells show a strong avidity for lipids and cholesterol, which they retrieve either from increased uptake of dietary lipids and/or cholesterol, or from enhanced lipogenesis and cholesterol synthesis [25, 26]. One of the most prominent features associated with increased dietary lipids, and hence dyslipidemia, is low-density lipoprotein (LDL) oxidation. Oxidative modifications of LDL can occur as a result of elevated oxidative stress, a metabolic derangement known to be common in cancer and MetS-related diseases.
Previously, it has been shown that increased levels of oxLDL have detrimental effects on healthy metabolic tissues, including the cardiovascular system [27] and the liver [28]. For instance, oxLDL impaired mitochondrial respiration in porcine aortic endothelial cells due to reduced activities of mitochondrial complexes [29], likely inducing a metabolic shift. Furthermore, overloading macrophages with oxLDL triggered a metabolic switch that resulted in high levels of HIF1-α [30, 31], which is a master regulator of metabolism that is constitutively expressed in cells. Normally, in the presence of oxygen, HIF-1α is destabilized and targeted for proteosomal degradation [32]. However, under abnormal conditions such as hypoxia, which is a common feature of cancer, HIF-1α is being stabilized, thereby mediating a switch towards glycolytic metabolism [33]. Alternatively, under normoxic conditions HIF-1α can also be stabilized in an oxygen-independent manner through the noncanonical HIF-1α activation pathway. Several factors such as oncometabolites (succinate/fumarate), heat shock proteins (Hsp90, Hsc70), regulators of tumor suppressor genes (MDM2 ligase) and changes in oxidative phosphorylation (OXPHOS) system are shown to affect HIF-1α stabilization. For example, tumorigenic factors are known to promote protein synthesis of HIF-1α via Akt pathway and Akt mediated synthesis of Hsp90 can further stabilize HIF-1α in an oxygen-independent fashion [34].
Given the fact that oxLDL stimulates the proliferation of a variety of cell types, including macrophages, endothelial and smooth muscle cells [35-37], oxLDL has been suggested to have pro-oncogenic properties. Indeed, in the context of cancer, it has been reported that increased accumulation of oxLDL, which is related to hyperlipidemia, is highly associated with the development of several types of cancer, including colon, breast and ovarian cancer [14, 38, 39]. Nevertheless, the exact mechanism of oxLDL on cancer cell metabolism has not yet been fully understood.
Previous studies showed that metabolic derangements, such as hypoxia, can cause a remarkable upregulation of HIF-1α [40], triggering glucose uptake in mouse granulosa tumor cells [41]. One way in which cancer cells become resilient or resistant, and thus enhance their capability to proliferate or metastasize, is by metabolically switching to glycolytic profiles and inducing cellular protective autophagy. Hence, several strategies, focused on reorienting cancer cell metabolism from glycolysis to oxidative phosphorylation, have demonstrated a beneficial decrease in cell survival, invasiveness and tumor growth. For instance, Poteet et al. showed that methylene blue was capable of increasing oxygen consumption while reducing lactate production, thereby reversing the glycolytic profile and decreasing proliferation of glioblastoma cell [42]. Another study showed that resveratrol increased ATP production and thus the oxidative capacity, while decreasing glycolysis, as shown by a reduction in the pentose phosphate pathway in colon cancer cells [43]. Moreover, they also demonstrated that resveratrol was able to modify the lipodomic profile of these cells, which is likely important for its metabolic effects on cancer energy metabolism. In the present study, we demonstrate for the first time that incubating KLM-1 pancreatic cancer cells with oxLDL, under normoxic conditions, leads to HIF-1α stabilization. These data are in line with others showing that oxLDL induced HIF-1α protein accumulation in human macrophages [30, 31]. Furthermore, these data also support the observed increase in basal ECAR levels, pointing towards oxLDL as an inducer of a metabolic switch towards glycolysis in cancer cells. Given that cancer cells, even under non-hypoxic conditions, are capable of inducing HIF-1α and can actively shift their energy production from mitochondrial to glycolytic sources [44], our data suggest that treatment of oxLDL mimics a 'Warburg'-like effect in KLM-1 pancreatic cancer cells. This characteristic of oxLDL which promotes the switching to glycolytic metabolic profiles could potentially explain how oxLDL increases cancer cell survival and in so doing promote tumorigenesis & proliferation. In addition to this, another known method cancer cells utilize to increase survival and in so doing promote tumorigenesis & proliferation is by inducing cellular protective autophagy.
One of the most conserved physiological processes present in eukaryotes is autophagy, a catabolic pathway that is known to involve lysosomal degradation of organelles and cytoplasmic contents that are recycled for sustaining cellular energy requirements [45]. It is known that under some circumstances, autophagy may be utilized as a cellular 'suicide' mechanism, thereby inducing cell death [46]. Recent studies also discovered several protective functions for autophagy, including the regulation of intracellular lipid stores and a role in immunity, cell death and inflammation [47, 48]. For instance, Kwanten et al demonstrated absence of fasting-induced steatosis, enhanced serum lipid profile, decreased glycemia using autophagy deficient mice. These findings underscored a role for the autophagy process in pathophysiology of diseases such as NAFLD/NASH among other liver diseases [49]. In the context of MetS, we demonstrated that oxLDL disturbs lysosomal function [28] and induces autophagy [50], which was correlated with hepatic inflammation, whereas others found that elevated levels of oxLDL enabled the selection and survival of cancer cells due to its involvement in protective autophagy [51], suggesting a multifaceted role of oxLDL. Previous studies have also shown that metabolic derangements, such as hypoxia, can cause a substantial upregulation of HIF-1α [40], triggering cellular autophagy in mouse granulosa tumor cells [41]. In line with these studies, we showed that oxLDL treatment resulted in an increase in the autophagosomal marker LC3B-II, which is a central protein in the regulation of autophagy [52]. Hence, our data suggest that oxLDL stabilizes HIF-1α, thereby activating the autophagic pathway, as shown by the concomitant increase in LC3B-II protein. Based on other studies [53, 54], it is very likely that these changes are responsible for the oxLDL-induced metabolic switch towards glycolysis observed in KLM-1 pancreatic cancer cells, which is crucial for cell survival.
As previously discussed oxLDL has stabilizing effects on HIF-1α and activates the autophagic pathway, both of which are known to be involved in pro-oncogenic processes. In the current study, we also explored the ability of natural IgM antibodies against oxLDL (referred to as EO6) to prevent the changes in metabolic profile due to oxLDL in the context of cancer cell metabolism. Our results showed that EO6, by scavenging oxLDL, is capable of reducing the ability of oxLDL to stabilize HIF-1α and the subsequent increase in autophagy. Given that oxLDL is involved in many aspects of metabolic diseases, it has become a promising target in chronic inflammatory diseases, in which lipid metabolism is disturbed. Exercise training [55, 56], lowering dietary fat intake [57] as well as antioxidant [58, 59], statin [60] and fenofibrate therapies [61] are therapeutic strategies that have been involved in lowering oxLDL levels. Relevantly, these strategies have also been shown to reduce cancer risk [62] or exhibit anticancer properties [63-65], of which some therapeutic modalities have been already used in clinical trials. In the context of MetS, several studies have demonstrated that circulating levels of oxLDL can also be effectively targeted in a more direct manner without inducing side effects, as unfortunately often seen with adjuvant therapies [66]. Recent evidence indicates that immunizing hyperlipidemic mice with Streptococcus pneumoniae is an effective way of triggering a natural immune response (i.e., inducing an increase in monoclonal IgM autoantibodies) against oxLDL, thereby reducing atherosclerotic lesions [18]. Similarly, in the context of NASH, we previously demonstrated that specific targeting of oxLDL by means of pneumococcal immunization also reduced hepatic inflammation [17], suggesting that the scavenging of oxLDL by anti-oxLDL autoantibodies might be a potent therapeutic strategy in cancer treatment. Obviously, chronic inflammation is also strongly associated with malignant diseases [67, 68] and tumor-associated macrophages are known to represent the major inflammatory cell population in tumors. Hence, several studies explored the effects of anti-inflammatory strategies, such as nonsteroidal anti-inflammatory drugs, and showed their ability to prevent early onset of cancer [69] and reduce the risk of cancer [70], thus underscoring the significance of inflammation during neoplastic progression.
Conclusions
In the present study, we demonstrate for the first time that in addition to the known beneficial metabolic effects of the tumor microenvironment, oxLDL has a direct effect on pancreatic cancer cells by promoting a metabolic shift towards a more glycolytic phenotype. Furthermore, our data revealed that the addition of EO6 IgM antibodies against oxLDL were able to reverse these oxLDL-mediated metabolic effects. While there are several studies looking into the effects of oxLDL in other types of cancer, little is known about its effect in pancreatic cancer. As oxLDl is likely to also play a role in pancreatic cancer, future studies should therefore look into the effect of oxLDL in pancreatic cancer and evaluate the potential of targeting oxLDL as a novel therapeutic option in cancer.
Abbreviations
Cq: chloroquine; ECAR: extracellular acidification rates; HIF-1α: hypoxia-inducible factor 1-alpha; LDL: low-density lipoprotein; MetS: metabolic syndrome; NASH: non-alcoholic steatohepatitis; oxLDL: oxidized low-density-lipoproteins; OCR: oxygen consumption rates.
Supplementary Material
Supplementary figures.
Acknowledgements
We acknowledge the contribution of all authors cited. We thank all people from the Shiri-Sverdlov and Theys research groups for their contribution, technical assistance and discussions. We acknowledge and thank Prof. Dr. Thorsten Cramer for his technical assistance and discussions.
Funding
This research was supported by the CVON IN-CONTROL grant (CVON2012-03), the Dutch Organisation for Scientific Research (NWO; Vidi grant no. 016.126.327), ASPASIA (grant no. 015.008.043), TKI-LSH (grant no. 40-41200-98-9306), Dutch Cancer Society (KWF grant no. 10820) and VCK (grant no. Swu16.0057-VT). The funding bodies had no role in the preparation of this manuscript or the decision to publish.
Authors' contributions
All authors contributed to conception and design of the study. A.V.B., Y.O., L.D., T.Y., A.W., T.H., R.D.W.V. performed the experiments. A.V.B., Y.O., T.H., R.D.W.V., J.T., R.S.S. analyzed and interpreted the data. Y.O. and A.V.B. wrote the manuscript with support from S.W.M.O.D., J.T. and R.S.S. All authors read and approved the final manuscript.
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians. 2021;71:209-49
2. Friedrich MJ. Global Obesity Epidemic Worsening. JAMA. 2017;318:603
3. Doerstling SS, O'Flanagan CH, Hursting SD. Obesity and Cancer Metabolism: A Perspective on Interacting Tumor-Intrinsic and Extrinsic Factors. Front Oncol. 2017;7:216
4. Chen Y, Wang X, Wang J, Yan Z, Luo J. Excess body weight and the risk of primary liver cancer: an updated meta-analysis of prospective studies. Eur J Cancer. 2012;48:2137-45
5. Genkinger JM, Spiegelman D, Anderson KE, Bernstein L, van den Brandt PA, Calle EE. et al. A pooled analysis of 14 cohort studies of anthropometric factors and pancreatic cancer risk. Int J Cancer. 2011;129:1708-17
6. Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. 2012;35:2402-11
7. Micucci C, Valli D, Matacchione G, Catalano A. Current perspectives between metabolic syndrome and cancer. Oncotarget. 2016;7:38959-72
8. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185
9. Brahmkhatri VP, Prasanna C, Atreya HS. Insulin-like growth factor system in cancer: novel targeted therapies. Biomed Res Int. 2015;2015:538019
10. Capper CP, Rae JM, Auchus RJ. The Metabolism, Analysis, and Targeting of Steroid Hormones in Breast and Prostate Cancer. Horm Cancer. 2016;7:149-64
11. Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M. et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7:271ps1
12. Di Pietro N, Formoso G, Pandolfi A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascul Pharmacol. 2016;84:1-7
13. Walenbergh SM, Koek GH, Bieghs V, Shiri-Sverdlov R. Non-alcoholic steatohepatitis: the role of oxidized low-density lipoproteins. J Hepatol. 2013;58:801-10
14. Suzuki K, Ito Y, Wakai K, Kawado M, Hashimoto S, Toyoshima H. et al. Serum oxidized low-density lipoprotein levels and risk of colorectal cancer: a case-control study nested in the Japan Collaborative Cohort Study. Cancer Epidemiol Biomarkers Prev. 2004;13:1781-7
15. Nicholls DG, Darley-Usmar VM, Wu M, Jensen PB, Rogers GW, Ferrick DA. Bioenergetic profile experiment using C2C12 myoblast cells. J Vis Exp. 2010
16. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-8
17. Bieghs V, van Gorp PJ, Walenbergh SM, Gijbels MJ, Verheyen F, Buurman WA. et al. Specific immunization strategies against oxidized low-density lipoprotein: a novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology. 2012;56:894-903
18. Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS. et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. 2003;9:736-43
19. Redmann M, Benavides GA, Berryhill TF, Wani WY, Ouyang X, Johnson MS. et al. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017;11:73-81
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
21. Zhang Y, Yang JM. Altered energy metabolism in cancer: a unique opportunity for therapeutic intervention. Cancer Biol Ther. 2013;14:81-9
22. Feinberg T, Herbig J, Kohl I, Las G, Cancilla JC, Torrecilla JS. et al. Cancer metabolism: the volatile signature of glycolysis-in vitro model in lung cancer cells. J Breath Res. 2017;11:016008
23. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG. et al. Lactate Metabolism in Human Lung Tumors. Cell. 2017;171:358-71 e9
24. Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun. 2018;504:582-9
25. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358-65
26. Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013;6:1353-63
27. Galle J, Hansen-Hagge T, Wanner C, Seibold S. Impact of oxidized low density lipoprotein on vascular cells. Atherosclerosis. 2006;185:219-26
28. Bieghs V, Walenbergh SM, Hendrikx T, van Gorp PJ, Verheyen F, Olde Damink SW. et al. Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int. 2013;33:1056-61
29. Xie X, Chowdhury SR, Sangle G, Shen GX. Impact of diabetes-associated lipoproteins on oxygen consumption and mitochondrial enzymes in porcine aortic endothelial cells. Acta Biochim Pol. 2010;57:393-8
30. Shatrov VA, Sumbayev VV, Zhou J, Brüne B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood. 2003;101:4847-9
31. Poitz DM, Augstein A, Weinert S, Braun-Dullaeus RC, Strasser RH, Schmeisser A. OxLDL and macrophage survival: essential and oxygen-independent involvement of the Hif-pathway. Basic Res Cardiol. 2011;106:761-72
32. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464-8
33. Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1. Cold Spring Harb Symp Quant Biol. 2011;76:347-53
34. Iommarini L, Porcelli AM, Gasparre G, Kurelac I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front Oncol. 2017;7:286
35. Zettler ME, Prociuk MA, Austria JA, Massaeli H, Zhong G, Pierce GN. OxLDL stimulates cell proliferation through a general induction of cell cycle proteins. Am J Physiol Heart Circ Physiol. 2003;284:H644-53
36. Seibold S, Schurle D, Heinloth A, Wolf G, Wagner M, Galle J. Oxidized LDL induces proliferation and hypertrophy in human umbilical vein endothelial cells via regulation of p27Kip1 expression: role of RhoA. J Am Soc Nephrol. 2004;15:3026-34
37. Liu J, Ren Y, Kang L, Zhang L. Oxidized low-density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int J Mol Med. 2014;33:1341-7
38. Delimaris I, Faviou E, Antonakos G, Stathopoulou E, Zachari A, Dionyssiou-Asteriou A. Oxidized LDL, serum oxidizability and serum lipid levels in patients with breast or ovarian cancer. Clin Biochem. 2007;40:1129-34
39. Chen KC, Liao YC, Wang JY, Lin YC, Chen CH, Juo SH. Oxidized low-density lipoprotein is a common risk factor for cardiovascular diseases and gastroenterological cancers via epigenomical regulation of microRNA-210. Oncotarget. 2015;6:24105-18
40. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343-54
41. Zhou J, Li C, Yao W, Alsiddig MC, Huo L, Liu H. et al. Hypoxia-inducible factor-1alpha-dependent autophagy plays a role in glycolysis switch in mouse granulosa cells. Biol Reprod. 2018;99:308-18
42. Poteet E, Choudhury GR, Winters A, Li W, Ryou MG, Liu R. et al. Reversing the Warburg effect as a treatment for glioblastoma. J Biol Chem. 2013;288:9153-64
43. Saunier E, Antonio S, Regazzetti A, Auzeil N, Laprevote O, Shay JW. et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci Rep. 2017;7:6945
44. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325-37
45. Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542-52
46. Lin L, Baehrecke EH. Autophagy, cell death, and cancer. Mol Cell Oncol. 2015;2:e985913
47. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35
48. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5
49. Kwanten WJ, Vandewynckel YP, Martinet W, De Winter BY, Michielsen PP, Van Hoof VO. et al. Hepatocellular autophagy modulates the unfolded protein response and fasting-induced steatosis in mice. Am J Physiol Gastrointest Liver Physiol. 2016;311:G599-g609
50. Hendrikx T, Bieghs V, Walenbergh SM, van Gorp PJ, Verheyen F, Jeurissen ML. et al. Macrophage specific caspase-1/11 deficiency protects against cholesterol crystallization and hepatic inflammation in hyperlipidemic mice. PLoS One. 2013;8:e78792
51. Zabirnyk O, Liu W, Khalil S, Sharma A, Phang JM. Oxidized low-density lipoproteins upregulate proline oxidase to initiate ROS-dependent autophagy. Carcinogenesis. 2010;31:446-54
52. Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77-88
53. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187-97
54. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705-13
55. Shern-Brewer R, Santanam N, Wetzstein C, White-Welkley J, Parthasarathy S. Exercise and cardiovascular disease: a new perspective. Arterioscler Thromb Vasc Biol. 1998;18:1181-7
56. Takashima A, Ise T, Yagi S, Iwase T, Kimura S, Ueda Y. et al. Cardiac rehabilitation reduces serum levels of oxidized low-density lipoprotein. Circ J. 2014;78:2682-7
57. Silaste ML, Rantala M, Alfthan G, Aro A, Witztum JL, Kesaniemi YA. et al. Changes in dietary fat intake alter plasma levels of oxidized low-density lipoprotein and lipoprotein(a). Arterioscler Thromb Vasc Biol. 2004;24:498-503
58. Harris WS. The prevention of atherosclerosis with antioxidants. Clin Cardiol. 1992;15:636-40
59. Najafpour Boushehri S, Yusof RM, Nasir Mohammad Taib M, Mirzaei K, Yazdekhasti N, Akbarzadeh S. Effect of vitamin supplementation on serum oxidized low-density lipoprotein levels in male subjects with cardiovascular disease risk factors. Iran J Basic Med Sci. 2012;15:958-64
60. Singh U, Devaraj S, Jialal I, Siegel D. Comparison effect of atorvastatin (10 versus 80 mg) on biomarkers of inflammation and oxidative stress in subjects with metabolic syndrome. Am J Cardiol. 2008;102:321-5
61. Rosenson RS, Wolff DA, Huskin AL, Helenowski IB, Rademaker AW. Fenofibrate therapy ameliorates fasting and postprandial lipoproteinemia, oxidative stress, and the inflammatory response in subjects with hypertriglyceridemia and the metabolic syndrome. Diabetes Care. 2007;30:1945-51
62. Prentice RL, Thomson CA, Caan B, Hubbell FA, Anderson GL, Beresford SA. et al. Low-fat dietary pattern and cancer incidence in the Women's Health Initiative Dietary Modification Randomized Controlled Trial. J Natl Cancer Inst. 2007;99:1534-43
63. Borgquist S, Bjarnadottir O, Kimbung S, Ahern TP. Statins: a role in breast cancer therapy? J Intern Med. 2018;284:346-57
64. Lian X, Gu J, Gao B, Li Y, Damodaran C, Wei W. et al. Fenofibrate inhibits mTOR-p70S6K signaling and simultaneously induces cell death in human prostate cancer cells. Biochem Biophys Res Commun. 2018;496:70-5
65. Thyagarajan A, Sahu RP. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr Cancer Ther. 2018;17:210-6
66. Shapiro CL, Recht A. Side effects of adjuvant treatment of breast cancer. N Engl J Med. 2001;344:1997-2008
67. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
68. Multhoff G, Molls M, Radons J. Chronic inflammation in cancer development. Front Immunol. 2011;2:98
69. Baron JA, Sandler RS. Nonsteroidal anti-inflammatory drugs and cancer prevention. Annu Rev Med. 2000;51:511-23
70. Garcia-Rodriguez LA, Huerta-Alvarez C. Reduced risk of colorectal cancer among long-term users of aspirin and nonaspirin nonsteroidal antiinflammatory drugs. Epidemiology. 2001;12:88-93
Author contact
![]() Corresponding authors: Ronit Shiri-Sverdlov (T: +31-43-388-1746 M: r.sverdlovnl,) and Jan Theys (T: +31-43-388-2975 M: jan.theysnl).
Corresponding authors: Ronit Shiri-Sverdlov (T: +31-43-388-1746 M: r.sverdlovnl,) and Jan Theys (T: +31-43-388-2975 M: jan.theysnl).