Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(24):7498-7506. doi:10.7150/jca.64833 This issue Cite
Review
Aberrant lipid metabolism in cancer cells and tumor microenvironment: the player rather than bystander in cancer progression and metastasis
Xiujing Yu1#, Shuyi Mi2#, Jun Ye2 ![]() , Guochun Lou2
, Guochun Lou2 ![]()
1. Department of Endoscopy Center, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, China.
2. Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, China.
# These authors contribute equally to this manuscript.
Received 2021-7-11; Accepted 2021-10-20; Published 2021-11-4
Abstract

As the primary cause of cancer-induced fatality and morbidity, cancer metastasis has been a hard nut to crack. Existing studies indicate that lipid metabolism reprogramming occurring in cancer cells and surrounding cells in TME also endows the aggressive and spreading properties with malignant cells. In this review we describe the lipid metabolic reprogramming of cancer cells at different steps along the metastatic process, we also summarize the altered lipid metabolism of non-cancer cells in TME during tumor metastasis. Additionally, we reveal both intrinsic and extrinsic factors which influence the cellular lipid metabolism reprogramming.
Keywords: lipid metabolism remodeling, tumor microenvironment, cancer metastasis, intrinsic factors, extrinsic factors
Introduction
As the primary cause of cancer-induced fatality and morbidity, cancer metastasis has been a hard nut to crack [1]. It involves a complex process by which a specific subgroup of cancer cells detaches from the primary tumor and colonizes into a non-local environment [2]. Recently, it has been suggested that cancer cells are able to adjust their metabolism selectively at different steps along the metastatic cascade, indicating that metabolism is not just a bystander, but it is a necessary part of the process of metastasis [3].
Reprogramming of cellular metabolism has been considered as a major hallmark of cancer, it fuels fast cell growth as well as provides tumor cells with several benefits, including macromolecule biosynthesis, adaptation to the microenvironment, and an ability to cope with oxidative stress [4-6]. Warburg effect is a kind of metabolism rewiring that cancer cells shift from oxidative phosphorylation to aerobic glycolysis and many studies have reported Warburg effect plays a significant role in cancer metastasis [7, 8]. Relatively, lipid metabolism dysregulation in cancer cells has received less concern but is gradually recognized one of the significant characteristics of cancer cell metabolism which contributes to the progression of various cancers such as colorectal cancer (CRC) and hepatocellular cancer (HCC) [9, 10].
Lipids, also known as fats, contain thousands of different types of molecules, including phospholipids, fatty acids (FAs), triglycerides, sphingolipids, cholesterol and cholesteryl easters (CE) [11]. Besides their role as energy storage and critical components of all membranes, lipids could serve as mediators of cancer-relevant phenotypes that promote transformation and tumor growth [12]. Increasing numbers of studies have provided evidence that the altered lipid metabolism in malignant cells is capable of being benefit to cancer cell migration, invasion and metastasis [13]. However, the precise role of lipid metabolism remodeling in tumor metastasis and the direct mechanistic link between the two still remains to be clarified.
It is also worth noting that in addition to cancer cells, the surrounding cells in tumor microenvironment (TME) such as fibroblasts, immune cells and adipocytes may undergo lipid metabolism reprogramming as well [14-16]. The metabolic cooperativity among cancer cells or between cancer cells and stromal cells are likely to benefit cancer cell fitness in anoxic, nutrient-deficient, acidic microenvironment, which promotes the metastasis finally [17]. Also, as the common condition of TME, acidosis and hypoxia play a part in the control of lipid anabolism and catabolism via transcriptional regulation [18, 19]. On this basis, understanding the factors introducing the lipid metabolism remodeling may help find out new auxiliary therapeutic targets for cancer. Alterations in oncogenes as internal factors directly lead to cellular metabolism modulation, which is important for primary cancer cells to metastasize [20]. It is also worth noting that systemic metabolism and gastrointestinal microbiota are able to serve as extrinsic factors to regulate many metabolic processes in the host, which may finally have an effect on cellular metabolism remodeling [21, 22].
The association between lipid metabolism reprogramming and cancer development has been well established, but the global lipid metabolism reprogramming in cancer metastasis still remains elusive. Therefore, in this review we describe the altered lipid metabolism of cancer cells occurring in cancer metastasis and unravel the underlying mechanisms. We also summarize the altered lipid metabolism occurring in TME which may cooperate with cancer cells to induce progression of cancer. In addition, the possible regulations of both extrinsic and intrinsic factors on lipid metabolism remodeling in cancer cells are also discussed in this paper. We believe that revealing lipid metabolism reprogramming in cancer cells and TME will provide more opportunities for clinically directing against cancer metastasis.
Altered lipid metabolism in cancer cells occurring in metastasis
The altered lipid metabolism in cancer cells involves a series of altered enzyme activities and were considered to be associated with the metastasis of various cancers to a certain extent [3, 11]. For instance, the main enzymes participating in de novo lipogenesis include fatty acid synthase (FASN), ATP citrate lyase (ACLY) and Acetyl-CoA Carboxylase (ACC), and studies have shown that the altered expression levels of these enzymes are closely related to increased migration and invasion of cancer cells in various cancers, such as pancreatic cancer, breast cancer (BC), HCC and so on [23-26]. Also, it is reported that FASN which is highly expressed in primary CRC and liver metastases are capable of upregulating expression of a transmembrane glycoprotein implicated in cancer metastasis called CD44, thus exerts a critical effect on CRC metastasis [27]. Among the proteins participated in FAs uptake, CD36 is a dominating player in metabolic tissues which increases long chain fatty acids uptake and has a unique role of initiating the metastasis in many types of cancers [28]. The key enzyme for the fatty acid oxidation (FAO) pathway carnitine palmitoyl transferase (CPT), the abnormal expression of which has been found to be associated with cancer cells proliferation in BC, prostate cancer (PC), lung cancer and so on [29]. Inside cells, acyl-CoA cholesterol acyltransferase (ACAT) is able to esterify excess free cholesterol into CE and store it in lipid droplets (LDs), which results in high metabolic activity and avoidance of lipotoxicity from free cholesterol [30]. Additionally, accumulating CE via ACAT-1 are found to be linked with metastasis in PC, for CE may keep signaling pathways active by maintaining a low free cholesterol environment [31].
Known as the first step of metastasis, epithelial-to-mesenchymal transition (EMT) which is characterized by losing epithelial markers together with enhanced mesenchymal markers confers to cancer cells increased survival, stemness and metastasis-initiation ability [32]. It has been reported that phosphorylation-dependent ACC inhibition is a common but critical event in EMT of human solid carcinomas [25]. Cellular acetyl-CoA levels are significantly upregulated due to the inhibition of ACC and directly promote acetylation and nuclear translocation of Smad transcription factor to mediate EMT [33]. In salivary adenoid cystic carcinoma (SACC), the mesenchymal cancer cells were found to secrete more free fatty acids (FFAs) at the invasive front. While the accumulation of FFAs significantly boosts Src and matrix metalloproteinases (MMP) 9 expression, both of which induce an increase of Stat5‐DNA to enable tumor migration and invasion in mouse model [34]. In CRC, it has been demonstrated that upregulation of de novo lipogenesis plays a crucial role in rendering the capabilities of adhesion, migration and invasion to CRC cells via upregulation of sphingolipid metabolism [35]. Also, inhibition of FASN and sphingolipid metabolism significantly decreases tumor markers associated with adhesion and migration such as pMET, pFAK, PAX in mouse model [35].
Lipid metabolism remodeling occurring in cancer cells and cancer related cells.
| Cells | Lipid metabolism | Effects |
|---|---|---|
| Cancer cells | lipogenesis | CD44↑, anoikis resistance |
| Lipid uptake | Initiate metastasis | |
| FAO | Maintain redox homeostasis | |
| Cholesterol esterification | Keep signaling pathway active | |
| TAMs | Lipid uptake | immunosuppression |
| Lipid biosynthesis | IL-6↑, TNF-α↑, ROS↑ | |
| CAFs | Synthesis/catabolism↑ | Secrete FAs |
| Lipid oxidation | CCL2↑, VEGF-A↑, MMP2↑ | |
| CAAs | Release FFAs | Reprogram the metabolism of cancer cells |
CAAs, cancer-associated adipocytes; CAFs, cancer-associated fibroblasts; FAO, fatty acid oxidation; IL-6, interleukin-6; MMP2, matrix metalloproteinases 2; ROS, reactive oxygen species; TAM, tumor-associated macrophage; TNF-α, tumor necrosis factor-α; VEGF-A, vascular endothelial growth factor-A
Conceivably, after acquiring the capability of migration, metastasizing cancer cells must constantly adapt their metabolism to the distinct environment of the blood or lymph based on the nutrients available in corresponding microenvironment as long as they leave the primary tumor site [20, 36]. It was reported that the initial step of various cancers metastasis is more likely to occur though LN which may not be required for metastasis through the blood [37]. Actually, LN-metastatic tumors exhibit higher accumulation of FAs as fuel in the lipid-rich LN niche and adapted cells prefer to depend on FAO rather than glucose or glutamine oxidation as a main pathway for generating energy [38]. As for metastasizing cancer cells in circulation, it is supposed that FA metabolism is also capable of supporting the survival of circulating cancer cells for exogenous lipids could be used and metabolized by these cells [3]. It has been confirmed that oxidative stress induced by reactive oxygen species (ROS) is proved to be harmful to circulating cancer cells in vivo [39]. As the key enzyme of the FAO pathway, CPT1A plays a critical role in generating cellular NADPH to eliminate ROS by activating FAO pathway, thus grants the detached cancer cells with ability of anoikis resistance. Suppressing CPT1A by etomoxir was demonstrated to decrease metastatic formation in vivo, which provides a potential target for cancer metastatic treatment [40].
Finally, these metastasizing cancer cells extravasate and colonize into a new organ to complete the metastasis [41]. It was supposed that cancer stem cell (CSC) or metastasis-initiating cell (MIC) which owns ability of maintaining self-renewal and proliferation plays an indispensable role in this final step of metastasis [33]. When it comes to MICs, it is worth mentioning cancer cells overexpressing CD36 which are able to initiate metastasis with recapitulation of their molecular and cellular heterogeneity from the primary origin [42]. Although CD36+ cells have the function of lipid uptake, a recent study has shown that their potential for initiating cancer metastasis requires the involvement of CD36-induced FAO activation [43]. When inhibiting the function of CD36, the endogenously synthesized, unmetabolized lipids accumulate continuously in cancer cells, finally leads to metastatic lipotoxicity and cell death [43].
The lipidomic reprogramming of surrounding cells in TME
Currently, TME become to be believed as an arena where tumor cells constantly interact with various microenvironmental components, giving rise to a protean landscape in which the tumor cells, the host cells and other tumor-associated cells acquire phenotypic alterations [44]. Cancer cells are able to bypass the bloodstream and acquire nutrients by scavenging macromolecules from TME [45]. Therefore, the microenvironment becomes hypoxic when vasculatures are inadequate or cancer cells are highly proliferating [46]. Apart from cancer cells, TME is populated by highly heterogeneous groups of cells, such as cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), endothelial cells, adipose cells, myeloid-derived suppressor cells (MDSCs), and other immune and inflammatory cells [47]. In this case, cancer cells may compete or cooperate with other cells in TME for nutrients, and other cells, like cancer cells, will undergo a similar lipid metabolic remodeling to a certain extent to meet the needs of their own growth just like cancer cells, ultimately promoting metastasis and increasing invasiveness [17].
It has become clear that various kinds of immune cells accelerate metastasis, partially due to their involvement of the establishment of an immunosuppressive microenvironment within primary lesions [48]. Emerging studies have supposed that TAMs are the key cells in promoting cancer metastasis by suppressing tumor immune surveillance [49, 50]. It was discovered that in a fatty acid-enriched niche induced by cancer cells, lipid droplets (LDs) originated FAs polarize the infiltrating monocytes into M2-like pro-tumoral macrophages, facilitating cancer escape from immune surveillance [49]. Compared with control macrophages, TAMs tended to have a higher level of lipid uptake and accumulation via CD36, which accordingly promotes the FAO of TAMs to produce more energy [51]. Additionally, as a response to signals from tumor cells in TME, TAMs upregulate the lipid biosynthesis to generate more ROS and produce higher levels of extracellular cytokines like interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) and so on, which ultimately promotes cancer cell survival, metastasis, angiogenesis and immune suppression [52]. Consistently, it has been found that increased lipid biosynthesis in TAMs contribute to the production of inflammatory phenotype and ROS production which are correlated with their protumoral functions, while inhibiting key enzymes of lipid biosynthesis in the TAMs could reverse the enhanced inflammatory cytokines and ROS, reducing the capacity of promoting cancer progression [51].
As a kind of specialized fibroblasts, CAFs are considered to be the principal non-cancerous cell type within TME, which actively promotes tumor cell differentiation and support metastasis by promoting matrix remodeling and EMT [53]. In order to fit into the deficiency of nutrients and oxygen in TME, CAFs undergo metabolic reprogramming [54]. A study has revealed enhanced fatty acid synthesis and reduced catabolism in CAFs of CRC tissues, and CAFs are able to secret FAs that is took up by CRC cells for the synthesis of other lipids to accelerate the migration of CRC cells, which suggests there possibly exists a novel lipidomic interaction between CAFs and cancer cells [55]. In addition, it has been demonstrated that under the influence of CAFs, which have a higher capacity of synthesizing lipids, breast cancer cells are inclined to uptake rather than synthesize lipids, thus increasing the accumulation lipids [56]. It is also worth mentioning that CAFs in colon cancers enhance the expression of CPT-1A to actively oxidize FAs, thus enhancing the ability of CAFs to secrete cytokines such as CCL2, VEGF-A, and MMP2 which are associated with angiogenesis and metastasis of tumor cells. Recent investigation was found that blocking FAO in CAFs which overexpress CPT-1 with etomoxir obviously inhibits migration and invasion in vitro and decreases tumor growth and intraperitoneal dissemination in vivo [57].
Furthermore, it has been confirmed that adipocytes in TME affected by tumor-secreted factors leading to activated phenotypes are called cancer-associated adipocytes (CAAs) [58]. Recently, emerging studies have underlined the bilateral interactions based on adipokines and lipids, between CAAs and tumors as well as their role in tumor development, which is also considered as part of a vicious circle [59]. Researchers have demonstrated that, upon prolonged coculture with breast cancer cells, adipocytes loss almost all LDs, resulting in morphological changes toward a fibroblast-like phenotype, suggesting that FFAs could be released from these cells and transferred to cancer cells [60]. The presence of CAAs may reprogram the metabolic activity of cancer cells from glycolysis to lipid-dependent energy production [61]. Thus, after the initial interaction with lipid-loaded CAAs, tumor cells further liberate FFAs for the sake of maintaining invasive activity, which will be crucial for circulating cancer cells to reach distant organs [62].
Lipid metabolism reprogramming of cancer cells and surrounding cells in TME. In cancer cells, there is an increase in lipid metabolism such as fatty acid synthesis (FAS), fatty acid oxygen (FAO), lipid uptake, cholesteryl easters (CE) accumulation etc., which promotes cancer cell proliferation, migration, anoikis resistance and so on. Also, resulting from the accelerated metabolism of cancer cells and cancer-associated cells, the hypoxia and acidosic characteristic of tumor microenvironment (TME) in turn reprogram the metabolism occurs in cancer cells and non-cancer cells. When exposed to chronic acidosis in TME, cancer cells may accumulate fatty acids (FAs) within lipid droplets (LDs) to reduce lipotoxicity. Furthermore, tumor-associated macrophage (TAM) tends to have a higher level of lipid uptake and accumulation via CD36, which accordingly promotes the FAO of TAMs to produce more energy. It also upregulates the lipid biosynthesis to generate more reactive oxygen species (ROS) and produce higher levels of extracellular cytokines like interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) and so on, which ultimately promotes cancer cell survival, metastasis, angiogenesis and immune suppression. As for cancer-associated fibroblast (CAF), it has been found that CAF can secrete FAs that is took up by cancer cells for the synthesis of other lipids, also CAF is able to enhance the expression of CPT-1A to actively oxidize FAs, thus enhancing the ability of CAFs to secrete cytokines such as CCL2, vascular endothelial growth factor-A (VEGF-A) and matrix metalloproteinases 2 (MMP2) which are associated with metastasis and angiogenesis of tumor cells. In addition, upon prolonged exposure to cancer cells, adipocyte losses almost all LDs, resulting in morphological changes toward a fibroblast-like phenotype and loads cancer cells with lipids, which contributes to maintaining invasive activity of cancer cells.
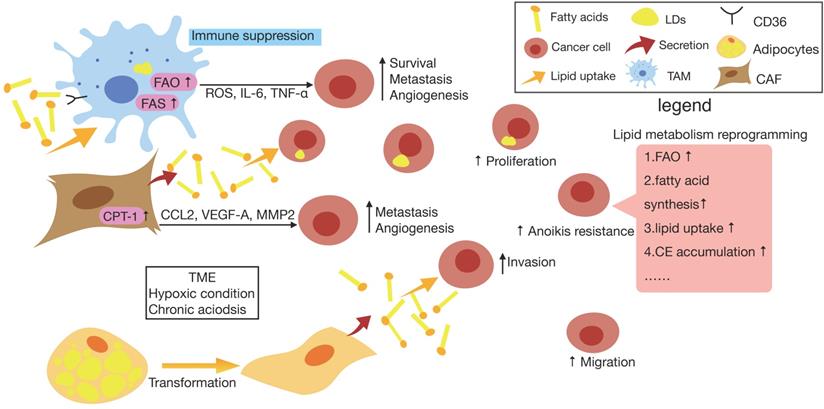
Lipid metabolism remodeling induction by extrinsic and intrinsic factors
It has been known that cancer cells are able to alter their metabolic state due to various intrinsic or extrinsic factors. Lipid metabolism reprogramming may occur in cancer cells as a result of intrinsic alterations like oncogenic mutations stimulate signal transduction components that either directly increases metabolic enzyme activity or transcription factors, thereby increasing expression of metabolic regulators [21].
Increased biosynthesis and uptake of lipids are supported by enhanced expression of the enzymes belonging to these pathways, which are regulated by a subclass of transcription factors called sterol regulatory-element binding proteins (SREBPs) [63]. Several studies have found that SREBPs are activated downstream of the oncogenic signaling pathways, primarily at the PI3K/AKT/mTORC1 and RAS/ERK/mTORC1 signaling axis [64]. The Myc oncogene and mutations of p53 tumor suppressor activate SREBP1 and SREBP2 respectively, also functioning as transcriptional coactivators for SREBP1 and SREBP2 to stimulate lipid synthesis [65, 66].
In fact, the physical and chemical nature of the TME may alter cellular lipid metabolism as a kind of extrinsic factor. Recently a study has reported that chronic acidosis in TME induces LDs formation in cancer cells and leads to more metastasis, with CD36 and diacylglycerol acyltransferase (DGAT) as key players to mediate LD biogenesis via the uptake of exogeneous FAs and triglyceride synthesis in a TGFβ2-dependent way [19]. Also, it was demonstrated that hypoxia in cancer tissue stabilizes hypoxia-inducible factor-1α (HIF-1α), which induces fatty acid uptake and lipid storage in cells, contributing to cell growth and survival [67].
Growing evidence suggests that systemic factors and their intracellular pathways may activate oncogenic signals to reshape the metabolism in cancer cells [68]. And these external signals fine-tune the cellular metabolism according to the availability of metabolites and the needs of the cell [21]. As one of the metabolic diseases, obesity generates risk factors for cancers have been identified including insulin/insulin-like growth factor (IGF) axis, adipokines and cytokines [69]. On the one hand, it was found that certain FFAs can be absorbed into cells in a CD36-dependent manner, resulting in the activation of mTOR to induce metabolic rewiring in obese-associated breast cancer cells [70]. For another, obesity tends to contribute to the development of insulin resistance (IR). Despite the fact that insulin stimulates lipogenesis and inhibits lipolysis via activating the PI3K/Akt/mTORC pathway under normal circumstances, it has been reported that in insulin-resistant states of obesity and type 2 diabetes, hepatic lipid production is increased in concert with increased hepatic glucose production, which indicate that lipogenesis persists in IR as well [71, 72]. Although signaling is impaired during IR conditions, mTORC1 which lies downstream of Akt still mediates SREBP-1c induction and function independently of Akt, therefore, lipogenic gene transcription is accelerated while glucogenic gene transcription remains alive [72].
Actually, it has been supposed that obesity and other metabolic diseases are associated closely with the gut microbiota which has been has considered as “metabolic organ”, plays a pivotal role in maintaining homeostasis of host lipid metabolism [73, 74]. Compared with conventionally raised mice, germ-free (GF) mice fed with high-fat diet are resistant to diet-induced obesity and increased lipid metabolism. In addition, conventionalization of GF mice with normal mouse microbiota is able to promote de novo hepatic lipogenesis, indicating an unneglectable role of gut microbiota in altering metabolic response [75].
On the one hand, the human gut is regarded as reservoir of lipopolysaccharide (LPS) which consists mainly of outer membrane of gram-negative bacteria, and it has been reported that the enhanced LPS levels is related to increased adipose macrophage infiltration and IR [76]. After LPS administration given to mice overnight, researchers found that the main proteins transporting FAs, CD36 and fatty acid transporter protein 4 (FATP4) decreased in response to LPS, suggesting that LPS inhibits FA absorption in vivo. And the effect of LPS on lipid metabolism was then demonstrated to be resulted from LPS stimulation induced overexpressed TNF-α in macrophages as TNF-α was able to activate caspase-3 which downregulates the expression levels of CD36 and FATP4 [77]. On the another hand, microbial processing of bile acid is supposed to result in a more hydrophilic bile acid pool and promotes excretion in the feces, while the bile acids lost in the fecal excretion also facilitates de novo synthesis from cholesterol [78]. Also, it has been confirmed that gut microbiota is able to regulate the accumulation of hepatic natural killer T cells (NKT cells) and antitumor immunity in the liver to protect against primary and metastatic tumor by utilizing bile acid as messengers [79].
Factors that introduce cellular lipid metabolism remodeling.
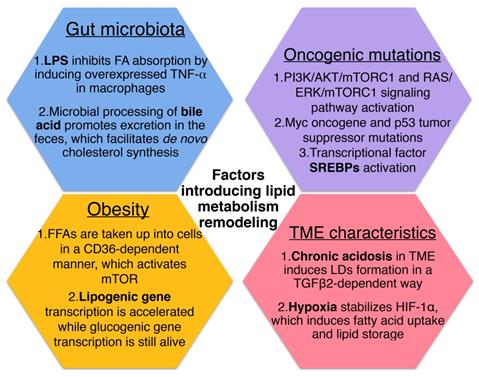
Prospective
Specific molecular features of cancers that distinguish from the normal ones were called “hallmarks of cancer” and it is supposed that metabolism reprogramming has been considered one of the hallmarks of cancer [80]. Of note, increasing studies have pointed out that a metabolic regulation of tumor progression affects many phenotypic traits of malignancy, including metastasis [81]. It has been widely recognized that cancer cells prioritize aerobic glycolysis (Warburg effect) as the main energy source and convert excess pyruvate to lactate independently from oxygen availability [82]. Actually, in addition to glycolysis, tumor cells can be expected to rewire their energy metabolism towards increased lipid synthesis to overcome therapy, an adaptation that further confers a more aggressive phenotype [83].
The so-called lipid metabolism remodeling in cells actually involves a series of altered enzyme activities which are likely to be associated with metastasis of various cancers [12]. In lipid metabolism, FA metabolism including synthesis, uptake, further processing and catabolism plays an important role in controlling cancer progression [40, 84-86]. Apart from FAs, it has been found that liver colonization of metastatic CRC cells required cholesterol biosynthesis pathways which involves key gene SREBP2 and its downstream target gene HMGCS and HMGCR, and knocking down SREBP2 significantly inhibited cancer liver metastasis [87]. While, in breast cancer, decreased intracellular cholesterol was related to metastatic behavior of advanced stage of tumor cells and low expression of SREBP2 and 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) evidently was associated with high EMT of the primary tumors [88]. These data indicate that also as an important component of lipids, the role played by metabolism of cholesterol in cancer metastasis is still controversial and warrants further research.
In recent years, the inhibitors targeting major participants in lipid metabolism such as fatty acids synthesis, FAO, and cholesterol metabolism have been broadly tested in cancer treatment [89]. As described in above content, CD36 is closely associated with tumor progression and metastatic initiation, based on which plenty of numerous studies and clinical trials targeting CD36 are being performed [90]. In a mouse model of advanced stage epithelial ovarian cancer (EOC), pretreatment of thrombospondin-1 type I repeats (3TSR) which binds to the receptor CD36 could result in tumor regression, normalized tumor vasculature, and improved uptake of chemotherapy drugs [91]. Besides, researchers have demonstrated that statins which are HMGCR inhibitors and previously used to diminish low-density lipoprotein cholesterol are also able to decrease the risk of metastatic PC and PC mortality [92].
Furthermore, exploring the factors influencing lipid metabolism remodeling may help find out new auxiliary therapeutic targets for cancer. On the one hand, it is thought that the metabolic demands of tumor cells were enhanced due to the mutational alterations in oncogenes and functional loss of onco-suppressors [93]. As mentioned in previous section, oncogenic mutations stimulate signal transduction components which can directly increase transcription factors SREBPs, in turn increase expression of metabolic regulators. Fatostatin which is a recently discovered SREBP chemical inhibitor was found to downregulate the expression of SREBP-regulated enzymes for lipid metabolism, and decrease prostate tumor growth as well as distant lymph node metastasis [94]. On the other hand, it is noteworthy that complexity of the metastatic process to some extent comes from interplays between cellular populations with various metabolic phenotypes within a given tumor, leading to metabolic symbiosis [95]. TME is an assemble of tumor cells, fibroblasts, endothelial cells, immune cells, and other stromal cells recruited by tumor cells within the surrounding extracellular matrix [17]. Resulting from the accelerated metabolism of cancer cells and cancer-associated cells, the hypoxia and acidosic characteristic of TME in turn reprogram the metabolism occurs in cancer and non-cancer cells, forming a pro-tumorigenic milieu around tumor cells [17, 96]. Possessing the ability to decrease tumor acidity, the proton-pump inhibitor (PPI) such as esomeprazole was found to inhibit tumor growth in mouse models and improve acid-related chemoresistance [97]. Interestingly, another PPI omeprazole was shown to block the FASN and dose-dependently suppress breast cancer cell metastasis [33].
To sum up, we review aberrant lipid metabolism occurring in cancer cells which plays a role in cancer metastasis. We also describe lipid metabolism alterations of surrounding cells in TME, which further contribute to cancer cell EMT, migration invasion and metastasis. This review can shed some light on the mechanisms underlying lipid metabolic reprogramming during tumor metastasis and reveal the intrinsic and extrinsic factors influencing the lipid metabolism reprogramming, and ultimately result in the identification of new therapeutic targets for cancer metastasis and improvement of patients' prognosis.
Abbreviations
ACAT: acyl-CoA cholesterol acyltransferase; ACC: Acetyl-CoA Carboxylase; ACLY: ATP citrate lyase; BC: breast cancer; CAAs: cancer-associated adipocytes; CAFs: cancer-associated fibroblasts; CE: cholesteryl easters; CPT: carnitine palmitoyl transferase; CRC: colorectal cancer; CSC: cancer stem cell; DGAT: diacylglycerol acyltransferase; EMT: epithelial-mesenchymal transition; FAs: fatty acids; FAO: fatty acid oxidation; FASN: fatty acid synthase; FATP4: fatty acid transporter protein 4; FFAs: free FAs; GF: germ-free; HCC: hepatocellular cancer; HIF-1α: hypoxia-inducible factor-1α; HMGCR: 3-hydroxy-3-methylglutaryl-CoA-reductase; IGF: insulin-like growth factor; IL-6: interleukin-6; IR: insulin resistance; MDSCs: myeloid-derived suppressor cells; MIC: metastasis-initiating cells; MMP: matrix metalloproteinases; NKT cell: natural killer T cell; LDs: lipid droplets; LN: lymph node; LPS: lipopolysaccharide; PC: prostate cancer; ROS: reactive oxygen species; SPF: specific pathogen-free; SREBPs: sterol regulatory-element binding proteins; TAM: tumor-associated microphage; TICs: tumor-initiating cells; TME: tumor environment; TNF-α: tumor necrosis factor-α.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81773065); Natural Science Foundation of Zhejiang Province (No. LY21H160023).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wan L, Pantel K, Kang Y. Tumor metastasis: moving new biological insights into the clinic. Nat Med. 2013;19:1450-64
2. Benzarti M, Delbrouck C, Neises L, Kiweler N, Meiser J. Metabolic Potential of Cancer Cells in Context of the Metastatic Cascade. Cells. 2020 9
3. Bergers G, Fendt SM. The metabolism of cancer cells during metastasis. Nat Rev Cancer. 2021;21:162-80
4. Yu L, Lai Q, Feng Q, Li Y, Feng J, Xu B. Serum Metabolic Profiling Analysis of Chronic Gastritis and Gastric Cancer by Untargeted Metabolomics. Front Oncol. 2021;11:636917
5. Li Y, Kasim V, Yan X, Li L, Meliala ITS, Huang C. et al. Yin Yang 1 facilitates hepatocellular carcinoma cell lipid metabolism and tumor progression by inhibiting PGC-1beta-induced fatty acid oxidation. Theranostics. 2019;9:7599-615
6. Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2016;73:377-92
7. Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015;356:156-64
8. Lu J. The Warburg metabolism fuels tumor metastasis. Cancer Metastasis Rev. 2019;38:157-64
9. Wang H, Xi Q, Wu G. Fatty acid synthase regulates invasion and metastasis of colorectal cancer via Wnt signaling pathway. Cancer Med. 2016;5:1599-606
10. Hu B, Lin JZ, Yang XB, Sang XT. Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Prolif. 2020;53:e12772
11. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38:27
12. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020;31:62-76
13. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L. et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16:76
14. Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T. et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene. 2019;38:4887-901
15. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N. et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28
16. Wen YA, Xing X, Harris JW, Zaytseva YY, Mitov MI, Napier DL. et al. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017;8:e2593
17. Gupta S, Roy A, Dwarakanath BS. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front Oncol. 2017;7:68
18. Mylonis I, Simos G, Paraskeva E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells. 2019 8
19. Corbet C, Bastien E, Santiago de Jesus JP, Dierge E, Martherus R, Vander Linden C. et al. TGFbeta2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat Commun. 2020;11:454
20. Kreuzaler P, Panina Y, Segal J, Yuneva M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol Metab. 2020;33:83-101
21. Dey P, Kimmelman AC, DePinho RA. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021;11:1067-81
22. Sonnenburg JL, Backhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535:56-64
23. Swierczynski J, Hebanowska A, Sledzinski T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J Gastroenterol. 2014;20:2279-303
24. Che L, Pilo MG, Cigliano A, Latte G, Simile MM, Ribback S. et al. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle. 2017;16:499-507
25. Rios Garcia M, Steinbauer B, Srivastava K, Singhal M, Mattijssen F, Maida A. et al. Acetyl-CoA Carboxylase 1-Dependent Protein Acetylation Controls Breast Cancer Metastasis and Recurrence. Cell Metab. 2017;26:842-55 e5
26. Qiao C, Huang W, Chen J, Feng W, Zhang T, Wang Y. et al. IGF1-mediated HOXA13 overexpression promotes colorectal cancer metastasis through upregulating ACLY and IGF1R. Cell Death Dis. 2021;12:564
27. Zaytseva YY, Rychahou PG, Gulhati P, Elliott VA, Mustain WC, O'Connor K. et al. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012;72:1504-17
28. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S. et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics. 2019;9:5359-73
29. Wang J, Xiang H, Lu Y, Wu T, Ji G. The role and therapeutic implication of CPTs in fatty acid oxidation and cancers progression. Am J Cancer Res. 2021;11:2477-94
30. Zhu Y, Chen CY, Li J, Cheng JX, Jang M, Kim KH. In vitro exploration of ACAT contributions to lipid droplet formation during adipogenesis. J Lipid Res. 2018;59:820-9
31. Li J, Gu D, Lee SS, Song B, Bandyopadhyay S, Chen S. et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016;35:6378-88
32. Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019;19:716-32
33. Wei Q, Qian Y, Yu J, Wong CC. Metabolic rewiring in the promotion of cancer metastasis: mechanisms and therapeutic implications. Oncogene. 2020;39:6139-56
34. Jiang YP, Tang YL, Wang SS, Wu JS, Zhang M, Pang X. et al. PRRX1-induced epithelial-to-mesenchymal transition in salivary adenoid cystic carcinoma activates the metabolic reprogramming of free fatty acids to promote invasion and metastasis. Cell Prolif. 2020;53:e12705
35. Jafari N, Drury J, Morris AJ, Onono FO, Stevens PD, Gao T. et al. De Novo Fatty Acid Synthesis-Driven Sphingolipid Metabolism Promotes Metastatic Potential of Colorectal Cancer. Mol Cancer Res. 2019;17:140-52
36. Elia I, Doglioni G, Fendt SM. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018;28:673-84
37. Ubellacker JM, Morrison SJ. Metabolic Adaptation Fuels Lymph Node Metastasis. Cell Metab. 2019;29:785-6
38. Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I. et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science. 2019;363:644-9
39. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z. et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186-91
40. Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM. et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene. 2018;37:6025-40
41. Yang J, Ren B, Yang G, Wang H, Chen G, You L. et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305-21
42. Vilahur G. New role for CD36 in metastasis through fat intake. Cardiovasc Res. 2017;113:e16-e7
43. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41-5
44. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18:359-76
45. Finicle BT, Jayashankar V, Edinger AL. Nutrient scavenging in cancer. Nat Rev Cancer. 2018;18:619-33
46. Peck B, Schulze A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer. 2019;5:693-703
47. Cheng HS, Lee JXT, Wahli W, Tan NS. Exploiting vulnerabilities of cancer by targeting nuclear receptors of stromal cells in tumor microenvironment. Mol Cancer. 2019;18:51
48. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell. 2017;32:135-54
49. Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C. et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. 2019;11:e10698
50. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12:76
51. Su P, Wang Q, Bi E, Ma X, Liu L, Yang M. et al. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020;80:1438-50
52. Rabold K, Aschenbrenner A, Thiele C, Boahen CK, Schiltmans A, Smit JWA. et al. Enhanced lipid biosynthesis in human tumor-induced macrophages contributes to their protumoral characteristics. J Immunother Cancer. 2020 8
53. Lee D, Ham IH, Son SY, Han SU, Kim YB, Hur H. Intratumor stromal proportion predicts aggressive phenotype of gastric signet ring cell carcinomas. Gastric Cancer. 2017;20:591-601
54. Wu D, Zhuo L, Wang X. Metabolic reprogramming of carcinoma-associated fibroblasts and its impact on metabolic heterogeneity of tumors. Semin Cell Dev Biol. 2017;64:125-31
55. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X. et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020;11:267
56. Lopes-Coelho F, Andre S, Felix A, Serpa J. Breast cancer metabolic cross-talk: Fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol Cell Endocrinol. 2018;462:93-106
57. Peng S, Chen D, Cai J, Yuan Z, Huang B, Li Y. et al. Enhancing cancer-associated fibroblast fatty acid catabolism within a metabolically challenging tumor microenvironment drives colon cancer peritoneal metastasis. Mol Oncol. 2021
58. Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019;129:3006-17
59. Dumas JF, Brisson L. Interaction between adipose tissue and cancer cells: role for cancer progression. Cancer Metastasis Rev. 2021;40:31-46
60. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B. et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455-65
61. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189
62. Wang YY, Attane C, Milhas D, Dirat B, Dauvillier S, Guerard A. et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017;2:e87489
63. Xue L, Qi H, Zhang H, Ding L, Huang Q, Zhao D. et al. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front Oncol. 2020;10:1510
64. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. 2016;35:1250-60
65. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R. et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244-58
66. Gouw AM, Margulis K, Liu NS, Raman SJ, Mancuso A, Toal GG. et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019;30:556-72 e5
67. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM. et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9:349-65
68. Hopkins BD, Goncalves MD, Cantley LC. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol. 2020;16:276-83
69. Chen J. Multiple signal pathways in obesity-associated cancer. Obes Rev. 2011;12:1063-70
70. Madak-Erdogan Z, Band S, Zhao YC, Smith BP, Kulkoyluoglu-Cotul E, Zuo Q. et al. Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR Signaling. Cancer Res. 2019;79:2494-510
71. Kojta I, Chacinska M, Blachnio-Zabielska A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients. 2020 12
72. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015;16:678-89
73. Wei M, Huang F, Zhao L, Zhang Y, Yang W, Wang S. et al. A dysregulated bile acid-gut microbiota axis contributes to obesity susceptibility. EBioMedicine. 2020;55:102766
74. Ko CW, Qu J, Black DD, Tso P. Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol. 2020;17:169-83
75. Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. The role of the microbiome in NAFLD and NASH. EMBO Mol Med. 2019 11
76. Caesar R, Fak F, Backhed F. Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J Intern Med. 2010;268:320-8
77. Liu H, Kai L, Du H, Wang X, Wang Y. LPS Inhibits Fatty Acid Absorption in Enterocytes through TNF-alpha Secreted by Macrophages. Cells. 2019 8
78. Schoeler M, Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. 2019;20:461-72
79. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018 360
80. Shuvalov O, Daks A, Fedorova O, Petukhov A, Barlev N. Linking Metabolic Reprogramming, Plasticity and Tumor Progression. Cancers (Basel). 2021 13
81. Nimmakayala RK, Leon F, Rachagani S, Rauth S, Nallasamy P, Marimuthu S. et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene. 2021;40:215-31
82. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019;38:2223-40
83. Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R. et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015;34:5128-40
84. Wang R, Tao B, Fan Q, Wang S, Chen L, Zhang J. et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine. 2019;45:108-23
85. Sun L, Yang X, Huang X, Yao Y, Wei X, Yang S. et al. 2-Hydroxylation of Fatty Acids Represses Colorectal Tumorigenesis and Metastasis via the YAP Transcriptional Axis. Cancer Res. 2021;81:289-302
86. Kuo CY, Ann DK. When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun (Lond). 2018;38:47
87. Zhang KL, Zhu WW, Wang SH, Gao C, Pan JJ, Du ZG. et al. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics. 2021;11:6560-72
88. Zhao Z, Hao D, Wang L, Li J, Meng Y, Li P. et al. CtBP promotes metastasis of breast cancer through repressing cholesterol and activating TGF-beta signaling. Oncogene. 2019;38:2076-91
89. Li H, Feng Z, He ML. Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells. Theranostics. 2020;10:7053-69
90. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9:4893-908
91. Russell S, Duquette M, Liu J, Drapkin R, Lawler J, Petrik J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significantly improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J. 2015;29:576-88
92. Van Rompay MI, Solomon KR, Nickel JC, Ranganathan G, Kantoff PW, McKinlay JB. Prostate cancer incidence and mortality among men using statins and non-statin lipid-lowering medications. Eur J Cancer. 2019;112:118-26
93. Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Sacca M, Ciliberto G. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37:2367-78
94. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E. et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018;50:206-18
95. Porporato PE, Payen VL, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cell Mol Life Sci. 2016;73:1349-63
96. Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F, Feron O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016;24:311-23
97. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. 2017;17:577-93
Author contact
![]() Corresponding authors: Dr. Guochun Lou, Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: 2319169edu.cn; Dr. Jun Ye, Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: wzmcyejunedu.cn
Corresponding authors: Dr. Guochun Lou, Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: 2319169edu.cn; Dr. Jun Ye, Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: wzmcyejunedu.cn