Impact Factor ISSN: 1837-9664
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
“Cancer is caused by...
Changes at some higher levels of...
There are many theories that...
Cellular differentiation may be...
Does mutation have anything to...
Our manipulations can only...
Only the neoplastic morphology...
We still have no way of directly...
There are two types of...
Immortality and autonomy may...
Benignity and malignity may be...
Above-described mouse...
Concluding remarks
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(9):2810-2843. doi:10.7150/jca.72628 This issue Cite
Review
Mutation or not, what directly establishes a neoplastic state, namely cellular immortality and autonomy, still remains unknown and should be prioritized in our research
Shengming Zhu1#, Jiangang Wang2#, Lucas Zellmer3, Ningzhi Xu4, Mei Liu4, Yun Hu5, Hong Ma6 ![]() , Fei Deng7
, Fei Deng7 ![]() , Wenxiu Yang8
, Wenxiu Yang8 ![]() , Dezhong Joshua Liao8,9
, Dezhong Joshua Liao8,9 ![]()
1. Department of Oncology, Taihe Hospital, Hubei University of Medicine, Shiyan 442000, Hubei Province, P.R. China.
2. Department of Health Management Center, The Third Xiangya Hospital, Central South University, 138 Tong-Zi-Po Road, Changsha 410013, Hunan Province, P. R. China.
3. Department of Medicine, Hennepin County Medical Center, 730 South 8th St., Minneapolis, MN 55415, USA.
4. Laboratory of Cell and Molecular Biology & State Key Laboratory of Molecular Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
5. Department of Pathology, School of Stomatology, Guizhou Medical University, 9 Beijing Road, Guiyang 550004, Guizhou Province, P.R. China.
6. Department of Oral and Maxillofacial Surgery, School of Stomatology, Guizhou Medical University, 9 Beijing Road, Guiyang 550004, Guizhou Province, P.R. China.
7. Department of Pathology, The Third Affiliated Hospital, Zunyi Medical University, Zunyi City 563000, Guizhou Province, P.R. China.
8. Department of Pathology, The Affiliated Hospital, Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China.
9. Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou Province 550004, P. R. China.
#These authors contributed equally to this work.
Received 2022-3-5; Accepted 2022-6-17; Published 2022-7-4
Abstract

Although the concept that cancer is caused by mutations has been widely accepted, there still are ample data deprecating it. For example, embryonic cells displaced in non-embryonic environments may develop to cancer, whereas cancer cells placed in embryonic environments may be reverted to phenotypic normal. Although many intracellular or extracellular aberrations are known to be able to initiate a lengthy tumorigenesis, the molecular or cellular alterations that directly establish a neoplastic state, namely cellular immortality and autonomy, still remain unknown. Hereditary traits are encoded not only by gene sequences but also by karyotype and DNA or chromosomal structures that may be altered via non-mutational mechanisms, such as post-translational modifications of nuclear proteins, to initiate tumorigenesis. However, the immortal and autonomous nature of neoplasms makes them “new” organisms, meaning that neoplasms should have mutations to distinguish themselves from their host patients in the genome. Neoplasms are malignant if they bear epigenetic or genetic alterations in mutator genes, i.e. the genes whose alterations accelerate other genes to mutate, whereas neoplasms are benign if their epigenetic or genetic aberrations occur only in non-mutator genes. Future mechanistic research should be focused on identifying the alterations that directly establish cellular immortality and autonomy. Benign tumors may have many fewer alterations and thus be much better models than cancers for such research. Future translational research should be aimed at identifying the cellular factors that control cancer cells' phenotypes and at establishing approaches of directing cancer cells towards differentiation, which should be a promising therapeutic tactic.
Keywords: Epigenetic, Genetic, Neoplastic transformation, Stem cell, Tumor classification
Introduction
An adult person has about 1-3 × 1013 cells [1, 2], with 50-70 billion cells supplanted by the newly minted ones every day [3]. However, there probably are not any two of these 1013 cells having exactly identical DNA sequences in the nuclear genome, which consists of 3.0-3.2 billion nucleotides in a diploid cell [4-6]. This is because development from a fertilized egg to an adult person involves numerous rounds of cell division, and during these divisions a huge number of genetic alterations have occurred, leastways many changes in single nucleotides [7-11]. These genetic alterations may occur via programs that had been evolutionarily entrenched in the genome, or may occur desultorily. An important piece of biological information that is less known to folks and even many biologists is that many genetic alterations, especially those occurring through genome-encoded programs, are beneficial [12-14] and required for the normal development and normal life of human beings [15, 16]. For example, sister chromatid exchanges select good genes and pass them to daughter cells while purging detrimental alleles from the genome [17]; meiosis creates haploidies so that the best parts of chromosomal DNA can be passed to the next generation [18-20], which, as already being noticed by Muller and others in the early 1930's [21], is an advantage of sexual reproduction. Moreover, establishment of acquired immunity involves genetic alterations to establish various strains of T and B lymphocytes that can respond to various pathogens [15, 22-24], and a large number of aneuploid hepatocytes are established in the liver to facilitate not only liver regeneration but also hepatic metabolism of various xenobiotics [25, 26]. Different neurons in the same brain undergo different genetic alterations during the early ages of life to specify their functions [27-30], which may be a reason why some persons are smarter than others [31].
All genetic alterations are under close surveillance and strict control by the cells because their aberrances will likely lead to pathologies, typically cancer. Indeed, a 2013 Nature paper that analyzed mutations in over 7,000 cancers averred that “all cancers are caused by somatic mutation” [32], which projects a fact that neoplasms are widely perceived as diseases brought about by genetic aberrations [33]. Actually, since 1956 mutations have been adopted by various nations' governments as a yardstick to assess cancer risk [34, 35], despite that soon afterwards it has been questioned [36-38]. A poser our body faces is that many genetic alterations are beneficial and needed, and thus should be permitted and even encouraged, but this increases the chance for the bad alterations, which are usually dubbed as “mutations”, to mistakenly pass the surveillance and remain uncorrected. If evolution had not programmed genetic alterations in our genome and had not permitted any genetic change to happen, we would have many fewer worries about various repercussions (such as cancer), but, meanwhile, we would not have an exquisite brain with so diversified neurons and would not be able to live a happy life in the Mother Nature that is fraught with pathogens and harmful materials. For instance, gene fusions often occur in leukemia and lymphoma, which in our opinion is related to the fact that their normal parental cells perform genetic modifications to evolve themselves into functional immune cells. Unfortunately, human beings face too many “buts”.
“Cancer is caused by mutation” has become a “cancer 101”
Classically, genetic alterations are stratified into three different levels, i.e., the cellular level shown as changes in the chromosome number, the chromosomal level manifested as alterations in the chromosomal structure such as a deletion or an amplification of a DNA region, and the DNA sequence level exhibited as changes in single nucleotides. Changes in the chromosome number, either hypoploidy, hyperploidy, or aneuploidy, can be regarded as an enlarged version of deletion or amplification of DNA sequences. For simplicity, these three levels of alterations are collectively coined herein as “mutation”, although in many publications “mutation” is only used to indicate single nucleotide changes. A huge number of hereditary diseases, such as Down syndrome (trisomy 21 syndrome), have been causally linked to alterations at one or more of these levels, making it clear that hereditary traits are encoded not only by DNA sequences (i.e. genes) but also by chromosomal structures and chromosome number. Heng et al have further pointed out that, while genes encode “parts” of inheritance, chromosomal structure and karyotype encode “system inheritance”, which usually is 'fuzzy” and does not follow Mendelian genetic law [39-42].
Ever since an abnormal number of chromosomes was observed in cancer cells in 1875 [43-45] and had later, during the turn from the 19th to 20th centuries, been propounded by Hansemann and Boveri as a cancer cause [44, 46-50], the idea that formation and progression of cancer are attributed to mutations has gradually become the orthodox doctrine of carcinogenesis research [51-57]. With accumulation of clinical and experimental evidence, Nordling formally proffered the first mutation theory of carcinogenesis in 1952 (published in 1953) [58] and, since then, there have been many discourses on the causal relations of various types of mutations to tumorigenesis or carcinogenesis, which are herein referred collectively to as “mutation theory”. The Science issue of November 22, 1991 was devoted to this mutation concept [59], making it even more popular in the cancer research fraternity in recent decades. In 1976 Nowell proffered a stepwise concept of mutation-caused carcinogenesis [60, 61]: A cell's genome somehow goes awry and becomes unstable, thus randomly resulting in more and more mutations when cells replicate. These mutations serve as raw materials for the cells to select the beneficial ones to become fitter mutant clones [62, 63]. In most cases cancer cells in the same patient are greatly heterogeneous in their morphology and comportment, which has also been imputed to their accrual of various mutations [62, 63]. In this “bi-phase” process, i.e. random mutations followed by clonal selection and expansion of the fitter mutant(s), genetic instability as the initial cause often results in chaotic karyotypes, at least in the cells that bear mutations in the p53 gene [40-42, 64-66].
Changes at some higher levels of genetic control may also initiate tumor formation
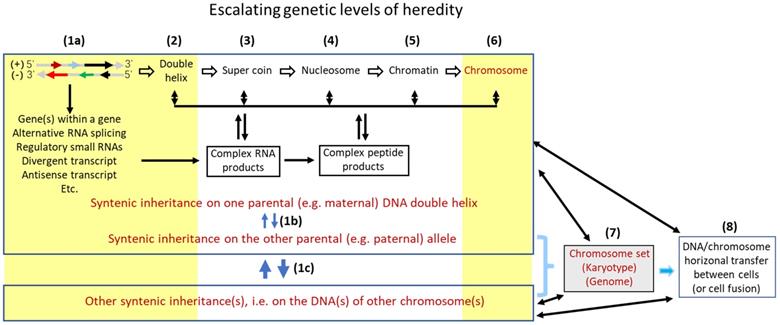
In our opinion, there are at least eight tiers of genetic control that escalate in the complexity and the influence on inheritance, with the DNA sequence as the bottom level (Fig. 1). Each higher tier encodes a set of hereditary traits that is more complex and probably “fuzzier”, compared to the set controlled by a lower tier. Karyotype studies, which analyze the structure and number of chromosomes and were popular in cancer research during the 1960's-2000's, had provided a profusion of data for the establishment of mutation theories [67], although, as pointed out by Heng et al. [41, 42], it is pity that this line of research has seemed to fade out in the past 20 years or so. The swift promulgation of DNA sequencing technology in recent decades has provided deep mechanistic insights into the effects of altered DNA sequences on tumor formation. However, in our opinion the intermediate levels between the gene sequences and the chromosomal structures shown in Figure 1 remain much understudied for their effects of alterations on tumor formation. Abnormalities at these intermediate tiers of genetic control, e.g. changes in nucleosomes, may have more systemic and thus more complicated contributions to formation of tumors, especially the malignant ones, compared with altered DNA sequences. Besides certain types of DNA mutations such as gene deletion or amplification, certain aberrant post-translational modifications of nuclear proteins, such as aberrant phosphorylations of histones, may alter some of these intermediate genomic structures, in turn altering hereditary traits or predisposing the afflicted cells to neoplastic transformation.
An oversimplified representation of multilevel structures of genomic control of inheritance. The lowest level (level 1a) is the two genomic DNA sequences that harbor genes, i.e. the plus and minus strands of DNA. On the (-) strand, the gene indicated by the short black arrow is embedded in the long-black-arrow gene on the (+) strand [68], while the gene indicated by the green arrow partly overlaps, reverse-complementarily, with the long-black-arrow gene. Another gene on the (-) strand (the long red arrow) not only has an antisense RNA (the short red arrow) but also has a divergent transcript (the short blue arrow), both on the (+) strand. The many introns of the transcripts from this genomic locus are also processed to different small regulatory RNAs (e.g. siRNA and microRNA) that are not shown in the figure to avoid overwhelming it. These genes may interact with their counterpart allele on the other parental chromosome, which constitutes another sub genetic level (1b). Furthermore, one gene on one chromosome may collaborate with another gene on another chromosome, as having been shown by many bitransgenic or double knockout models of animals, which also constitutes an additional sub level (1c). Changes at these three sub levels that involve DNA sequences have been extensively studied for their roles in tumorigenesis (denoted with a yellow-shaded area). However, several higher levels (levels 2, 3, 4 and 5) of genetic controls, i.e. the levels at the formation of double helix, super coin, nucleosome, and chromatin, each of which encodes a set of hereditary traits that is more complex than the set controlled at a lower level, have been much understudied for their effects of alterations on tumorigenesis in part due to technical constraints. Fortunately, alterations at the two higher levels (levels 6 and 7) that deal with chromosomal structure and number, such as chromosomal translocation, aneuploidy, etc., have received relatively-more extensive studies for their contribution to tumorigenesis (denoted with a yellow-shaded area), although it seems that these lines of studies have gradually faded in the past 20 years or so. Studies on the level 8, i.e. the roles of DNA horizonal transfer (including fusion of two cells' genomes) in tumorigenesis have, in general, been understudied as well.
There are many theories that dissent from the mutation one
While there are voluminous data undergirding the aforementioned mutation theory, there also are profuse biological phenomena and experimental data suggesting that mutations may not necessarily be required for cancer formation [56, 69-77] and even for the heterogeneity of cancer cells [78], as first broached by Rous in 1947 [79]. Actually, this constellation of data has led to formation of many theories and models of tumorigenesis or carcinogenesis that are collectively referred to as “non-mutation theories” herein [56, 80-94]. These divergent non-mutation theories include (but are not limited to) the “tissue organization field” theory (TOFT) [56, 91, 95-99], the “dynamic developmental disorder” theory [53, 100], the “population dynamics of cancer” theory [74, 101], the “dynamical non-equilibrium systems” theory [102-104], the “embryonic morphogenetic field” theory [94], the self-disorganization theory [105, 106], the eco-evolution, speciation, or atavism theory [107-112], the systemic-evolutionary theory [113, 114], the “cell reversal” theory [115], the karyotypic theory [116], the chaos theory [117], the pericyte hypothesis [118], the “activity paradigm” theory [119], etc. These deprecatory theories usually overlap with, or are complementary to, one another [120]. Many of them do not completely foreclose the mutation theory [121] but, instead, attempt to integrate with it to form a conflated theory, such as the “emergence framework of carcinogenesis” theory [122-126], the molecular theory [121], etc. The data as the raison d'être of these deprecatory theories are synopsized below, with the relevant history provided to our best knowledge and with our somewhat provocative perspectives elaborated. The terms “tumorigenesis” and “carcinogenesis” are used in different places in part because the described process sometimes implicates formation of benign tumors.
Dissenting evidence 1: Some biological phenomena are paradoxical to the mutation theory
Species of larger animals are supposed to have higher cancer incidences than the smaller ones, because a larger body has experienced more rounds of cell replication and thus has encountered more chances for spontaneous mutations to occur (Fig. 2). However, this is not shown in reality, as was pointed out by Peto in 1975 and thus is dubbed as Peto's paradox [127-131]. Usually, cell proliferation is quicker and more robust during the early part of life, and thus mutations and cancers should occur more often in the young if mutations are directly responsible for cancer formation. However, cancers occur more often in the elderly [132, 133], although very old individuals often have decreased cancer incidences [134-137]. Of course, there are pediatric cancers, which are likely initiated during the embryonic stage [138, 139] and occur via different mechanisms from those for the sporadic cancers in adults [140]. A related perplexity is called “the proliferation paradox” [141]: Some cell types that have the fastest turnover in the human body have the lowest cancer incidences [141], epitomized by the epithelial cancers in the hair follicles and the small intestine. Epithelial cells of the hair follicles grow so fast that men need to cut their hair once a month, but these cells rarely develop to cancer [142, 143]. Small intestine makes up 75% of the length and 90% of the mucosal surface area of the digestive tract [144], and the lifespan of its mucosal cells is so short that the cells are supplanted by the newly minted ones every 3-4 days [3, 145]. However, the incidences of epithelial cancer in the small intestine, especially in the jejunum and ileum, are extremely low [144, 146-148], in stark contrast to the cancer incidence in the colon and rectum that are much shorter in length with their liner epithelia having a much longer (5-21 days) lifespan [3, 145].
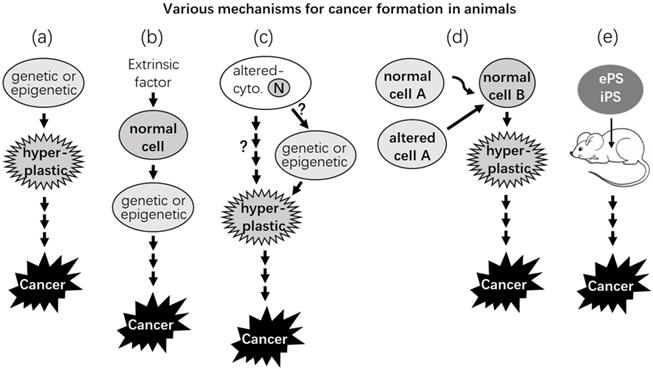
Almost any aberration inside or outside a cell, mutational or not, may initiate cancer formation. (a) A primary cell may bear certain epigenetic or genetic alteration(s), such as one inherited from a parent, that enable the cell to proliferate and form a hyperplastic lesion while gradually becoming cancerous. (b) Certain extrinsic (extracellular) factors, such as radiation, a chemical, a virus, or an abnormal endocrine or paracrine signal, can cause genetic or epigenetic change(s) in the nucleus of a cell, either directly or via altering certain cytoplasmic factor(s), and make the cell cancerous, as in (a). (c) Hypothetically (the question marks), certain factors in the cytoplasm may become abnormal, due to such as an unhealthy lifestyle or aging, which renders the cell hyperplastic either directly or by causing genetic or epigenetic alteration(s) in the nucleus (N), driving evolution of the cell to a cancer. (d) Some cytoplasmic or nuclear alterations of some cell(s) (such as stromal cells) may alter their communications and interactions with other (such as epithelial) cell(s). The alterations may direct evolution of the latter cell(s) to cancers while the former cells remain phenotypically normal. (e) A normal embryonic or induced pluripotent stem cell (ePS or iPS) may develop into a cancer at an ectopic (i.e., extrauterine) place in adult animals.
There are many genotoxic agents that are not carcinogens [149], whereas a large percentage of known chemical carcinogens are non-genotoxic, such as chloroform and p-dichlorobenzene [150, 151]. Endogenous hormones can beget benign or malignant tumors when they are present in an aberrant amount, which can be achieved using simple surgeries such as partial thyroidectomy [138, 152, 153], gonadectomy, and transplantation of gonads to an ectopic body site (such as to the spleen) [154-157], as we have reviewed before [138, 158-160]. Obviously, endogenous hormones and simple surgeries cannot be considered mutagenic. There are too many other factors that are not mutagenic but can increase risk for cancer, such as obesity and certain unhealthy lifestyles.
There are some cancers in which no recurrent mutations could be identified [161, 162], and there has not been any proven set of mutations known to transform a normal cell to a cancerous one [163]. More bewilderingly, there are some oncogenic driver mutations appearing in benign diseases at a high frequency, sometimes even much higher than in malignant tumors [164-166]. There also are cancer-driver mutations that are found in normal cells or culminate in only clonal proliferation of normal cells, but not cancers [132, 133, 167, 168]. A conjecture on these observations is that, besides causing neoplastic transformation, these mutations can also improve fitness of relatively old cells and thus extend their life span; therefore, there is no need for the mutations to drive these fitter cells to a neoplastic state [132, 165, 168]. All of the observations enumerated above do not seem to be consonant with the mutation theory, although there may be other explanations.
Dissenting evidence 2: Altered extracellular milieu may initiate carcinogenesis
There have been several lines of experimental data intimating that abnormal extracellular signals from the matrix or from other cells may initiate neoplastic transformation [169, 170], which occurs even in evolutionarily very low animals like metazoan Hydra [171, 172]. Actually, there is a theory opining that cancer is a problem in intercellular communication [173]. One line of advocating data is derived from many animal studies showing that implantation of various foreign bodies can cause tumors, mainly sarcomas [174-177], which was first reported by Turner in 1941 who fortuitously found that subcutaneous implantation of Bakelite disks in the rat caused sarcoma at the site of implantation [178]. Implantation or chronic injections into animals' peritoneal cavity of different non-mutagenic materials, such as solid plastics, mineral oil, and certain immunological adjuvants, can induce plasmacytomas, which has been reviewed by Potter decades ago [179-181]. These implanted materials fall into various categories, including metal, plastic, polymers, millipore filters, nitrocellulose, etc., and are insoluble and not toxic [182-188]. Moreover, the carcinogenesis does not seem to correlate with the amount (dose) of the implanted materials, but rather is related to their physical shape or surface [176, 188-190]. Therefore, the carcinogenesis does not seem to occur via mutations caused by the intake of the materials into the cells but rather occurs due to disturbances to the extracellular milieu (Scenarios b and d in Fig. 2). It needs to be emphasized that such foreign-body-caused carcinogenesis has its human relevance. For example, there have been over 800 cases of “breast implant-associated large cell lymphoma” reported in the literature [191]. Moreover, soft tissue malignancies caused by foreign bodies derived from shotgun blasts have also been reported [192]. Relevant mechanistic studies in the past 80 years suggest that the carcinogenesis is likely to be ascribed to the chronic inflammation ignited by the implanted materials, such as the involvement of macrophages, plasma cells, and other inflammatory cells as well as various cytokines and other factors released by these cells [174, 189, 193]. Actually, Miller has already shown in 1931 that injections of tuberculo-proteins into the peritoneal cavity of rabbits can induce nodules in the omentum that contain “undifferentiated cells” [194], which are neoplastic in pathology term. In our opinion [138], this carcinogenesis is likely elicited via chronic inflammation, but not mutations, caused by the bacterial proteins, and supports Rudolf Virchow's theory that cancer results from chronic irritation [195-198], mainly inflammation [199-201].
Another line of espousing data is derived from experiments of transplantation of tumor tissues into normal animals [202-204]. According to Staab [205] and Goldenberg [206], during 1905-1907 Ehrlich, Apolant, Loeb, Bashford, and a few others had reported in German language that transplantation of mouse mammary carcinoma into other mice could culminate with sarcomatous transformation of the recipient's stromal cells. Actually, in the 1902 Loeb had already noticed that the regrowing tumors in the recipient animals were sarcomas although the original tumors implanted were carcinomas [207], which suggests a possibility that the tumors occurring in the recipient animals may not really be a regrowth of the grafted tumor but may rather be a new tumor derived from a different cell lineage. Unfortunately, the importance of these earlier observations had been ignored for decades, and it was only in the 1970's was it shown in a series of studies that inoculation of surgically-removed human cancer tissues into immunodeficient mice, rats or hamsters, followed by poly-passages of the transplanted tumor tissues from one animal to another, can cause sarcomatous transformation of the rodents' stromal cells within the grafted human tumor tissues [205, 206, 208-213]. The detailed mechanisms underlying this horizontal transformation of malignancy remain nebulous even now. Possible explanations include that certain transforming-genes have been horizontally transferred from the primary tumor cells to some recipients' cells [213-219], that spontaneous mutations have occurred in some recipients' cells, and that immunodeficient animals have already borne certain mutations that drive malignant transformation of their stromal cells. However, in our musing these possibilities are improbable, partly because inoculation of well-established cancer cell lines [206, 220], such as several subclones of Hela cells [221-223], into nude mice culminate only in metaplasia, typically bone or cartilage formation, and not neoplastic transformation, of the recipients' cells. These discrepancies between inoculation of a cancer tissue and inoculation of a single cancer cell line intimate that heterogeneous populations of cancer cells and/or connective tissue components in the donor cancer may contribute to the neoplastic transformation of the recipients' stromal cells.
Ever since 1951, another set of tissue graft studies has also reached the conclusion described above: Billingham et al repeatedly painted some areas of mouse skin with 20-methylcholanthrene, a chemical carcinogen, and then removed the epidermis and implanted it onto an untreated area of dermis with the epidermis pre-removed [224]. Unlike other painted areas that developed many tumors, no tumors developed at the transplanted epidermis. Conversely, if implanting a pad of epidermis from an untreated area to a treated area with the original epidermis pre-removed, tumors would develop at the untreated epidermis. Obviously, the tumorigenesis in the graft of untreated epidermis is begotten by the deeper, carcinogen-treated tissues [203, 224, 225]. Similarly, non-tumorigenic COMMA-D cells inoculated into a mouse mammary fat-pad that was previously irradiated and cleared of epithelial cells developed to cancer [226-229]. Normal rat mammary epithelial cells inoculated into a mammary fat-pad of a rat that was previously treated with the chemical carcinogen N-nitrosomethylurea developed to cancers as well [56, 92, 98, 230]. In these experiments, mutations may still contribute to the tumorigenesis, but in such a way that a cell or cells bear mutation(s) and therefore keep providing a disturbing signal to other cells, eventually making the latter neoplastic (Fig. 2). Supporting this conjecture, normal ovaries grafted into the spleen [154, 156, 231] or tubal eggs grafted into the testis [232] have since the 1940s been shown to develop tumors. The ectopic site, i.e., the spleen or the testis, should not be mutagenic, but it provides a long-lasting disturbance to the grafted cells. Mention should be made of similar results from many unethical (likely criminal by today's law) studies involving inoculation of cells directly into human bodies performed mainly during the 1940's-1960's [233-237]. For instance, it had already been reported in Science in 1956 that subcutaneous inoculations of cultured human epithelial cell lines into forearms of human “volunteers” could lead to tumor formation, although the tumors eventually regressed [238, 239], likely due to immune clearance by the recipients [240]. The normal recipients' forearms are not mutagenic but can still neoplastically transform the inoculated epithelial cells.
Dissenting evidence 3: Primary rodent cells readily immortalize themselves in vitro
Normal somatic cells have allegiance to the host's body and are mortal, as they have lifespans. A neoplastic state of a cell means that the cell has lost its allegiance to the host's body [111, 138, 139, 241, 242]. In other words, a neoplastic cell, benign or malignant, has become autonomous and maintains itself as a unicellular organism by interminable symmetric division, namely becoming immortal [243, 244], just like a bacterial cell that keeps symmetrical division to maintain its strain [111, 138, 241]. Unfortunately, many studies on neoplasms do not stick with this “immortality and autonomy” definition of neoplasia [138] but describe tumors in various ways, as summarized by Gatenby et al [245] and Soto et al [246]. Some lesions that are described as benign tumors in pathology textbooks are not actually neoplastic because they are not immortal. For instance, many osteochondromas cease growth and even diminish after skeletal maturity [247-250], and thus should be considered as developmental malformations, but not neoplasms. A caveat is that in reality, every sizable tumor mass has a sheer number of neoplastic cells that are either dying or already dead (mortal) or have already committed to mortality, i.e., have lost the ability of interminable self-renewal, due to variegated reasons such as desultory development of lethal mutations or insufficient supply of oxygen or nutrients. This reality should not disqualify immortality and autonomy as the cannons for neoplasia. Unfortunately, as we have pointed out and discoursed recently [138], it has led to a wide misconception in cancer research that only a tiny number of cells in a cancer mass encompass the self-renewing ability and these cells should thus be specifically classified as “cancer stem cells” to be distinguishable from the remaining vast majority of cancer cells.
The neoplastic nature of cells in vitro is usually referred to as “neoplastic transformation” or just “transformation”, which is equivocal as it does not clearly announce whether the “transformed” cells are immortal and/or autonomous [251]. Cells in culture dishes can only be evaluated for their immortality with their ability to be passaged endlessly, whereas their autonomy cannot be assessed, unfortunately, because no allegiance to the host animals is involved [111, 138, 241]. Actually, even “unlimited passage” is difficult to assess as it requires continuous passage for a long period of time, and currently we still lack a feasible approach to determine the turning-point from mortality to immortality of cells in culture.
Carrel and his associates Burrows and Ebeling had since the 1910's presented a series of publications claiming their successful in vitro immortalization of chick embryonic fibroblasts by continuing adding chick embryo extracts into the culture of chick embryonic heart tissue, although many contemporaries questioned this world's first success in transforming cells in vitro [252-255]. Nevertheless, there have since 1940's been many studies showing that in vitro culture can easily transform primary cells of small rodent origins [71], especially the hamster and mouse [256-260], as the cultured cells can form tumors when injected into syngeneic animals. A so-called “3T3 protocol”, namely transferring 3 x 103 cells from one flask to another every 3 days, had been established in the 1960's as an effective procedure to immortalize primary mouse fibroblasts, especially those from early embryos [261-263]. Rodent epithelial cells can easily transform themselves in vitro as well, which has been postulated to be due in part to the disruption of their interactions and communications with stromal cells. Moreover, isolation of epithelial cells detaches them from the basement membrane, which has been known for decades to facilitate immortalization [106]. In general, immortalization or neoplastic transformation of primary cells is much more efficient, once estimated to be 1010 times better [264], in vitro than in vivo [105, 106]. Treatment with various non-mutagenic agents can facilitate in vitro immortalization and neoplastic transformation. For example, a low dose of hydrogen peroxide can cause a transformation [265].
The cell culture situations enumerated above are stressful to the cells, which is likely to cause chaos of the karyotype, especially when the p53 gene is also mutated, as observed by Heng et al. [40-42]. However, in our opinion the stress itself may not be mutagenic; epigenetic alterations may more likely be the initial events occurring in the cultured cells whereas genetic changes, if they occur, may be the secondary and spontaneous ones. Indeed, it has been known that many spontaneously-established cell lines show deletion in the INK4a/ARF locus [266-268], besides methylation of the p16 gene within this locus [269, 270].
Dissenting evidence 4: Mutations rarely transform cells in animals
Although some inherited mutations are associated with higher tumor incidences [271-273], one particular inherited mutation culminates with only one or several tumor masses in only one or several cell or tissue types. For instance, an inherited mutation in the Rb gene may cause retinoblastoma, but often only one tumor is developed, although not only all of the retinal cells in both eyes but also all of the cells in the patient's body bear the mutation [274]. This means that there are inherent factors in the body that prevent the vast majority of the incriminated cells from the mutation-initiated carcinogenesis [132]. This phenomenon can also be discerned in most animal models of solid tumors with genetically manipulated mice, as we have repeatedly pointed out before [6, 138, 275]: In these models, although all animals may develop the anticipated tumor, usually each animal develops only one or several overt tumors during the life span, despite that the target organ of the animal, such as the liver or the mammary glands, have trillions of cells that bear the same genetic modification, as having been noticed decades ago [276, 277]. For instance, only 4 or 5 islets in the pancreas of SV40-LT transgenic mice develop β-cell tumors [278], and only 1 among 10 mammary glands in c-myc transgenic mice develops a tumor [279, 280], although we did occasionally find two or three mammary tumors in a mouse in our lab (empirical experience of DJ Liao). To our knowledge, these two transgenic lines are already ones of the best models of carcinogenesis as they produce the highest tumor incidences. The fact that only one to several out of trillions of targeted cells in the same animal are transformed early enough for the cells to develop to overt tumors signifies that the genetic manipulation as an artificial mutation has negligible efficacy in neoplastic transformation [6, 138, 275]. The discrepancy between 100% tumor penetrance at the animal level and the negligible transformation efficacy at the cellular level is reminiscent of the situation in the human being that “cancer is so common a disease yet so rare at a cellular level”, as pointed out by Ferrell Jr et al [281]. Indeed, one of five people will likely develop cancer in his/her lifespan [142, 143], which is horrible. However, since one person has 1-3 x 1013 cells with 50-70 million cells supplanted by newly minted ones every day [2, 3], this still means that the rate of cellular neoplastic transformation is negligibly low, much lower than 1/1013.
Dissenting evidence 5: Mutations are not required for showing neoplastic properties
Benign neoplasms are already immortal and autonomous, and malignant neoplasms have three additional features, i.e., 1) encroachment into their normal adjacent tissue, which can be considered as local metastases, 2) consumption of their normal surrounding tissue, which can be regarded as a cannibalism at the cellular level [282-286], and 3) metastasis to distant body site(s). The mutation theory contends that a normal cell develops mutation(s) to evolve to a neoplasm, and then develop more mutations to acquire the three malignant features. However, none of the five neoplastic properties are unique to malignant cells, and not even to benign cells, as these cellular properties are developed along with evolution from prokaryotic to eukaryotic and then to multicellular organisms. In other words, the genomes of animals (including the human being) encode these cellular properties and thus do not need mutations for their occurrence [62, 287, 288]: First, a normal human body consists of not only somatic cells, which are mortal and may undergo symmetric division, but also germline cells that are immortal and undergo asymmetric division [289, 290]. Actually, there are some plants and animals that are immortal as well [290, 291]. Therefore, immortality has been evolutionarily built within, or encoded by, our genomes although normally the program is derelict in somatic cells. An intriguing but unsolved question is how autonomy is related to immortality and whether it is also evolutionarily built within the genomes of multicellular organisms. In our cogitation, immortality and autonomy are the two sides of the same coin for neoplastic cells, meaning that they are controlled by the same factors that are currently unknown to us [138], as there is no evidence showing extrication of immortality from autonomy in human tumors. Second, invasion is an evolutionarily-developed cellular comportment seen widely in normal cells of animals and plants [292]. For instance, normal trophoblasts are highly invasive [293, 294] and can make inroads into the uterine wall to establish gestation and may even encroach into blood vessels and home in on the lungs of the mother and many organs of the newborn [295]. Third, macrophages and even some other cell types like epithelia can engulf other cells and materials in their surroundings; osteoclasts function to eat up bone tissue [296, 297]. Fourth, many bone-marrow-derived or thymus-derived cells can enter, i.e., “metastasize”, into the blood or lymphatic circulation and home in on almost anywhere in the body. Probably because of this property, in all pathology textbooks neoplasms of the bone-marrow and lymphatic origins are all classified as malignancy without exception. Fortunately, this property seems to have its benefits: Because these neoplasms, which usually are liquid cancers, do not need to experience additional cellular or molecular changes to be metastatic, many of them have fewer alterations and are easier to cure, compared to many solid tumors [298]. Actually, during embryonic development many cells migrate, with an instructive embodiment already described by Markert in 1968: “…melanoblasts originating in the neural crest migrate through many tissues of the body before reaching the terminal locations in which they complete their differentiation into nondividing, nonmigrating melanocytes” [288]. As we have described before [111, 138, 139] and in this essay, carcinogenesis is an atavistic process and cancer cells resemble embryonic cells in morphology and comportment, and metastasis of cancer cells may be considered as showing behavior of embryonic cells. Actually, for this reason pathologists use embryological terms, such as “undifferentiated”, “poorly differentiated”, “differentiated”, etc., to describe neoplasia [299].
According to the systemic-evolution theory of Mazzocca et al [113, 114, 300-302], other neoplastic properties, such as fermentative glycolysis, are also entrenched in the genomes of eukaryotes that evolve from fusion of two different types of prokaryotes, with one of the erstwhile types now being represented by the nucleus and the other being represented by the mitochondria. This explains why cancer cells sometimes, but not always, manifest fermentative glycolysis. Many intracellular or extracellular disturbances, including epigenetic perturbations, may reactivate some of these derelict programs, making the cell stay at or return to an embryonic (or stem) stage to become neoplastic [303-305]. From a vantage point of logic, even very egregious cancer properties do not need to be derived from mutations [56], because they have already been entrenched in the normal genome, mostly in the genomic DNA and, probably, mildly in the mitochondrial DNA. Of course, some mutations may bestow these properties upon cells while some other mutations (such as a deletion) may make them disappear. This point of view is not just a logical inference but has actually been buttressed by many experimental data, as have been summarized by Pierce in 1983 [306].
Dissenting evidence 6: Pluripotent stem cells may develop into cancer at extrauterine sites in adult animals
According to Needham [307], Belogolowy showed in 1918 that morulae and blastulae of anuran amphibia implanted into tissues or body-cavities of adult frogs developed into “round-celled sarcoma” that penetrated into the surrounding tissue and metastasized to the liver and lungs. Also according to Needham [307], Skubiszewski reported in 1926 that injection of chick embryonic tissue into chicken muscle or other tissues produced similar “round celled sarcoma”. In the 1930s, both Needham and Thomas observed oocyte-caused tumors in adult worms [307]. Witschi in the 1930s showed that if frog eggs were kept for a prolonged period of time before fertilization by sperms, which is referred to as over-ripeness, the eggs would produce teratomas or teratocarcinomas [307-309]. In 1960's, Steven et al showed that, when germinal stem cells from early embryos of male mice of the 129-strain were transplanted into testicles of adult mice, the cells developed into teratomas or teratocarcinomas (Fig. 2) [232, 310, 311]. As reviewed by many pundits [312-327], many other researchers have later confirmed that early embryonic pluripotent stem (ePS) cells, including those of human origin [316], placed into several extrauterine sites of adult animals can indeed develop into teratomas or teratocarcinomas [315, 328-332]. Sobis et al [333-340] and Hirai et al. [341] have also shown that displacement of yolk sac cells in fetectomized placenta induce teratomas and teratocarcinomas in small rodents.
A host of studies in the past decade or so have confirmed and extended the earlier findings mentioned above on the development of teratomas or teratocarcinomas from induced pluripotent stem (iPS) cells [312, 342-347]. It is now clear that either ePS or iPS cells may develop into teratomas and even teratocarcinomas if the cells are placed ectopically, i.e., at an extrauterine site of adult animals (Fig. 2). The tumorigenic mechanisms, according to Rose's work in 1955 with embryonic and adult frogs, may involve inhibition of differentiation of the pluripotent cells by the adult tissue matrix [348]. As extrauterine sites in animals should not be mutagenic, this tumorigenesis or carcinogenesis may not involve mutations. Moreover, the tumor formation can be greatly minimized or prevented by various manipulations [312, 344, 345, 347, 349], which also favors the perception that the tumorigenesis is mainly precipitated by the non-mutagenic microenvironment.
Dissenting evidence 7: Embryonic environment may revert cancer cells back to normal
Mutations are in general considered irreversible [92], although sometimes polyploidy of cancer cells may be reversible [350, 351] and in some rare cases single nucleotide mutations may mutate back to the wild type [22], which had been described already in 1940's [352] and coined as “reverse mutation” or “back mutation” [353-357]. The irreversibility of mutations dovetails with the fact that human cancers rarely regress spontaneously. Of course, there are some rare cancer subtypes showing high frequencies of spontaneous regression with unclear reasons, such as the stage IV-S of neuroblastoma [358-360], some indolent histologic subtypes of non-Hodgkin's lymphoma [361], and some subtypes of cutaneous malignant melanoma [362, 363]. A caveat is that many precursor lesions in animal models of carcinogenesis [138] and certain outgrowing lesions of humans [105, 106] can regress via apoptosis because these lesions are still mortal and have not yet become authentic neoplasms [138].
While ePS cells may develop into cancer in extrauterine matrices, ever since 1907 [364, 365] a myriad of animal studies have also shown that cancer cells may be reverted back to normal in an embryonic environment as well (Fig. 3). When teratocarcinoma cells were injected into mouse blastocysts, the cells became incorporated into the developing embryos; organs and tissues of the adult mice developed from such embryos consisted of cells from both the normal blastocyst and the cancer (Fig. 3) [94, 306, 366-376]. Actually, a similar observation was already made in 1907 by Askanazy [377] who, according to Telerman [378], showed that ovarian teratoma cells could differentiate to normal tissues that contained embryonic germinal layers. Since the late 1950s, Pierce and his colleagues have further shown that a single cell of teratocarcinoma or some other cancer types can develop to the three major germ-cell layers of embryos [326, 379-385]. After being frozen-and-thawed in vitro for many times, cells of teratocarcinomas that were derived from mouse embryonic cells could still be made to develop to gametes, and the oocytes or sperms could generate normal progeny [386, 387]. Cells of other tumor types such as leukemia and neuroblastoma have been shown to be regulated by certain embryonic fields as well [306, 388, 389]. However, it seems that the embryonic environment's control over malignant phenotypes has its specificity, since the blastocyst fails to control certain leukemia and sarcoma cells [376] and only tumor cell types with a normal cellular counterpart in the blastocyst could be well controlled [306]. More interestingly, treatment with Zebrafish embryo extracts, alone or in combination with certain chemo-drugs, has been shown to have therapeutic effects on liver cancer and breast cancer both in the lab and in clinical trials [390-394]. The microenvironment of mammary tissue can direct differentiation of breast cancer cells as well as normal cells of certain tissue origins (such as testes and nerves) [395, 396]. A regenerating mammary gland can also provide a special milieu in which human breast cancer cells can be reverted to mammary epithelial cells [397, 398]. These mammary-gland-related data are somewhat related to the effects of embryonic environments, because the mammary gland is special as it starts to develop only at the pubertal age and becomes mature only after parturition.
Several modes of reverting cancer cells back to normal. (a) If a mouse cancer cell is injected into a mouse blastocyst, it would develop together with the embryonic cells into an embryo and then to a fetus. (b) If a nucleus isolated from a Lucké cancer cell of frog origin is injected into a denucleated frog egg, the chimeric egg can hatch a normal tadpole, showing that the normal cytoplasm of the egg overrides the cancerous nucleus in controlling the cellular and organic phenotypes. (c) Fusion of a normal cell with a cancer cell may make the hybrid phenotypically normal. However, removal of certain chromosome(s) from the hybrid that has already been normalized may revert it back to the cancer phenotype again [415-424].
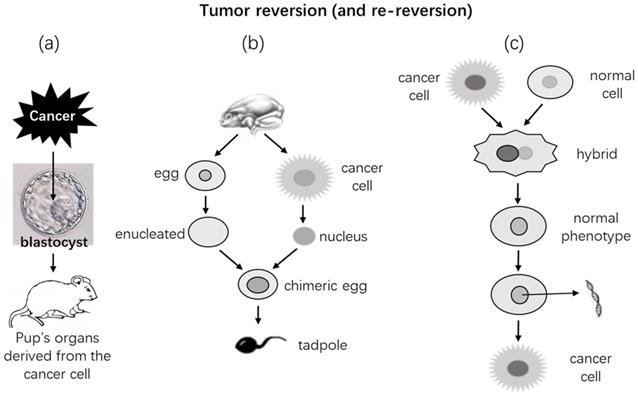
It had been shown in the 1960's that if nuclei isolated from the Lucké renal cancer cells of frog origin [399-402] were injected into enucleated frog eggs, the chimeric eggs could hatch phenotypically normal tadpoles (Fig. 2) [403-413]. Further transplantation of tissues from these tadpoles into normal recipients produced phenotypically normal tissues as well [407]. Similarly, if nuclei isolated from cells of mouse medulloblastoma are injected into enucleated mouse oocytes, the chimeric eggs can develop to embryos in recipient female mice, and the embryos can survive for 8.5 days of the embryonic stage with various normal embryonic tissues and without showing any neoplastic features [414]. These observations further extend the aforementioned in vivo findings by suggesting that an extranuclear milieu, i.e., the cytoplasm, of normal embryonic cells or eggs can override the nuclear genome in controlling the cellular phenotype. Therefore, leastways in these experimental settings, even if the nuclear genome bears oncogenic mutations, the mutations may not inevitably lead to neoplastic phenotypes. Many experiments have also shown that fusion of a cancer cell with a normal cell can make a phenotypically normal hybrid [415-424]. However, mention should be made of that in many other occasions the hybrid cells may turn out to be more malignant [425]. Actually, according to the reviews by Pawelek [426, 427] and Dittmar [428], already in the 1911 the pathologist Otto Aichel had proposed in a German language paper that fusion of tumor cells with leukocytes rendered the hybrid cells aneuploid and metastatic. Indeed, many studies have later shown that cell fusion may be a mechanism for tumor initiation and progression. It is worth mentioning that cell fusion is also a physiological event programmed in the mammalian genomes and occurring more often during embryonic stages [427-429]. Since such hybrid cells contain both normal and neoplastic nuclei, whether and how this complicated system is related to embryonic milieu are unclear. The Parrondo's paradox, which says that losing strategies can work together to produce winning outcomes [425, 430], leads us to wonder whether the hybrid that doubles its number of chromosomes is fitter than the two original cells.
Mention should also be made of the plant evidence of the reversion, which has already been thoroughly reviewed by Braun in 1981 [431]. It has been shown, ever since 1926, that tumor cells in some plants can be reverted to normal plant cells and that tumor cells grafted onto another plant can develop into a normal plant which can bloom and produce seeds; the seeds can then germinate and grow to normal plants [432-453]. What remains unknown is whether (or how) these observations on plants relate to the above-described data from embryonic environments of animals.
The above-described antithetic relationship between ePS or iPS cells and embryonic matrices, i.e. that stem cells in non-embryonic environment develop to tumors whereas embryonic environment reverts tumor cells back to phenotypical normal, extends the “seed and soil” theory that was initially proposed by Paget in 1889 to explain tumor metastasis [454]: ePS or iPS cells as “the seeds” develop to normal tissues in one soil (embryonic environment) but to tumors in another soil (non-embryonic environment). Moreover, when the soil has changed (to an embryonic environment), the product of the seed (i.e. the tumor) may be changed (back to normal). These extended explanations of the theory favor the non-mutation theory as it is the environment (the “soil”), but not the cell (the “seed”) itself, decides whether the cell should develop into a tumor and, if it has already become a tumor, whether it should return back to normal again. The antithesis also dovetails with the initial “cancer stem cell” theory described by Julius Cohnheim in the 1870's [455, 456], which proposes that cancers are derived from stem cells in the normal tissues. Mention should be made of that there is a different “cancer stem cell” concept proffering that a tumor mass contains some cells that encompass properties of normal stem cells, such as self-renewal ability [138].
Dissenting evidence 8: Immortality can be disengaged from transformation and other neoplastic properties in the lab
Some researchers have shown that cellular immortalization occurs before, and is a prerequisite of, neoplastic transformation [256-260, 457-461], which is the punditry of some other cancer wizards as well [258, 462-466]. Ample animal studies have accentuated that tumor development undergoes a two-step procedure of initiation and promotion; in some peers' opinions, “initiation” immortalizes normal cells whereas “promotion” transforms the immortalized cells [467, 468]. However, in 1983, Land et al showed that a mutant ras gene could transform embryonic fibroblasts in vitro, as these ras-expressing cells could form colonies in soft agar, but the transformed cells were still mortal because they could not grow indefinitely in the culture; their immortalization required concomitant expression of the c-myc or a viral oncogene [469]. Similarly, mouse embryonic fibroblasts transformed with the SV40 large T antigen can efficiently form colonies in soft agar, but most of the cells will eventually die [470, 471]. Concomitant expression of the CDK4 gene and a ras mutant can confer upon primary cells the ability to form colonies in agar and to develop into invasive tumors in animals, but the transformed cells remain mortal as evidenced by their limited passages in culture [472]. These data suggest that in vitro neoplastic transformation can occur before, and thus can be extricated from, immortalization. Other studies have also shown this segregation [473], and there are data showing that simian virus 40 can transform human cells without immortalizing them [474]. Telomerase alone has been shown to prod primary cells into growing in agar and in animals, which together is a well-accepted emblem of a neoplastic state, but these effects of telomerase are independent from immortalization [475-478] and transformation [467, 479]. In some animal experiments, epithelial cells can be manipulated to invade, disseminate, and enter into the bloodstream before they can form primary tumors [480, 481]; mammary epithelial cells can be manipulated to metastasize and colonize in the lungs before they are malignantly transformed [482, 483]. All of the abovementioned laboratory data seem to suggest that immortality, transformation, invasion, and metastasis as key neoplastic properties can occur independently of one other and in any order, depending on the experimental setting. Because epigenetic alterations are reversible and occur more easily than mutations, it is much more easily fathomable if each of these key neoplastic features is caused by epigenetic alterations, and not by mutations, and thus can occur earlier or later than other neoplastic features. Of course, as aforesaid, other plausible explanations exist.
Dissenting evidence 9: A neoplasm is a unicellular species and is somewhat genetically stable
As described earlier in this essay, the mutation theory says that cancer cells are genetically unstable and thus continuously accumulate mutations while endlessly replicating, leading to genetic heterogeneity [62, 63]. It is therefore envisioned that over a long period of time each tumor lineage should have accumulated too many mutations that are too huge a burden for it to survive, meaning that no lineage of tumor, especially a very malignant one, can survive for a long time. However, a canine transmissible venereal tumor has survived for 11,000 years [484], and the Hela cervical cancer cell line has survived for seven decades [485]; yet their genomes are still stable enough to maintain their lineages [484]. The atavistic nature of carcinogenesis connotes that each cancer lineage is a new or semi-new species of unicellular organism [111, 112, 138]. This “new species” concept denotes two important but often neglected notions: While “species” insinuates that a tumor lineage has a stable genome to forever maintain itself, “new” means that its genome has enough mutations to distinguish itself from its erstwhile one, i.e. the genome of its normal progenitor, because a species is defined by the specificity of the genome. It is possible that genomic instability of cancer cells, even at a chaotic extent, affects only certain parts of the genome while leaving certain other parts undamaged, and that once a mutant clone is selected, genomic stability resumes the hegemony until its cells enter into a new round of “mutations and clonal selections”, likely driven by new stress, and yield a newer mutant clone as a “newer species”. This is also to say that instability-caused mutations in cancer cells are not completely random and stochastic as they do not touch certain currently characterized core(s) of the genome that can later keep the “new” genome relatively intact. We envision that, if the Hela cell line was continuously cultured in dishes for another-thousand years, it would still be alive and be the Hela cells, although having millions of additional mutations. It seems that cancer researchers have emphasized enough the genomic instability of cancer but have put insufficient attention onto the aspect of their genomic stability. It remains unknown but very intriguing to us how a newly formed mutant clone, likely more malignant, turns from genomic instability to genomic stability.
Cellular differentiation may be a mechanism for tumor reversion
In many (if not most) cases [306, 369-371, 376, 388, 389, 398, 486-491], reversion of cancer cells back to a normal state in an embryonic microenvironment occurs mechanistically via cellular differentiation [490, 492-497]. With models of chick embryo and Zebrafish embryo, or with an intrauterine injection approach in mice, a slew of studies has shown that human malignant melanoma cells in an embryonic microenvironment do not develop to tumors but, instead, differentiate to neural-crest-like cells [498-501]. Actually, earlier studies have shown that when the SRC oncogene is inactivated, the SRC-induced myosarcoma cells will differentiate into mature myocytes [502-504], and this inactivation-caused differentiation is actually a common event for SRC-caused transformation [505]. Emphasis should be given to a study by Pierce and Wallace in 1971, in which some cells of squamous cell carcinomas were shown to differentiate into mature keratinized cells as squamous pearls [380]. This observation is of significance as it shows that the squamous carcinoma cells highly resemble normal skin stem cells that divide asymmetrically to one stem cell (equivalent to a cancer stem cell) and one keratinocyte, and the latter continues both maturation and symmetrical division towards stratum corneum (equivalent to the other cancer cell that replicates and differentiates to the squamous pearl). Similar cellular differentiation has also been observed for the cells of chondrosarcoma as well as the cells of breast and colon cancers, which leads Pierce to conclude that the rules learned from teratocarcinoma govern the behavior of neoplasms in general [304, 306].
Certain extracellular matrices other than the embryonic milieu can also control cancer cells' phenotypes in vivo. The BAG2-GN6TF cells of rat hepatocyte origin may quickly develop into tumors or develop into normal hepatocytes in rats, depending on the sites and routes of the cell inoculation and on the age of the recipient rats [488, 506]. S. Meryl Rose had also reported in 1948 that after frog kidney cancer cells were transplanted into and well grew in a salamander limb and then the limb was amputated through the tumor site, the limb could regenerate and some cancer cells within the regenerate differentiated into muscle and cartilage before they eventually died [507, 508]. These earlier observations suggest that xenografted tumors can grow persistently in an alien animal species if the tumor cells remain undifferentiated, but if they differentiated to be more and more foreign, they will eventually be eliminated by the host. Shvemberger et al have in a series of publications reported that inoculation of mouse or rat malignant cells into an eye's anterior chamber of syngeneic animals can reduce the malignancy and increase differentiation of the tumor cells in association with a trend to normalizing the karyotype to diploidy, which presumably occur via selection of the subclones of cells that are relatively less malignant, more differentiated, and less aneuploid [90]. However, these observations are partly incongruous with the seminal findings of Greene et al in the 1940's [138, 240]. In a series of experiments, Greene et al. found that some cancers were transplantable to eyes' anterior chambers of those syngeneic animals that bear a tumor, but not of those without tumors [243, 509, 510]. Greene et al also found that only those human tumors capable of metastasizing (but not those incapable) could be transplantable to eyes' anterior chambers of animals of a different species [509, 511-513]. What remains vague is whether the various extracellular microenvironments described above are in a way related to an embryonic milieu.
Ever since almost a century ago [514-516], there have already been a battery of studies showing that certain extrinsic factors, such as some drugs or nucleic acids [385, 517-529], can facilitate the reversion of cancer cells back to normal via differentiation or maturation [89, 514-516, 530-532] in culture dishes, in animals, or in patients [468, 496, 533-536]. Neural differentiation of the PC12 rat pheochromocytoma cell line induced by nerve growth factors or some chemicals is among the best-studied examples [537-539]. A dietary supplement methylsulfonylmethane [540], which is also a normal oxidation product of dimethyl sulfoxide (DMSO) [541], can obviate metastatic properties of a few different cancer cell lines via differentiating the cells [542-546]. Actually, there have been some clinical successes as proof in the remission of acute promyelocytic leukemias via differentiation induced by treatment with retinoic acid [547] or arsenic trioxide [548, 549], alone or in combination with other chemotherapeutic agents, although relapses from extant cells often ensue later [107]. Some of these chemicals, with the arsenic trioxide being best studied, are known to effect via driving cells towards differentiation [548, 549]. Cell lines from the abovementioned teratocarcinomas have been well studied for the molecular mechanisms of the reversion [550, 551]. Other studies have suggested that reversion of leastways certain malignancies to a normal state may entail over 300 genes [378, 552-554].
Mention should be made of spontaneous regression of certain human cancers [90, 138], certain tumors in fish and amphibians [410, 555-563], and the canine transmissible venereal sarcoma [140, 564, 565]. Reversion seen in some of these human and animal cases may in part be ascribable to differentiation and ensuing senescent death of the differentiated cells.
Does mutation have anything to do with tumor reversion?
In Pierce's apercu [318], the above-described tumor reversion challenges the dictum of “once a cancer cell, always a cancer cell”. Simple explanations for the reversion include that the reversible tumors are not caused by mutations but by reversible epigenetic alterations [566] or, alternatively, that they are caused by mutations but the mutations are readily reversed back to normal [567]. Actually, a “cell reversal theory” opines that carcinogenesis may start with reversal of a differentiated cell to a less differentiated epigenetic status, such as a stem cell status, whereas a stem cell or a cell at a stem status that does not dwell in the stem cell niche is very chaotic and will enter into uncontrolled proliferation [115]. However, although all cancer researchers likely agree that epigenetic alterations are instrumental to the formation and progression of tumors [568, 569], whether or not such alterations alone are sufficient to cause tumors, especially the malignant ones, remains as an enchanting but fiendish puzzle. On the other hand, there are other equally plausible explanations for the tumor reversion, such as the three scenarios proposed by Telerman and Amson [378].
If tumors are caused by mutations as most cancer researchers believe, it seems improbable that the mutations would disappear later from live cells (lethal mutation may disappear along with the death of the cell [570]). Therefore, a possibility is that the reverting pathway activated by the extrinsic reverting factors, such as an embryonic milieu or a chemical like retinoid acid or arsenic trioxide, is a different one from the mutations-initiated tumorigenic process and is not impeded by the mutations. Alternatively, the mutations may hinder the reversion but the extrinsic reverting factors can override the impediment, since correction of one or two signaling pathways has been shown to be capable of reverting cancer cells [496]. In either scenario, the reverted cells are perceived to still retain the mutations [529, 571-574]. In Harris' words, “the malignant phenotype may be held in an abeyance during the reversion” [418], which insinuates that the malignant phenotype can still reappear. Indeed, the animals developing from cancer-cell-derived gametes have a high chance to develop cancers late [575]. Therefore, unless the normalized cells eventually die of senescence like all terminally differentiated cells [576, 577], thus purging mutations and cancer cells from the patient, the patient still faces a peril of tumor recurrence because, as aforementioned, the mutated genome still retains the right to control the phenotype. Today, with the feasibility of whole genome sequencing, repeating the early experiments described above and sequencing the whole genome of the cells before and after the reversion should help clarify these scenarios and provide us with information on what mutations the cells have that hinder the extrinsic-factor-driven differentiation of malignant cells.
Our manipulations can only coerce primary cells into showing neoplastic features, but cannot directly transform the cells
We have previously realized a few attributes of experimental tumorigenesis [112, 138, 139, 578]: 1) Lesions induced in most, if not all, animal models of tumorigenesis are inducer-dependent until terminal stages (for more early references, see [497, 579, 580]). The lesions, even if they manifest cancerous morphology and behavior, regress upon withdrawal of the inducer, although reintroduction of the inducer usually [138, 581, 582], but not always [497, 581, 583], induces quick recurrence of the lesions. Regression of these non-neoplastic lesions occurs via apoptosis and thus differs from the aforementioned regression of tumors that occurs via differentiation and ensuing senescent death. 2) Cancer induction in animals requires a long latent time, and usually only one to several tumor masses appear in an animal [138, 276, 277]. As aforesaid, these phenomena evince a negligible transformation efficacy of our manipulations at the cellular level. Besides these two properties, we have described three additional phenomena earlier in this essay: 1) Formation of tumors may not necessarily entail mutations. 2) Cells considered to be “transformed” may still be mortal. 3) Immortality, transformation, invasion, and metastasis as key neoplastic attributes can be segregated from one another in the lab and can occur in different orders, depending on the experimental setting.
In our opinion, which is partly similar to Harris' punditry [416], all of the five traits of experimental tumorigenesis described above suggest that our manipulations in cell culture or in animals are not able to directly cause the cellular or molecular alteration(s) that bestow immortality and autonomy upon the primary cells. In most, if not all, of our in vitro or in vivo systems, our manipulation, such as knockout of the p53 gene or ectopic expression of a k-ras mutant, is simply to coerce the primary cells into 1) replicating incessantly, 2) manifesting transformed morphology and/or behavior, 3) sustaining the cells' life, 4) causing or accelerating DNA damage, and 5) impairing DNA repair mechanisms [112, 138, 139, 241, 584]. The lesions produced are actually hyperplastic, and not neoplastic. Actually, the malignant behavior of these hyperplastic cells had already been observed in the world's first experiment of chemical tumorigenesis by Fischer in 1906 [585]. According to Braun [431], Fischer repeatedly injected Scharlach R into subcutaneous sites of rabbits' ears, which drove the local epithelial cells to proliferate and invade deeply into the blood and lymphatic vessels. In some animals the lesions metastasized distantly. However, the cells, although invasive and even metastatic, remained mortal as they regressed upon withdrawal of the Scharlach R. The cellular alterations directly responsible for the immortality and autonomy, which are still unknown to us even now (Fig. 4), can only occur spontaneously in a random and stochastic manner during the incessant cell replication under the duress from our manipulations. This is why when the transforming agents, such as oncoviruses, are withdrawn or lost, the “transformed” cells may revert back to normal, a phenomenon that has already been discerned for over 50 years [586-588] and reviewed many years ago [497]. Actually, sometimes our manipulations can just confer upon primary cells additional rounds of cell replication, as epitomized by additional 20-30 population doublings of primary cells offered by ectopic expression of the SV40 large T antigen, during which a few cells acquire spontaneous cellular or molecular alterations that establish immortalization [589]. Pierce had once stated in 1983 [306]: “it is easy to show what cells can be made to do, and it is often difficult to know what cells do.” We should remind ourselves that what we have observed in our experiments is what cells are forced by us to do, but what we actually want to know is what cells, and even the organism (such as a human being) as a whole, would like to do in a given physiological or pathological situation [6, 68, 112, 138, 578, 584]. Probably, we often put the cart before the horse in our research [112].
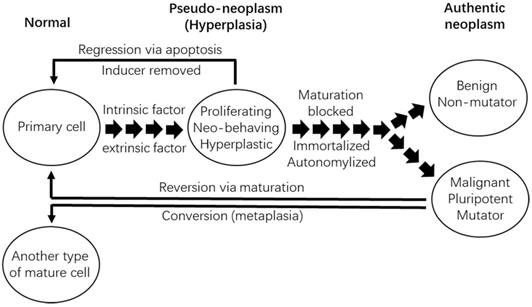
Depiction of our coercion hypothesis and of tumor reversion or conversion. Certain intrinsic factors (such as an inherited mutation) or extrinsic factors (such as our manipulation in an experimental animal) may drive proliferation of a primary cell to form an outgrowing lesion. If the factor disappears, such as due to the manipulator withdrawal, the lesion will regress via cellular apoptosis, suggesting that the lesion, which may have already exhibited cancerous features, has not yet become immortal and thus is still hyperplastic, and not yet neoplastic. At this stage, its proliferation and its possible manifestation of cancerous features are actually sustained under the coercion of the intrinsic or extrinsic factor. However, if the factor lasts much longer, the lesion will evolve to an authentic neoplasm by acquiring cellular immortality, autonomy, and maturation obstruction that are caused by epigenetic or genetic changes in non-mutator genes (in this case the neoplasm is benign) or in mutator genes (in this case it is malignant). The neoplasm, benign or malignant, may (or may not) be reverted back to normal like its normal counterpart tissue via cellular maturation, or may (or may not) be converted to another mature tissue type via metaplasia.
Our manipulations drive cell proliferation to form hyperplastic lesions, cells of which are redundant and still allegiant to the animal's body. This allegiance forces the cells to commit suicidal apoptosis and probably, to a lesser extent, also senescent death, because the animal's body wants to avoid cellular redundancy of the tissue or organ [111, 138, 139, 241, 546, 576, 590-595]. It is likely that our manipulations inhibit apoptosis and senescent death as components of their coercive mechanisms, but this inhibition disappears once our manipulations are withdrawn. Oncogene-withdrawal-caused regression of the-oncogene-induced outgrowths may involve modification of metabolism [596] and immune functions [597, 598], which is not surprising as the cells die via apoptosis and apoptotic cells are known to be eliminated via phagocytosis by macrophages, according to Kerr et al who created the word “apoptosis” [599]. Since in culture systems cells do not have to care about the cellular redundancy issue, a spellbinding but unaddressed question is whether, after withdrawal of the coercion, the proliferating cells die of senescent death or/and some other form(s) of programmed cell death [138, 577].
Knowing that there is no way of promptly immortalizing primary cells, researchers often perpetuate the manipulations, namely the coercions, by using such as stably-expressing cell clones or transgenic animals. However, for different research needs, many systems of “conditional immortality” or “conditional transformation” have also been created [600-605], including transgenic animals [606]. Accordingly, many conditional cell lines have been established [589, 606-615], like the temperature-controlled ones [612, 616], which show controllable immortalization or neoplastic transformation [612, 615, 617]. The words “conditional” and “controllable” already proclaim the nature of swift reversibility and accentuate that the immortality or the neoplastic transformation so created is not authentic because the cells are still mortal.
Bearing the manipulation-bestowed duress in mind, many “surprising findings” in animal models are actually not so surprising, such as the aforementioned observations that epithelial cells can evade, disseminate, and enter into the bloodstream before they can form primary tumors [480], that cancer cells can enter into the circulation before invading adjacent stroma [481], and that mammary epithelial cells can metastasize and colonize in the lungs before they are malignantly transformed [482, 483]. These results from manipulated animals show diversion from the “growth, invasion, and then metastasis” trajectory of epithelial carcinogenesis [62, 295]. These phenomena have not and will not be discerned in human situations, because withdrawal of the coercers will likely lead to the disappearance of these comportments of manipulated cells.
Most, if not all, of our manipulations in experimental systems of tumorigenesis have been designed to simulate epigenetic or genetic alterations identified in human tumors. For instance, we often ectopically express a k-ras mutant in pancreatic ductal cells to transform them because we know that most pancreatic cancers bear this mutation [251, 618, 619]. However, we need to bear several points in mind: 1) In human tumors, these alterations are not the intrinsic factors directly responsible for the tumor cells' immortality and autonomy, although they might have already caused, by kindling a cascade of molecular events, cellular immortality and autonomy at the time of diagnosis. 2) In many, if not most, experimental studies, the target cells may not have been immortalized but have already displayed transformed comportments and/or morphology, which may dupe us into discontinuing our manipulations and harvesting the lesions before they have experienced spontaneous immortalization and become genuine neoplasms. 3) The molecular or cellular aberrations we conferred onto primary cells, such as k-ras mutations, can transform the cells in culture dishes and in animals, but it does not mean that there actually is a patient whose tumor is caused by one of these anomalies. There are probably over 100 million genetic alterations of different types in human cancers, since there have been about 85 million point mutations identified [620, 621], pancreatic cancer alone has 857,971 genetic alterations identified [622], and the p53 gene alone has over 30,000 mutation types [6, 623]. A fact is that many cancer researchers endlessly use different combinations or different sequences of these alterations to efficiently transform primary cells in culture or to precipitate tumors in animals, and then claim identification of novel carcinogenic pathways. However, researchers are still unable to pinpoint any of these alterations, these combinations of alterations, or these orders of alterations, which together make innumerable permutations, as the cause for the tumor formation in a patient [112]. Even worse, it remains possible that these alterations, or these combinations or sequences of alterations, are just the results or byproducts, but not the causes, of the tumor formation in patients.
Only the neoplastic morphology and behavior caused by intrinsic factors are authentic
The above-described “coercion hypothesis” signifies an important fact learned from over a century of experimental tumorigenesis research: In vitro colony formation, neoplastic morphologies, as well as invasive and metastatic behaviors can all be caused by both extrinsic and intrinsic factors. The currently-unidentified cellular or molecular alterations responsible for immortality and autonomy are intrinsic factors, and the neoplastic morphology and behavior caused by them reflect an authentically neoplastic state (Fig. 5). On the contrary, those neoplastic morphology and behaviors occurring under the duress from our manipulations, which are extrinsic factors, do not reflect a neoplastic state.
Illustration of the relationship between the “tumor-initiating factors” and their downstream “immediate tumor-causing factors”. A cell (cell A) may have an epigenetic or genetic alteration in the nucleus (α) that occurred spontaneously, was inherited from a parent, or was caused by an altered factor in the cytoplasm (β) or by an extracellular factor (γ, such as a radiation, a chemical, or a virus). A similar alteration in the nucleus (δ) or cytoplasm (ε) may also occur in another cell (cell B) nearby or even in a distant body site, which alters the communications and interactions with cell A, in turn causing α or β. All of these alterations may mutually affect each other (between the two cells, between the nucleus and the cytoplasm of a cell, as well as between the intracellular and extracellular environments of a cell). α or β is defined herein as a “tumor-initiating factor” as it triggers a cascade (referred to as a, b, c, etc.) of molecular events in the nucleus (e.g., epigenetic or genetic changes) and/or the cytoplasm, culminating in one or some currently-unknown cellular or molecular alterations (question mark) that establish cellular immortality (Immor.) and autonomy (Auton.), namely a neoplastic state, and thus are coined herein as “immediate-tumor-causing factors”.
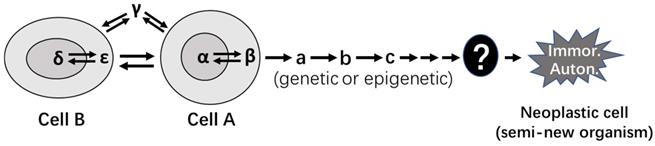
The notion that only intrinsic-factor-caused neoplastic properties are authentic repudiates extrinsic-factor-caused spuriousness, and thus is of importance and has clinical relevance. Many things, such as chronic viral or bacterial infections, treatments with certain drugs, exposures to certain environmental pollutants, etc., may be such extrinsic factors that coerce cells into outgrowing and manifesting neoplastic features. For example, chronic infection by Helicobacter pylori (HP) can result in low-grade lymphomas [624-630], chronic infection by human T cell lymphotropic virus type I (HTLV-1) can cause lymphoma or leukemia [631-633], and infection by parasite theileria can transform bovine leukocytes into disseminating tumors [634-636]. However, therapeutic removal of these causal pathogens can cure these tumors, leastways at an early stage. For another example, hepatomas and hepatocellular carcinomas had been reported frequently during the 1970's-1980's among women chronically using estrogen-rich oral contraceptives, but the tumors could regress upon cessation of the contraceptives [637-643]. In these instructive cases, the cure of the tumors upon removal of the extrinsic factors is reminiscent of the withdrawal of our manipulations in experimental systems. In our opinion, the tumor cells caused by the HP, HTLV-1, theileria parasite, or excessive estrogen may not have been immortal and autonomous at the time of diagnosis and thus may not be authentically neoplastic, albeit their morphology denotes a pathological diagnosis of malignancy and they, if left untreated, will eventually evolve to genuine neoplasms.
We still have no way of directly transforming primary cells
In all experimental systems established so far, our manipulation can make primary cells of small rodent origins truly immortal only after weeks in cell culture or months in animals [138], obviously as a secondary event. Some plant cells may be exceptions, as some early studies showed that some plant cells could be transformed after only 34-48 hours of manipulation [440, 443, 453], with a few more days of manipulation creating more aggressive cells [431, 441, 442, 444-446, 449-451]. This is to say that in our in vitro and in vivo models, the cellular alterations responsible for immortality and autonomy occur only spontaneously during the enduring cell replication caused by the duress from our manipulations (Fig. 5). The aforementioned fact that usually only one to several of the cells in an animal develops into tumors signifies that normal cells guard firmly their mortality program to ensure that all cells will die eventually, which is the will of a higher eco-system as we expounded before [241]. We still hitherto have had no way of breaking through this guard of normal cells and thus have to wait until the cells themselves give up this guard to adapt to the stressful milieu. Fortunately, our manipulations as extrinsic factors can accelerate this giving-up not only by imposing a stressful environment but also by sustaining the cells' life, accelerating cell replication, damaging DNA, impairing DNA repair, etc.
There are two types of differentiation obstructed by two types of cellular alterations
All neoplastic cells, benign or malignant, are less differentiated than their normal counterparts, as pointed out decades ago by Markert [288], betokening that cellular maturation has been blocked during tumorigenesis [644, 645]. Of course, in benign tumor cells this blockade may be set at a point near the terminal differentiation, allowing the cells to highly resemble their normal counterparts. For example, uterine leiomyoma cells are highly similar to, and thus basically indistinguishable from, uterine muscle cells in cellular morphology; these tumors are pathologically diagnosed mainly based on their histological features.
There are three different sets of antithetical cellular properties that pertain to the obstruction of maturation (Table 1). First, a normal cell can be well-differentiated but still possesses a strong proliferation potential [646, 647], evincing that differentiation and proliferation are not incompatible [44, 149, 415], although cells that proliferate robustly are usually less differentiated. As noted by Harris [415, 648], it has become an “ancient question of whether a tumor grows rapidly because it does not differentiate or does not differentiate because it grows rapidly, but this is a false question.” For instance, after partial hepatectomy, the remaining hepatocytes that are highly differentiated can robustly proliferate to produce new well-differentiated hepatocytes [6, 649]. Second, immortality, which can be considered as an extreme of proliferation potential, and differentiation are not incompatible either. For instance, many benign tumor cells not only are immortal but also are well-differentiated, with uterine leiomyoma cells as an epitome. Third, even very malignant cells from the same patient can differentiate into a diversity of tissue types [650-658], which is a phenomenon often dubbed as “metaplasia” or “transdifferentiation” [385], as in pathology textbooks “metaplasia” means conversion from one differentiated cell type to another, such as squamous metaplasia and osseous metaplasia. Actually, benign tumor cells from a given patient may exhibit multiple types of metaplasia as well [659-661]. This betokens that tumor cells may retain pluripotency, although they are blocked somewhere towards the terminal maturation. Therefore, there are two different types of cellular differentiation, one being maturation towards the parental cell or tissue type, and the other being metaplasia towards some other cell or tissue type(s). This fact further annunciates that the currently-unidentified molecular or cellular alterations which militate against cellular differentiation can be dichotomized into two categories, i.e., 1) those that prevent tumor cells from maturation without stymieing their pluripotency and thus allowing the cells to differentiate into one or more other cell types, and 2) those that not only interdict maturation but also cancel pluripotency. A captivating question oblivious of by many researchers is whether immortality, autonomy, and maturation interdiction are three different facets of the same dice, i.e., whether these three neoplastic properties are controlled by the same cellular factor(s).
Three sets of opposing cellular properties relevant to neoplasms
| Cell type | Maturity | Opposing properties |
|---|---|---|
| Normal | Mature | Proliferating and differentiated |
| Benign | Blocked at late differentiation stage | Immortal (endlessly proliferating) and differentiated |
| Malignant | Blocked at early stage | Undifferentiated and pluri-differentiating potency |
One important concept learned from the above introduction is the existence of three cellular antitheses, i.e., 1) well-differentiated status vs proliferation potential of normal cells, 2) immortal status vs well-differentiated status of benign tumor cells, and 3) maturation blockade vs pluripotency, or maturation disability vs metaplasia ability (Table 1). Another important notion is that, germane to tumorigenesis, one type of cellular or molecular aberration is those stymieing only cellular maturation and another type is those impeding both maturation and pluripotency. Being cognizant of these two concepts is of importance, because we may consider developing some approaches or extrinsic factors as remedies for directing cancer cells towards certain types of metaplasia as an alternative, if it is difficult or impossible to direct the cells towards maturity such as in the situation where maturation genes are severely impaired [467]. Either type of differentiation should be followed by senescent death of the cells [576, 577].
Immortality and autonomy may entail one set of genes, while malignant morphology may involve another set
A prodigious number of publications deliver, to many cancer biologists and molecular biologists who lack clinical experience in oncology and surgical pathology, a convoluted message about the demarcation between benign and malignant neoplasms. For instance, most of the “cancer hallmarks” described by Hanahan and Weinberg [662, 663] are actually not unique to malignancy, and certainly are not unique to every cancer cell in the same cancer mass. They are, in fact, hallmarks of “any growing tissue”, in Llambi's words [664], including benign neoplasms, as pointed out first by Lazebnik [665] and later by us [112]. In Blagosklonny's words, “…hallmarks can be observed without cancer” [475]. Today, there have not been any molecular markers available for us to distinguish malignity from benignity, and morphological features are still the main clinical criteria for this differentiation. However, morphological criteria are not flawless, as has been pointed out by the superlative surgical pathologist Harry S. N. Greene in 1948 [509] and has been reviewed by us [138]. Concerns about pathological criteria include overdiagnosis [666, 667], such as overdiagnosis of thyroid cancer [668-671]. Therefore, we may need to find a better way to classify tumors or to reset the demarcation between malignity and benignity so as to better explain a tumor's prognosis.
Both benign and malignant cells are immortal and autonomous. However, many benign cells highly resemble, whereas most malignant cells differ greatly from, their normal counterparts in morphology. This disparity connotes that there are some epigenetic or genetic alterations establishing only immortality and autonomy without significantly affecting cellular morphology, whereas there are some other alterations that specifically establish malignant morphology and do not occur in benign tumors. We surmise that there may be a set of genes, or one or more genomic structures depicted in Figure 1, that govern not only cellular mortality but also the loyalty of cells to their host body; their alterations, epigenetic or genetic, establish cellular immortality and autonomy. We herein call these genes or genomic structures “mortal and loyal factors” and call their alterations “immediate tumor-causing factors”, so as to distinguish them from well-studied oncogenes or tumor suppressor genes that have well-known roles in initiating a lengthy tumorigenesis (Table 2 and Fig. 5). Conversely, there may be another set of “malignant morphology genes or genomic structures”, dubbed herein as “malignant morphology factors” for simplicity, whose anomalies are responsible only for the establishment of malignant morphology (Table 2). An enthralling but unaddressed question is whether the “malignant morphology factors” are also those controlling cellular maturation, since maturation pertains not only to morphology but also to function.
Classification of genetic factors relevant to key properties of tumor biology
| Category | Features/Functions | Current state | |
|---|---|---|---|
| Tumor-initiating factors | Oncogenes | Well studied | |
| Tumor suppressor genes | |||
| Mortal and loyal factors | Block maturation and establish immortality and autonomy | Hypothetical; unidentified | |
| Tumor morphology factors | Benign (similar to normal) | Hypothetical; unidentified | |
| Malignant (greatly divergent from normal) | |||
| Tumor progression factors | Non-mutators related to benign tumors | Some identified as oncogenes or tumor suppressor genes | |
| Mutators related to malignant tumors |
Note: The “genetic factors" may be canonically defined genes on the genomic DNA sequences but can also be higher genomic structures depicted in Figure 1.
So far, there has not been any “mortal and loyal factor” or “malignant morphology factor” established, although whole genome sequencing has been performed on thousands of tumors. Probably, as Heng et al has frequently pointed out before [39, 41, 42, 672], one of the reasons is that these factors or some of them are not genes shown as the level 1a in Figure 1 but entail higher genetic levels. Moreover, in our opinion, one tactical mistake cancer researchers have made for many decades is to dwell mainly in the research of malignancy and hardly set foot onto research of very benign neoplasms. Very benign neoplasms, typified by uterine leiomyoma, are likely to have many fewer and much stabler genetic and epigenetic alterations, compared to their malignant counterparts. Therefore, they serve as much simpler and thus better models for us to identify the critical alterations immediately behind cellular immortalization, autonomization, and probably also maturation interdiction.
The benignity or malignity, and even the neoplastic state, of cells transformed in vitro require much more attention. Some cell lines such as MCF10AT [673, 674] can form colonies in soft agar, which is considered an insignia of a neoplastically transformed state [251, 675], but in animals they can form only benign tumors, judged by the histology of the xenograft tumors. On the other hand, some other cell lines, like NMuMG (ATCC website, [676], and our experience), cannot efficiently form colonies in agar but can often form benign tumors in animals. These and other dissonant lab data lead us to consider that both colony formation in culture and xenograft tumor formation in animals are required to qualify a neoplastic transformation. Determining whether in vitro transformed cells are malignant is difficult, and currently we still lack convenient but reliable measures for this purpose [251], since the soft agar clonogenic assay initially developed by Hamburger and Salmon in 1977 [677] is not always reliable [678, 679]. Cell fusion studies have suggested that transformed and malignant phenotypes are under separate genetic control [680], which is fathomable because a transformed, i.e., neoplastic, state may be benign. As Lazebnik has pointed out, distant metastasis is currently the only reliable yardstick for malignancy [665], although this canon is still not flawless because in some rare cases histologically benign lesions also metastasize, such as in the cutaneous fibrous histiocytoma [681] and in the growing teratoma syndrome of the ovary [682, 683]. Unfortunately, most relevant studies employ only subcutaneous inoculation of in vitro transformed cells, whereas few cell lines at a subcutaneous site can metastasize distantly, according to the literature and our experience, although we suspect that some cell lines may metastasize if inoculated viscerally.
Benignity and malignity may be defined based on genomic alteration
Benign tumors in general do not progress but malignant ones are always on their way to more-wayward states, notably states of metastasis and therapeutic resistance. Tumor progression is perceived to be attributed chiefly to accumulation of more epigenetic or genetic alterations, which is in turn ascribed to certain initial alterations, such as those impairing DNA repair. Pertinent to progression, tumor-related genes can be dichotomized into 1) mutators that are defined herein as the genes or genomic structures whose epigenetic or genetic alterations can cause or accelerate alterations at others and 2) non-mutators whose epigenetic or genetic alterations do not cause alterations at others (Table 2). While the “evolvability” theory of Pienta et al suggests the involvement of the ability of evolution in tumorigenesis [141, 684], our “mutator” concept, which may entail any genomic level(s) illustrated in Figure 1 and not just canonically defined genes, emphasizes the ability of evolution in tumor progression to more-heinous states. With this dichotomy, neoplasms can be reclassified at the genomic level: Benign neoplasms are those bearing epigenetic or genetic anomalies at non-mutators and thus do not accumulate genetic abnormalities, whereas malignant neoplasms are those bearing epigenetic or genetic alteration(s) at the mutators and thus easily have accrued alterations (Table 3 and Fig. 4) as the bedrock for continuous progression towards more-diabolical states [62]. The essence of this reclassification is first to attribute accumulation of epigenetic or genetic alterations to the initial ones at certain mutator(s), then to attribute progression potential to the accrual of such alterations, and finally to utilize progression potential to demarcate the border between benignity and malignity. Of course, benign cells are also immortal and keep replicating, which increases the risk for new alterations to occur. Actually, this is a reason why some benign tumors are at peril for progression.
Tumor classification at the genomic level
| Type | Non-mutator | Mutator | Properties | ||
|---|---|---|---|---|---|
| Epigenetic | Mutation | Epigenetic | Mutation | ||
| I | with | without | without | without | benign, easily cure |
| II | without | with | without | without | benign, curable |
| III | with | with | without | without | benign, curable |
| IV | with/without | with/without | with | without | malignant, relatively better |
| V | with/without | with/without | without | with | malignant, bad |
| VI | with/without | with/without | with | with | malignant, worse |
Note: mutator and non-mutator include not only canonically defined genes but also higher structural level(s) shown in Figure 1.
Epigenetic aberrations more often change the expression level of the inflicted gene than confer new function onto it, whereas mutations may completely change the gene's function. Therefore, epigenetic changes may or may not resemble mutations. It is perceivable that epigenetic alterations of mutators may trigger epigenetic and genetic changes at other mutators and non-mutators and thus may drive tumor progression as well. With our reclassification, benign tumors can be further systemized into 1) those bearing epigenetic alterations only at non-mutators, 2) those bearing mutations only at non-mutators, and 3) those bearing both. Similarly, malignant tumors can be further stratified into several subgroups as those with or without epigenetic or genetic changes at non-mutators, besides the alterations in mutators (Table 3 and Fig. 6).
Propounded classification of tumors. Benign tumors are those bearing epigenetic alterations (green triangle) and/or mutations (black dot) in non-mutator genes or genomic structures. Malignant tumors are those bearing epigenetic alterations (red dot), mutations (green dot), or both in mutators, with or without epigenetic alterations and/or mutations in non-mutators.
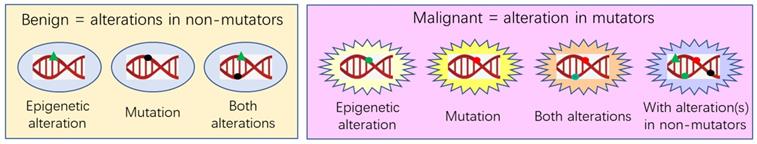
Currently, our classification approach can only be used in the study of neoplastic comportments and underlying mechanisms, and is inapplicable in clinical practice because we still do not have enough detail about which genes or genomic structures are mutators and what alterations they have that are implicated in establishing malignancy, although many mutations of many genes are already considered by some cancer pundits as cancer “drivers” [62, 685-689]. However, as aforesaid, our classification provides an explanation for the question as to why some rare tumors can regress spontaneously or can be cured easily: They may bear only alterations in non-mutators without involvement of epigenetic or genetic alterations in mutators, and are benign even if they manifest malignant morphology. Moreover, there is an obvious incongruity between morphology and prognosis seen now and then in the clinics. For example, most nasopharyngeal cancers are the undifferentiated type, but many of them actually have a good prognosis and can even be cured [690-692], which sharply contrasts with most other types of poorly differentiated or undifferentiated cancer that have very poor prognoses. It is captivating to know whether those undifferentiated but curable cancers bear alterations only in non-mutators.
Above-described mouse teratocarcinomas may be special with little human relevance
In clinics, teratoma and teratocarcinoma are usually pediatric pathologies, although teratoma in some males may be diagnosed as late as middle age [693]. Pediatric tumorigenesis has its inception at an embryonic stage [694] and may indeed occur as a repercussion of epigenetic aberration. In our rumination, teratomas occur simply because epigenetic or genetic changes occur to some early pluripotent cells and thwart their differentiation while the cells proliferate continuously, whereas teratocarcinomas occur because such alterations occur at an even earlier embryonic stage and the hindrance of differentiation makes the tumor cells less differentiated. Reiterated, if less-differentiated pluripotent cells are the tumor progenitors, teratocarcinomas would result, whereas if more-differentiated cells are the tumor progenitors, teratomas would result [318]. However, even if epigenetic alterations were the initial causes, in real life these tumors have likely developed mutations as secondary events and become irrevocable at the time of diagnosis. If other types of pediatric neoplasms are initiated by epigenetic aberrations alone as well, many of them may have also acquired some mutations at the time of diagnosis, or even before the child was born. The 40-week gestation is a long stint during which a single fertilized egg grows into a fetus of several kilograms, involving numerous rounds of cell replication and thus providing numerous opportunities for mutations to occur. Actually, if certain rare sporadic tumors in adults are also initiated by epigenetic alterations alone, the tumors have likely developed mutations at the time of diagnosis as well. Therefore, in real life the adult cancers that bear only epigenetic aberrations are probably as scarce as hen's teeth. We realize that there are some tumors without mutations detected, but several possibilities remain to be ruled out. First, some single nucleotide polymorphisms in these tumors may actually function as mutations. Second, mutations on the extrachromosomal DNA [695, 696] or alterations at the level(s) of genomic structures higher than the gene level (Fig. 1) have been neglected or are harder to discover. Third, technical issues may exist [620, 697, 698]. Actually, we still do not know what mutations are responsible for immortality and autonomy and thus do not know what we should specifically look for. However, tumors in small rodents may show epigenetic alterations alone, partly due to their much shorter lifespans and smaller body sizes, besides other disparities from humans [699]. Because cancer cells in the human and the mouse require similar time frames for completing one cell cycle, which is around 24 hours for those fast-proliferating cell lines according to the literature and our experience [6], a tumor of the same size in mice and in humans has a similar cell number, but a very small tumor in a human is already very large in a mouse. Therefore, tumors in mice are much smaller and have experienced far fewer rounds of cell replication, thus having far fewer chances to develop mutations, generally speaking. In reality there is no way of knowing whether or not clinically-diagnosed human cancers are solely caused by or bear only epigenetic alteration(s).
Concluding remarks
There have been many theories about tumorigenesis, disputing over the involvement of mutations. One extreme theory considers that mutations not only are the initial cause but also reach a chaotic extent, while the diametrical theory thinks that mutations are not necessary. Main evidence against the mutation theory includes that ePS or iPS cells displaced in a non-embryonic environment may develop to neoplasms, whereas neoplastic cells placed in an embryonic environment may be reverted back to phenotypic normal. Until now, the differential control by the embryonic environment in this antithesis still remains vague. In our opinion, both extremes and many, if not all, intermediate theories are correct as they describe formations of different types of neoplasms in different situations. We envision that a chaotic level of genomic changes can occur in highly stressful situations and can more efficiently establish a malignant state. For example, isolating a primary cell and putting it into a culture dish containing a medium with 10% fetal (but not adult) bovine serum make the cell highly stressed, because it is nourished abnormally and has lost all interactions with other cell types and lost normal neural and hormonal controls. Forcing the cell to ectopically express one or more oncogenes, which is a common approach to transform cells, further raises the stress level. However, it remains questionable whether in patients the genome still has to experience a chaotic mess for development of some benign tumors, such as uterine leiomyoma that is indistinguishable from normal uterine muscle in most cellular aspects and may be caused by mild hormonal imbalance. Theoretically, certain epigenetic changes in nuclear proteins, such as abnormal phosphorylations of histones, may alter some genomic structures such as nucleosomes and chromosomes, in turn initiating tumor formation. While carcinogenesis has been extensively studied, an important aspect of tumorigenesis, i.e. development of benign tumors in a slightly abnormal situation, has been much understudied. In turn, fewer discourses have been focused on the immediate tumor-causing factors, i.e., those molecular or cellular alterations that directly establish cellular immortality and autonomy. The “immortality and autonomy” definition of neoplasia connotes that a neoplasm resembles a new or quasi-new unicellular organism [700] and thus should have some mutations, because a new organism should have something new in the genome [141]. Therefore, wrangling over “whether epigenetic abnormality alone can establish cellular immortality and autonomy, namely establishing a neoplastic state”, is actually a debate on whether “difference(s) only at the epigenetic level are sufficient to define a new organism”, making this issue a general question of taxonomy. In our opinion, neoplasms are malignant if they bear epigenetic or genetic abnormalities in mutator genes or genomic structures, defined as those whose alterations accelerate others to change, whereas neoplasms bearing epigenetic or genetic abnormalities only in non-mutators are benign. Future mechanistic research should be devoted to identifying the abovementioned “immediate tumor-causing factors”. Very benign tumors may have many fewer alterations and thus be much simpler and better models than malignant ones for this line of research [701, 702]. Future therapeutic research should be focused on identifying the extracellular and intracellular factors (such as embryonic ones) that control tumor cells' phenotypes and on establishing approaches or drugs that can revert cancer cells to a differentiated state, either maturation or metaplasia.
Abbreviations
DMSO: dimethyl sulfoxide; ePS or iPS: embryonic or induced pluripotent stem (cells); HP: Helicobacter pylori; HTLV-1: human T cell lymphotropic virus type I; TOFT: tissue organization field theory; 3T3: transferring 3×103 cells from one flask to another every 3 days.
Acknowledgements
We would like to thank Dr. Fred Bogott at Austin Medical Center, Mayo Clinic in Austin, Minnesota, USA, for his excellent English editing of this manuscript.
Funding
This work was supported by grants from the Natural Science Foundation of China to Dezhong Joshua Liao (grant No. 81660501 and 82060489).
Data Availability Statement
Not applicable, as this is a review and no unpublished data and materials are involved. A preprint has previously been published [703], which differs greatly from the current version and contains many fewer references.
Author Contributions
SZ and JW drafted the manuscript. NZX, ML and YH prepared the figures and tables and participated in the discussion. FD, HM, WXY and DJL conceptualized the manuscript. LZ edited the manuscript and contributed to the discussions. DJL finalized the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci U S A. 2010 107 Suppl 1(Suppl 1): 1725-1730
2. Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016;14(8):e1002533-doi 10.1371/journal.pbio.1002533
3. Sender R, Milo R. The distribution of cellular turnover in the human body. Nat Med. 2021;27(1):45-48
4. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745-755
5. Belizario JE. The humankind genome: from genetic diversity to the origin of human diseases. Genome. 2013;56(12):705-716
6. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13(11):1677-1693
7. Mortezaei Z, Tavallaei M. Recent innovations and in-depth aspects of post-genome wide association study (Post-GWAS) to understand the genetic basis of complex phenotypes. Heredity (Edinb). 2021;127(6):485-497
8. Suh A, Dion-Côté AM. New Perspectives on the Evolution of Within-Individual Genome Variation and Germline/Soma Distinction. Genome Biol Evol. 2021;13(6):doi 10.1093/gbe/evab095
9. Queremel Milani DA, Chauhan PR. Genetics, Mosaicism. In. Treasure Island (FL): StatPearls Publishing. 2021 p. https://www.ncbi.nlm.nih.gov/books/NBK559193/
10. Mangiavacchi A, Liu P, Della VF, Orlando V. New insights into the functional role of retrotransposon dynamics in mammalian somatic cells. Cell Mol Life Sci. 2021;78(13):5245-5256
11. Mustjoki S, Young NS. Somatic Mutations in “Benign” Disease. N Engl J Med. 2021;384(21):2039-2052
12. Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K. et al. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell. 2008;135(5):879-893
13. Tenaillon O, Matic I. The Impact of Neutral Mutations on Genome Evolvability. Curr Biol. 2020;30(10):R527-R534
14. Sniegowski PD, Gerrish PJ. Beneficial mutations and the dynamics of adaptation in asexual populations. Philos Trans R Soc Lond B Biol Sci. 2010;365(1544):1255-1263
15. Pilzecker B, Jacobs H. Mutating for Good: DNA Damage Responses During Somatic Hypermutation. Front Immunol. 2019;10:438-doi 10.3389/fimmu.2019.00438
16. Thorpe J, Osei-Owusu IA, Avigdor BE, Tupler R, Pevsner J. Mosaicism in Human Health and Disease. Annu Rev Genet. 2020;54:487-510
17. Wilson DM, III, Thompson LH. Molecular mechanisms of sister-chromatid exchange. Mutat Res. 2007;616(1-2):11-23
18. Hörandl. A combinational theory for maintenance of sex. Heredity (Edinb). 2009;103(6):445-457
19. Brown MS, Bishop DK. DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb Perspect Biol. 2014;7(1):a016659-doi 10.1101/cshperspect.a016659
20. Humphryes N, Hochwagen A. A non-sister act: recombination template choice during meiosis. Exp Cell Res. 2014;329(1):53-60
21. Muller HJ. Some genetic aspects of sex. Am Nat. 1932;66(1):118-138.doi 10.1086/280418
22. Inaba T, Nagamachi A. Revertant somatic mosaicism as a cause of cancer. Cancer Sci. 2021;112(4):1383-1389
23. Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the Germinal Center Response. Front Immunol. 2018;9:2469-doi 10.3389/fimmu.2018.02469
24. Wilson IA, Stanfield RL. 50 Years of structural immunology. J Biol Chem. 2021;296:100745-doi 10.1016/j.jbc.2021.100745
25. Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB. et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467(7316):707-710
26. Lin YH, Zhang S, Zhu M, Lu T, Chen K, Wen Z. et al. Mice With Increased Numbers of Polyploid Hepatocytes Maintain Regenerative Capacity But Develop Fewer Hepatocellular Carcinomas Following Chronic Liver Injury. Gastroenterology. 2020;158(6):1698-1712
27. Verheijen BM, Vermulst M, van Leeuwen FW. Somatic mutations in neurons during aging and neurodegeneration. Acta Neuropathol. 2018;135(6):811-826
28. Costantino I, Nicodemus J, Chun J. Genomic Mosaicism Formed by Somatic Variation in the Aging and Diseased Brain. Genes (Basel). 2021;12(7):1071.-doi 10.3390/genes12071071
29. Avila J, Gómez-Ramos A, oriano E. Variations in brain DNA. Front Aging Neurosci. 2014;6:323-doi 10.3389/fnagi.2014.00323
30. Evrony GD. One brain, many genomes. Science. 2016;354(6312):557-558
31. Deary IJ, Cox SR, Hill WD. Genetic variation, brain, and intelligence differences. Mol Psychiatry. 2021: doi: 10.1038/s41380-021-01027-y.
32. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV. et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415-421
33. Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2(5):331-341
34. Calabrese EJ. The Mistaken Birth and Adoption of LNT: An Abridged Version. Dose Response. 2017;15(4):1559325817735478
35. Calabrese EJ. Origin of the linearity no threshold (LNT) dose-response concept. Arch Toxicol. 2013;87(9):1621-1633
36. Gelboin HV. Carcinogens, enzyme induction, and gene action. Adv Cancer Res. 1967;10:1-81
37. Rubin H. Carcinogenicity tests. Science. 1976;191(4224):241-245
38. Rubin H. On the nature of enduring modifications induced in cells and organisms. Am J Physiol. 1990;258(2 Pt 1):L19-L24
39. Heng HH, Stevens JB, Bremer SW, Liu G, Abdallah BY, Ye CJ. Evolutionary mechanisms and diversity in cancer. Adv Cancer Res. 2011;112:217-253
40. Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY. et al. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013;32(3-4):325-340
41. Heng HH, Horne SD, Chaudhry S, Regan SM, Liu G, Abdallah BY. et al. A Postgenomic Perspective on Molecular Cytogenetics. Curr Genomics. 2018;19(3):227-239
42. Heng J, Heng HH. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes (Basel). 2021;13(1):101.-doi 10.3390/genes13010101
43. Hirpara A, Bloomfield M, Duesberg P. Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers. Genes (Basel). 2018 9(8). doi: 10.3390/genes9080402
44. Bignold LP, Coghlan BL, Jersmann HP. Cancer morphology, carcinogenesis and genetic instability: a background. EXS. 2006(96): 1-24.
45. Duesberg P, Li R, Fabarius A, Hehlmann R. The chromosomal basis of cancer. Cell Oncol. 2005;27(5-6):293-318
46. Hardy PA, Zacharias H. Reappraisal of the Hansemann-Boveri hypothesis on the origin of tumors. Cell Biol Int. 2005;29(12):983-992
47. Bignold LP, Coghlan BL, Jersmann HP. Hansemann, Boveri, chromosomes and the gametogenesis-related theories of tumours. Cell Biol Int. 2006;30(7):640-644
48. Wunderlich V. [“He corrects my view and develops it further”. Comments by David von Hansemann on the monograph by Theodor Boveri Concerning the origin of malignant tumors (1914)]. Ber Wiss. 2011;34(3):263-283
49. Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl 1):1-84
50. Von Hansemann DP. On the asymmetrical division of cells in epithelial carcinomata and their biological importance. Virchows Arch. 890; 119: 299-326.
51. Tyzzer EE. A Series of spontaneous tumors in Mice with Observations on the Influence of Heredity on the Frequency of their Occurrence. J Med Res. 1909;21(3):479-518
52. Tyzzer EE. A Study of Heredity in Relation to the Development of tumors in Mice. J Med Res. 1907;17(2):199-211
53. Rubin H. Rethinking “cancer as a dynamic developmental disorder” a quarter century later. Cancer Res. 2009;69(6):2171-2175
54. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759-767
55. Lipsick J. A History of Cancer Research: Carcinogens and Mutagens. Cold Spring Harb Perspect Med. 2021 11(3): -doi: 10.1101/cshperspect.a035857
56. Sonnenschein C, Soto AM. Over a century of cancer research: Inconvenient truths and promising leads. PLoS Biol. 2020;18(4):e3000670-doi 10.1371/journal.pbio.3000670
57. Vendramin R, Litchfield K, Swanton C. Cancer evolution: Darwin and beyond. EMBO J. 2021: e108389-doi: 10.15252/embj.2021108389.
58. NORDLING CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7(1):68-72
59. Solomon E, Borrow J, Goddard AD. Chromosome aberrations and cancer. Science. 1991;254(5035):1153-1160
60. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23-28
61. Greaves M. Nothing in cancer makes sense except. BMC Biol. 2018;16(1):22-doi 10.1186/s12915-018-0493-8
62. Wang G, Chen L, Yu B, Zellmer L, Xu N, Liao DJ. Learning about the Importance of Mutation Prevention from Curable Cancers and Benign Tumors. J Cancer. 2016;7(4):436-445
63. Giraudeau M, Sepp T, Ujvari B, Renaud F, Tasiemski A, Roche B. et al. Differences in mutational processes and intra-tumour heterogeneity between organs: The local selective filter hypothesis. Evol Med Public Health. 2019;2019(1):139-146
64. Niu N, Zhang J, Zhang N, Mercado-Uribe I, Tao F, Han Z. et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis. 2016;5(12):e281-doi 10.1038/oncsis.2016.75
65. Tainsky MA, Bischoff FZ, Strong LC. Genomic instability due to germline p53 mutations drives preneoplastic progression toward cancer in human cells. Cancer Metastasis Rev. 1995;14(1):43-48
66. Bischoff FZ, Yim SO, Pathak S, Grant G, Siciliano MJ, Giovanella BC. et al. Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: aneuploidy and immortalization. Cancer Res. 1990;50(24):7979-7984
67. de GJ, de NC. A chromosomal theory of carcinogenesis. Ann Intern Med. 1968;69(2):381-391
68. Jia Y, Chen L, Ma Y, Zhang J, Xu N, Liao DJ. To Know How a Gene Works, We Need to Redefine It First but then, More Importantly, to Let the Cell Itself Decide How to Transcribe and Process Its RNAs. Int J Biol Sci. 2015;11(12):1413-1423
69. Berenblum I, SHUBIK P. An experimental study of the initiating state of carcinogenesis, and a re-examination of the somatic cell mutation theory of cancer. Br J Cancer. 1949;3(1):109-118
70. Burdette WJ. The significance of mutation in relation to the origin of tumors: a review. Cancer Res. 1955;15(4):201-226
71. Earle WR. Production of malignancy in vitro. J Natl Cancer Inst. 1943;4(1):131-248
72. Sanford KK. Malignant transformation of cells in vitro. Int Rev Cytol. 1965;18:249-311
73. Hayflick L. Oncogenesis in vitro. Natl Cancer Inst Monogr. 1967;26:355-385
74. Vineis P, Schatzkin A, Potter JD. Models of carcinogenesis: an overview. Carcinogenesis. 2010;31(10):1703-1709
75. Rous P. Surmise and fact on the nature of cancer. Nature. 1959;183(4672):1357-1361
76. Baker SG. The case for a cancer paradox initiative. Carcinogenesis. 2021;42(8):1023-1025
77. Gabor Miklos GL. The human cancer genome project-one more misstep in the war on cancer. Nat Biotechnol. 2005;23(5):535-537
78. Gonzalez-Garcia I, Sole RV, Costa J. Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci U S A. 2002;99(20):13085-13089
79. Rous P. Recent Advances in Cancer Research. Bull N Y Acad Med. 1947;23(2):65-78
80. Brash D, Cairns J. The mysterious steps in carcinogenesis. Br J Cancer. 2009;101(3):379-380
81. Brash D, Cairns J. The mysterious steps in carcinogenesis: addendum. Br J Cancer. 2009;101(8):1490. doi: 10.1038/sj.bjc.6605332
82. Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335(6186):142-145
83. Cairns J. Mutation and cancer: the antecedents to our studies of adaptive mutation. Genetics. 1998;148(4):1433-1440
84. Kennedy AR, Fox M, Murphy G, Little JB. Relationship between x-ray exposure and malignant transformation in C3H 10T1/2 cells. Proc Natl Acad Sci U S A. 1980;77(12):7262-7266
85. Kennedy AR, Cairns J, Little JB. Timing of the steps in transformation of C3H 10T 1/2 cells by X-irradiation. Nature. 1984;307(5946):85-86
86. Kennedy AR. Is there a critical target gene for the first step in carcinogenesis? Environ Health Perspect. 1991;93:199-203
87. Kennedy AR. Is a mutagenic event involved in radiation induced malignant transformation? Mutat Res. 1996;350(1):81-91
88. Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? Bioessays. 2006;28(5):515-524
89. Rubin H, Rubin AL. Phenotypic selection as the biological mode of epigenetic conversion and reversion in cell transformation. Proc Natl Acad Sci U S A. 2018;115(4):E725-E732
90. Shvemberger IN. Conversion of malignant cells into normal ones. Int Rev Cytol. 1986;103:341-386
91. Soto AM, Sonnenschein C. One hundred years of somatic mutation theory of carcinogenesis: is it time to switch? Bioessays. 2014;36(1):118-120
92. Brucher BL, Jamall IS. Somatic Mutation Theory - Why it's Wrong for Most Cancers. Cell Physiol Biochem. 2016;38(5):1663-1680
93. Baker SG. A cancer theory kerfuffle can lead to new lines of research. J Natl Cancer Inst. 2014;107(2):doi 10.1093/jnci/dju405
94. Bizzarri M, Cucina A, Biava PM, Proietti S, D'Anselmi F, Dinicola S. et al. Embryonic morphogenetic field induces phenotypic reversion in cancer cells. Review article. Curr Pharm Biotechnol. 2011;12(2):243-253
95. Sonnenschein C, Soto AM. The aging of the 2000 and 2011 Hallmarks of Cancer reviews: a critique. J Biosci. 2013;38(3):651-663
96. Sonnenschein C, Soto AM, Rangarajan A, Kulkarni P. Competing views on cancer. J Biosci. 2014;39(2):281-302
97. Sonnenschein C, Soto AM. Carcinogenesis explained within the context of a theory of organisms. Prog Biophys Mol Biol. 2016;122(1):70-76
98. Soto AM, Sonnenschein C. Emergentism as a default: cancer as a problem of tissue organization. J Biosci. 2005;30(1):103-118
99. Soto AM, Sonnenschein C. Paradoxes in Carcinogenesis: There Is Light at the End of That Tunnel!. Disrupt Sci Technol. 2013;1(3):154-156
100. Rubin H. Cancer as a dynamic developmental disorder. Cancer Res. 1985;45(7):2935-2942
101. Vineis P, Berwick M. The population dynamics of cancer: a Darwinian perspective. Int J Epidemiol. 2006;35(5):1151-1159
102. Davies PC, Demetrius L, Tuszynski JA. Cancer as a dynamical phase transition. Theor Biol Med Model. 2011;8:30-doi 10.1186/1742-4682-8-30
103. Frieden BR, Gatenby RA. Information dynamics in living systems: prokaryotes, eukaryotes, and cancer. PLoS One. 2011;6(7):e22085-doi 10.1371/journal.pone.0022085
104. Ellis RJ. Non-equilibrium thermodynamics and degenerate disease. Med Hypotheses. 1984;14(1):83-98
105. Clark WH. Tumour progression and the nature of cancer. Br J Cancer. 1991;64(4):631-644
106. Clark WH Jr. The nature of cancer: morphogenesis and progressive (self)-disorganization in neoplastic development and progression. Acta Oncol. 1995;34(1):3-21
107. Pienta KJ, Hammarlund EU, Axelrod R, Amend SR, Brown JS. Convergent Evolution, Evolving Evolvability, and the Origins of Lethal Cancer. Mol Cancer Res. 2020;18(6):801-810 doi: 10.1158/1541-7786.MCR-19-1158
108. Erenpreisa J, Giuliani A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. Int J Mol Sci. 2019;21(1):240-doi 10.3390/ijms21010240
109. de GJ. The malignant primate? Ann Genet. 1991;34(3-4):137-142
110. Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283-350
111. Zhang J, Lou XM, Jin LY, Zhou RJ, Liu SQ, Xu NZ. et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: clearance of a few misconceptions. Oncoscience. 2014;1(6):407-422
112. Zhang J, Lou X, Zellmer L, Liu S, Xu N, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1(9):580-590
113. Mazzocca A. The Systemic-Evolutionary Theory of the Origin of Cancer (SETOC): A New Interpretative Model of Cancer as a Complex Biological System. Int J Mol Sci. 2019;20(19):4885-doi 10.3390/ijms20194885
114. Mazzocca A, Fais S. New hypotheses for cancer generation and progression. Med Hypotheses. 2021;152:110614.-DOI 10.1016/j.mehy.2021.110614
115. Carvalho J. Cell Reversal From a Differentiated to a Stem-Like State at Cancer Initiation. Front Oncol. 2020;10:541-doi 10.3389/fonc.2020.00541
116. Nicholson JM. Will we cure cancer by sequencing thousands of genomes? Mol Cytogenet. 2013;6(1):57-doi 10.1186/1755-8166-6-57
117. Kwasniewski W, Stupak A, Kotarski J, Gozdzicak-Jozefiak A. Chaos and cancers. Theories concerning carcinogenesis. Ginekol Pol. 2021;92(4):318-321
118. Baker SG. Rethinking carcinogenesis: The detached pericyte hypothesis. Med Hypotheses. 2020;144:110056-doi 10.1016/j.mehy.2020.110056
119. Venerin AA, Venerina YA, Alexandrov YI. Cell functioning in norm and pathology in terms of the activity paradigm: Oncogenesis. Med Hypotheses. 2020;144:110240-doi 10.1016/j.mehy.2020.110240
120. Loser T. Process analysis of carcinogenesis: concept derivation of the tissue function “preservation of a homogeneous gene expression”. Theory Biosci. 2018;137(1):85-97
121. Blagosklonny MV. Molecular theory of cancer. Cancer Biol Ther. 2005;4(6):621-627
122. Sigston EAW, Williams BRG. An Emergence Framework of Carcinogenesis. Front Oncol. 2017;7:198-doi 10.3389/fonc.2017.00198
123. Rosenfeld S. Global consensus theorem and self-organized criticality: unifying principles for understanding self-organization, swarm intelligence and mechanisms of carcinogenesis. Gene Regul Syst Bio. 2013;7:23-39
124. Rosenfeld S. Are the somatic mutation and tissue organization field theories of carcinogenesis incompatible? Cancer Inform. 2013;12:221-229
125. Bedessem B, Ruphy S. SMT and TOFT Integrable After All: A Reply to Bizzarri and Cucina. Acta Biotheor. 2017;65(1):81-85
126. Bedessem B, Ruphy S. SMT or TOFT? How the two main theories of carcinogenesis are made (artificially) incompatible. Acta Biotheor. 2015;63(3):257-267
127. Tidwell TR, Soreide K, Hagland HR. Aging, Metabolism, and Cancer Development: from Peto's Paradox to the Warburg Effect. Aging Dis. 2017;8(5):662-676
128. Callier V. Core Concept: Solving Peto's Paradox to better understand cancer. Proc Natl Acad Sci U S A. 2019;116(6):1825-1828
129. Tollis M, Boddy AM, Maley CC. Peto's Paradox: how has evolution solved the problem of cancer prevention? BMC Biol. 2017;15(1):60-doi 10.1186/s12915-017-0401-7
130. Peto R, Roe FJ, Lee PN, Levy L, Clack J. Cancer and ageing in mice and men. Br J Cancer. 1975;32(4):411-426
131. Cagan A, Baez-Ortega A, Brzozowska N, Abascal F, Coorens THH, Sanders MA. et al. Somatic mutation rates scale with lifespan across mammals. Nature. 2022;604(7906):517-524
132. Laconi E, Marongiu F, DeGregori J. Cancer as a disease of old age: changing mutational and microenvironmental landscapes. Br J Cancer. 2020;122(7):943-952
133. DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2013;32(15):1869-1875
134. Tomasetti C, Poling J, Roberts NJ, London NR Jr, Pittman ME, Haffner MC. et al. Cell division rates decrease with age, providing a potential explanation for the age-dependent deceleration in cancer incidence. Proc Natl Acad Sci U S A. 2019;116(41):20482-20488
135. BURCH PR. NATURAL AND RADIATION CARCINOGENESIS IN MAN. I. THEORY OF INITIATION PHASE. Proc R Soc Lond B Biol Sci. 1965;162:223-239
136. Hanson HA, Smith KR, Stroup AM, Harrell CJ. An age-period-cohort analysis of cancer incidence among the oldest old, Utah 1973-2002. Popul Stud (Camb). 2015;69(1):7-22
137. Horiuchi S, Wilmoth JR. Deceleration in the age pattern of mortality at older ages. Demography. 1998;35(4):391-412
138. Dou X, Tong P, Huang H, Zellmer L, He Y, Jia Q. et al. Evidence for immortality and autonomy in animal cancer models is often not provided, which causes confusion on key issues of cancer biology. J Cancer. 2020;11(10):2887-2920
139. Shi M, Zhou H, Lei M, Chen L, Zellmer L, He Y. et al. Spontaneous Cancers, But Not Many Induced Ones in Animals, Resemble Semi-New Organisms that Possess a Unique Programmed Cell Death Mode Different from Apoptosis, Senescent Death, Necrosis and Stress-Induced Cell Death. J Cancer. 2018;9(24):4726-4735
140. Leroi AM, Koufopanou V, Burt A. Cancer selection. Nat Rev Cancer. 2003;3(3):226-231
141. Hammarlund EU, Amend SR, Pienta KJ. The issues with tissues: the wide range of cell fate separation enables the evolution of multicellularity and cancer. Med Oncol. 2020;37(7):62-doi 10.1007/s12032-020-01387-5
142. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A. et al. Cancer statistics for the year 2020: An overview. Int J Cancer. 2021: -doi: 10.1002/ijc.33588.
143. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7-33
144. Aparicio T, Zaanan A, Mary F, Afchain P, Manfredi S, Evans TR. Small Bowel Adenocarcinoma. Gastroenterol Clin North Am. 2016;45(3):447-457
145. Blanpain C, Horsley V, Fuchs E. Epithelial stem cells: turning over new leaves. Cell. 2007;128(3):445-458
146. Ocasio Quinones GA, Woolf A. Small Bowel Cancer. Treasure Island (FL): StatPearls Publishing. 2021 https://www.ncbi.nlm.nih.gov/books/NBK560725/
147. Lowenfels AB. Why are small-bowel tumours so rare? Lancet. 1973;1(7793):24-26
148. Manley J, Ibrahim Y, Ansell J, Alastal H, Rasheed A. Small bowel adenocarcinoma: Case reports and review of the literature. Arab J Gastroenterol. 2021;22(3):240-245
149. Bignold LP. The mutator phenotype theory of carcinogenesis and the complex histopathology of tumours: support for the theory from the independent occurrence of nuclear abnormality, loss of specialisation and invasiveness among occasional neoplastic lesions. Cell Mol Life Sci. 2003;60(5):883-891
150. Mally A, Chipman JK. Non-genotoxic carcinogens: early effects on gap junctions, cell proliferation and apoptosis in the rat. Toxicology. 2002;180(3):233-248
151. Nohmi T. Thresholds of Genotoxic and Non-Genotoxic Carcinogens. Toxicol Res. 2018;34(4):281-290
152. Bielschowsky F. Chronic iodine deficiency as cause of neoplasia in thyroid and pituitary of aged rats. Br J Cancer. 1953;7(2):203-213
153. Bielschowsky F, Hall WH. Carcinogenesis in the thyroidectomized rat. Br J Cancer. 1953;7(3):358-366
154. Biskind GR, Kordan B, Biskind MS. Ovary transplanted to spleen in rats: the effect of unilateral castration, pregnancy, and subsequent castration. Cancer Res. 1950;10(5):309-318
155. Biskind GR, Biskind MS. Atrophy of Ovaries Transplanted to the Spleen in Unilaterally Castrated Rats; Proliferative Changes Following Subsequent Removal of Intact Ovary. Science. 1948;108(2797):137-138
156. Li MH, Gardner WU. Tumors in Intrasplenic Ovarian Transplants in Castrated Mice. Science. 1947;105(2714):13-15
157. Biskind GR, Biskind MS. Experimental ovarian tumors in rats. Am J Clin Pathol. 1949;19(6):501-521
158. Liao DJ, Dickson RB. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J Steroid Biochem Mol Biol. 2002;80(2):175-189
159. Liao DJ, Dickson RB. Steroid hormone-growth factor interactions in proliferative controls of the mammary gland and breast cancer: a rapidly evolving perspective. J Steroid Biochem Mol Biol. 2002;80(2):135-136
160. Liao DZ, Pantazis CG, Hou X, Li SA. Promotion of estrogen-induced mammary gland carcinogenesis by androgen in the male Noble rat: probable mediation by steroid receptors. Carcinogenesis. 1998;19(12):2173-2180
161. Versteeg R. Cancer: Tumours outside the mutation box. Nature. 2014;506(7489):438-439
162. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82-93
163. Hirpara A, Bloomfield M, Duesberg P. Speciation Theory of Carcinogenesis Explains Karyotypic Individuality and Long Latencies of Cancers. Genes (Basel). 2018;9(8):402.-doi 10.3390/genes9080402
164. Adashek JJ, Kato S, Lippman SM, Kurzrock R. The paradox of cancer genes in non-malignant conditions: implications for precision medicine. Genome Med. 2020;12(1):16-doi 10.1186/s13073-020-0714-y
165. Venkatachalam A, Pikarsky E, Ben-Neriah Y. Putative homeostatic role of cancer driver mutations. Trends Cell Biol. 2021: S0962-8924(21)00142-2-doi: 10.1016/j.tcb. 2021 07.002
166. Kato S, Lippman SM, Flaherty KT, Kurzrock R. The Conundrum of Genetic “Drivers” in Benign Conditions. J Natl Cancer Inst. 2016;108(8):doi 10.1093/jnci/djw036
167. Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA. et al. The mutational landscape of human somatic and germline cells. Nature. 2021: -doi: 10.1038/s41586-021-03822-7.
168. Martincorena I. Somatic mutation and clonal expansions in human tissues. Genome Med. 2019;11(1):35-DOI 10.1038/d41586-020-02749-9
169. Baker SG, Soto AM, Sonnenschein C, Cappuccio A, Potter JD, Kramer BS. Plausibility of stromal initiation of epithelial cancers without a mutation in the epithelium: a computer simulation of morphostats. BMC Cancer. 2009;9:89-doi 10.1186/1471-2407-9-89
170. Brucher BL, Jamall IS. Cell-cell communication in the tumor microenvironment, carcinogenesis, and anticancer treatment. Cell Physiol Biochem. 2014;34(2):213-243
171. Domazet-Loso T, Klimovich A, Anokhin B, Anton-Erxleben F, Hamm MJ, Lange C. et al. Naturally occurring tumours in the basal metazoan Hydra. Nat Commun. 2014;5:4222. doi: 10.1038/ncomms5222
172. Rathje K, Mortzfeld B, Hoeppner MP, Taubenheim J, Bosch TCG, Klimovich A. Dynamic interactions within the host-associated microbiota cause tumor formation in the basal metazoan Hydra. PLoS Pathog. 2020;16(3):e1008375.-doi 10.1371/journal.ppat.1008375
173. Potter VR. Cancer as a problem in intercellular communication: regulation by growth-inhibiting factors (Chalones). Prog Nucleic Acid Res Mol Biol. 1983;29:161-173
174. Williams DF. Carcinogenicity of implantable materials: experimental and epidemiological evidence. Int Urogynecol J. 2014;25(5):577-580
175. Moizhess TG. Carcinogenesis induced by foreign bodies. Biochemistry (Mosc). 2008;73(7):763-775
176. Moizhess TG, VASILIEV JM. Early and late stages of foreign-body carcinogenesis can be induced by implants of different shapes. Int J Cancer. 1989;44(3):449-453
177. Brand KG, Johnson KH, Buoen LC. Foreign body tumorigenesis. CRC Crit Rev Toxicol. 1976;4(4):353-394
178. Turner FC. Sarcomas at sites of subcutaneously implanted bakelite disks in rats. J Natl Cancer Inst. 941; 2(1): 81-83.
179. Potter M. A resume of the current status of the development of plasma-cell tumors in mice. Cancer Res. 1968;28(9):1891-1896
180. Potter M. The developmental history of the neoplastic plasma cell in mice: a brief review of recent developments. Semin Hematol. 1973;10(1):19-32
181. Potter M. The early history of plasma cell tumors in mice, 1954-1976. Adv Cancer Res. 2007;98:17-51
182. Furst A, Haro RT. A survey of metal carcinogenesis. Prog Exp Tumor Res. 1969;12:102-133
183. Oppenheimer BS, Oppenheimer ET, Stout AP. Sarcomas induced in rats by implanting cellophane. Proc Soc Exp Biol Med. 1948;67(1):33-doi 10.3181/00379727-67-16195p
184. Oppenheimer BS, Oppenheimer ET, Stout AP. Sarcomas induced in rodents by imbedding various plastic films. Proc Soc Exp Biol Med. 1952;79(3):366-369
185. Oppenheimer BS, Oppenheimer ET, Ddanishefsky I, Stout AP, Eirich FR. Further studies of polymers as carcinogenic agents in animals. Cancer Res. 1955;15(5):333-340
186. Fitzhugh AF, Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I, Eirich FR. Malignant tumors and high polymers. Science. 1953;118(3078):783-784
187. Oppenheimer BS, Oppenheimer ET, Stout AP, Danishefsky I. Malignant tumors resulting from embedding plastics in rodents. Science. 1953;118(3063):305-306
188. Karp RD, Johnson KH, Buoen LC, Ghobrial HK, Brand I, Brand KG. Tumorigenesis by Millipore filters in mice: histology and ultrastructure of tissue reactions as related to pore size. J Natl Cancer Inst. 1973;51(4):1275-1285
189. Okada F. Beyond foreign-body-induced carcinogenesis: impact of reactive oxygen species derived from inflammatory cells in tumorigenic conversion and tumor progression. Int J Cancer. 2007;121(11):2364-2372
190. Iomhair MM, Lavelle SM. Effect of film size on production of foreign body sarcoma by perforated film implants. Technol Health Care. 1997;5(4):331-334
191. DeCoster RC, Clemens MW, Di NA, Lynch EB, Bonaroti AR, Rinker BD. et al. Cellular and Molecular Mechanisms of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Plast Reconstr Surg. 2021;147(1):30e-41e
192. Blue CT, Pospiech EA, Lewis JM, Nodit L. Soft Tissue Malignancy Due to Long-Standing Foreign Bodies after Shotgun Blast. Am Surg. 2017;83(7):e261-e263
193. Oppernheimer BS, Oppenheimer ET, Stout AP, Danishefsky I, Willhite M. Studies of the mechanism of carcinogenesis by plastic films. Acta Unio Int Contra Cancrum. 1959;15:659-663
194. Miller FR. The induced development and histogenesis of plam cells. J Exp Med. 1931;54(3):333-347
195. Schmidt A, Weber OF. In memoriam of Rudolf virchow: a historical retrospective including aspects of inflammation, infection and neoplasia. Contrib Microbiol. 2006;13:1-15
196. Cuddihy J. Rudolf Ludwig Carl Virchow. Cancer Cells. 1991;3(3):110-112
197. Morton LT. Rudolf Ludwig Carl Virchow (1821-1902): bibliography. J Med Biogr. 1993;1(1):46-47
198. Wullstein HL, Hellmer L. Rudolf Ludwig Carl Virchow (1821 to 1902). Arch Otolaryngol. 1970;92(3):299-301
199. Triolo VA. Nineteenth centruy foundations of cancer research. Origins of experimental research. Cancer Res. 1964;24:4-27
200. No Author. Retrospect: 1889. Br Med J. 1889;2(1513):1433-1471
201. Power D. Some effects of chronic irritation upon living tissues, being first steps in a rational study of cancer. Br Med J. 893; 2:830-834.
202. Billingham RE, Orr JW, Woodhouse DL. Epidermal transplantation during chemical carcinogenesis. Nature. 1950;166(4234):1080-doi 10.1038/1661080a0
203. Billingham RE. Transplantation: past, present and future. J Invest Dermatol. 1963;41:165-180
204. Marchant J, Orr JW. Further attempts to analyse the role of epidermis and deeper tissues in experimental chemical carcinogenesis by transplantation and other method. Br J Cancer. 1953;7(3):329-341
205. Staab HJ, Heilbronner H, Schrader M, Anderer FA. In vivo induction of neoplastic growth in nude mouse connective tissue adjacent to xenografted human tumors. J Cancer Res Clin Oncol. 1983;106(1):27-35
206. Goldenberg DM, Pavia RA. Malignant potential of murine stromal cells after transplantation of human tumors into nude mice. Science. 1981;212(4490):65-67
207. Loeb L. Further Investigations in Transplantation of tumors. J Med Res. 1902;8(1):44-73
208. Tveit KM, Fodstad O, Brøgger A, Olsnes S. Human embryonal carcinoma grown in athymic mice and in vitro. Cancer Res. 1980;40(3):949-953
209. Houghton JA, Taylor DM. Maintenance of biological and biochemical characteristics of human colorectal tumours during serial passage in immune-deprived mice. Br J Cancer. 1978;37(2):199-212
210. Popescu NC, Cioloca L, Liciu F, Encut I. A study of some tumours of human origin. I. Chromosomes of rat tumour (HR 18) and mouse tumour (HM 18) obtained by heterotransplantation of a human melanocarcinoma. Eur J Cancer. 1970;6(3):175-180
211. Huebner RJ, Fish DC, Djurickovic D, Trimmer RW, Bare AL, Bare RM. et al. Induction of rat sarcomas in rats treated with antithymocyte sera after transplantation of human cancer cells. Proc Natl Acad Sci U S A. 1979;76(4):1793-1794
212. Russell PJ, Brown J, Grimmond S, Stapleton P, Russell P, Raghavan D. et al. Tumour-induced host stromal-cell transformation: induction of mouse spindle-cell fibrosarcoma not mediated by gene transfer. Int J Cancer. 1990;46(2):299-309
213. Kompf J, Staab HJ, Heilbronner H, Anderer FA, Ritter H. Cell fusion responsible for horizontal oncogenesis by human tumors in nude mice. Biochem Biophys Res Commun. 1984;124(3):933-938
214. Goldenberg DM, Pavia RA. Horizontal transmission of malignant conditions rediscovered. N Engl J Med. 1981;305(5):283-284
215. Goldenberg DM, Pavia RA. In vivo horizontal oncogenesis by a human tumor in nude mice. Proc Natl Acad Sci U S A. 1982;79(7):2389-2392
216. Goldenberg DM, Zagzag D, Heselmeyer-Haddad KM, Berroa Garcia LY, Ried T, Loo M. et al. Horizontal transmission and retention of malignancy, as well as functional human genes, after spontaneous fusion of human glioblastoma and hamster host cells in vivo. Int J Cancer. 2012;131(1):49-58
217. Goldenberg DM. Horizontal transmission of malignancy by cell-cell fusion. Expert Opin Biol Ther. 2012;12(Suppl 1):S133-S139
218. Goldenberg DM, Gold DV, Loo M, Liu D, Chang CH, Jaffe ES. Horizontal transmission of malignancy: in-vivo fusion of human lymphomas with hamster stroma produces tumors retaining human genes and lymphoid pathology. PLoS One. 2013;8(2):e55324-doi 10.1371/journal.pone.0055324
219. Abdouh M, Floris M, Gao ZH, Arena V, Arena M, Arena GO. Colorectal cancer-derived extracellular vesicles induce transformation of fibroblasts into colon carcinoma cells. J Exp Clin Cancer Res. 2019;38(1):257-doi 10.1186/s13046-019-1248-2
220. Wlodarski KH, Reddi HA. Tumor cells stimulate in vivo periosteal bone formation. Bone Miner. 1987;2(3):185-192
221. Ostrowski K, Wlodarski K, Aden D. Heterotopic chondrogenesis and osteogenesis induced by transformed cells: use of nude mice as a model system. Somatic Cell Genet. 1975;1(4):391-395
222. Kochanowska IE, Wlodarski K, Wojtowicz A, Niemira K, Ostrowski K. Osteogenic properties of various HeLa cell lines and the BMP family genes expression. Ann Transplant. 2002;7(4):58-62
223. Kochanowska IE, Wlodarski K, Wojtowicz A, Kinsner A, Ostrowski K. BMP-4 and BMP-6 involvement in the osteogenic properties of the HeLa cell line. Exp Biol Med (Maywood). 2002;227(1):57-62
224. Billingham RE, Orr JW, Woodhouse DL. Transplantation of skin components during chemical carcinogenesis with 20-methylcholanthrene. Br J Cancer. 1951;5(4):417-432
225. Orr JW. The mechanism of chemical carcinogenesis, with particular reference to the time of development of irreversible changes in the epithelial cells. Br Med Bull. 1958;14(2):99-101
226. Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60(5):1254-1260
227. Barcellos-Hoff MH. It takes a tissue to make a tumor: epigenetics, cancer and the microenvironment. J Mammary Gland Biol Neoplasia. 2001;6(2):213-221
228. Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment - tumorigenesis and therapy. Nat Rev Cancer. 2005;5(11):867-875
229. Barcellos-Hoff MH. Stromal mediation of radiation carcinogenesis. J Mammary Gland Biol Neoplasia. 2010;15(4):381-387
230. Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117(Pt 8):1495-1502
231. Biskind MSBGR. Development of tumors in the rat ovary after transplntation into the spleen. Proc Soc Exp Biol Med. 1944;55:176-179
232. Stevens LC. The development of teratomas from intratesticular grafts of tubal mouse eggs. J Embryol Exp Morphol. 1968;20(3):329-341
233. Hornblum AM. Ethical lapses in dermatologic "research". Arch Dermatol. 1999;135(4):383-385
234. Hornblum AM. They were cheap and available: prisoners as research subjects in twentieth century America. BMJ. 1997;315(7120):1437-1441
235. Scnlon EF, Hawkings RA, Fox WW, Smith WS. Fatal homotransplanted melanoma: A case report. Cancer. 1965;18:782-789
236. Southam CM. Evidence for cancer-specific angigens in man. Prog Exp Tumor Res. 1967;9:1-39
237. Moore AE. Tumorigenic activity of cultures. Ann N Y Acad Sci. 1958;76(3):497-505
238. Moore AE, Rhoads CP, Southam CM. Homotransplantation of human cell lines. Science. 1957;125(3239):158-160
239. Moore AE, Southam CM, Sternberg SS. Neoplastic changes developing in epithelial cell lines derived from normal persons. Science. 1956;124(3212):127-129
240. Southam CM. Homotransplantation of human cell lines. Bull N Y Acad Med. 1958;34(6):416-423
241. Dou X, Chen L, Lei M, Zellmer L, Jia Q, Ling P. et al. Evaluating the Remote Control of Programmed Cell Death, with or without a Compensatory Cell Proliferation. Int J Biol Sci. 2018;14(13):1800-1812
242. Rose SM. Failure of self-inhibition in tumors. J Natl Cancer Inst. 1958;20(4):653-664
243. Greene HS. A conception of tumor autonomy based on transplantation studies: a review. Cancer Res. 1951;11(12):899-903
244. Tarabichi M, Antoniou A, Saiselet M, Pita JM, Andry G, Dumont JE. et al. Systems biology of cancer: entropy, disorder, and selection-driven evolution to independence, invasion and "swarm intelligence". Cancer Metastasis Rev. 2013;32(3-4):403-421
245. Gatenby RA, Avdieiev S, Tsai KY, Brown JS. Integrating genetic and nongenetic drivers of somatic evolution during carcinogenesis: The biplane model. Evol Appl. 2020;13(7):1651-1659
246. Soto AM, Sonnenschein C. The cancer puzzle: Welcome to organicism. Prog Biophys Mol Biol. 2021;165:114-119
247. Hakim DN, Pelly T, Kulendran M, Caris JA. Benign tumours of the bone: A review. J Bone Oncol. 2015;4(2):37-41
248. Wodajo FM. Top five lesions that do not need referral to orthopedic oncology. Orthop Clin North Am. 2015;46(2):303-314
249. Steffner R. Benign bone tumors. Cancer Treat Res. 2014;162:31-63
250. Qasem SA, DeYoung BR. Cartilage-forming tumors. Semin Diagn Pathol. 2014;31(1):10-20
251. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10(1):57-67
252. Witkowski JA. Carrel's cultures. Science. 1990;247(4949 Pt 1):1385-1386
253. Witkowski JA. Dr. Carrel's immortal cells. Med Hist. 1980;24(2):129-142
254. Carrel A. The Immortality of Animal Tissues and Its Significance. Can Med Assoc J. 1928;18(3):327-329
255. Carrel A. The Immortality of Animal Tissues and Its Significance. Edinb Med J. 1928;35(7):410-413
256. Kraemer PM, Travis GL, Ray FA, Cram LS. Spontaneous neoplastic evolution of Chinese hamster cells in culture: multistep progression of phenotype. Cancer Res. 1983;43(10):4822-4827
257. Kraemer PM, Ray FA, Brothman AR, Bartholdi MF, Cram LS. Spontaneous immortalization rate of cultured Chinese hamster cells. J Natl Cancer Inst. 1986;76(4):703-709
258. Newbold RF, Overell RW, Connell JR. Induction of immortality is an early event in malignant transformation of mammalian cells by carcinogens. Nature. 1982;299(5884):633-635
259. Ray FA, Bartholdi MF, Kraemer PM, Cram LS. Spontaneous in vitro neoplastic evolution: recurrent chromosome changes of newly immortalized Chinese hamster cells. Cancer Genet. Cytogenet. 1986;21(1):35-51
260. Cram LS, Bartholdi MF, Ray FA, Travis GL, Kraemer PM. Spontaneous neoplastic evolution of Chinese hamster cells in culture: multistep progression of karyotype. Cancer Res. 1983;43(10):4828-4837
261. Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299-313
262. Todaro GJ, Green H, Goldberg BD. Transformation of properties of an established cell line by SV40 and Polyoma virus. Proc Natl Acad Sci U S A. 1964;51:66-73
263. Nilausen K, Green H. Reversible arrest of growth in G1 of an established fibroblast line (3T3). Exp Cell Res. 1965;40(1):166-168
264. Parodi S, Brambilla G. Relationships between mutation and transformation frequencies in mammalian cells treated "in vitro" with chemical carcinogens. Mutat Res. 1977;47(1):53-74
265. Mori K, Shibanuma M, Nose K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004;64(20):7464-7472
266. Miyasaka K, Kawauchi S. Homozygous deletion of mouse homolog of p16/CDKN2 gene on chromosome 4 in mouse liver epithelial cells in culture. Biol Pharm Bull. 1996;19(3):345-349
267. Huschtscha LI, Reddel RR. p16(INK4a) and the control of cellular proliferative life span. Carcinogenesis. 1999;20(6):921-926
268. Murakami Y, Sekiya T. Accumulation of genetic alterations and their significance in each primary human cancer and cell line. Mutat Res. 1998;400(1-2):421-437
269. Huschtscha LI, Noble JR, Neumann AA, Moy EL, Barry P, Melki JR. et al. Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res. 1998;58(16):3508-3512
270. Foster SA, Wong DJ, Barrett MT, Galloway DA. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol. 1998;18(4):1793-1801
271. Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005;23(2):276-292
272. Frank TS. Hereditary cancer syndromes. Arch Pathol Lab Med. 2001;125(1):85-90
273. Lynch HT, Fusaro RM, Lynch J. Hereditary cancer in adults. Cancer Detect Prev. 1995;19(3):219-233
274. Vogel F. Genetics of retinoblastoma. Hum Genet. 1979;52(1):1-54
275. Zhang J, Lou X, Shen H, Zellmer L, Sun Y, Liu S. et al. Isoforms of wild type proteins often appear as low molecular weight bands on SDS-PAGE. Biotechnol J. 2014;9(8):1044-1054
276. Shvemberger IN, Ermilov AN. Some characteristics of neoplastic cell transformation in transgenic mice. Int Rev Cytol. 1996;164:37-90
277. Berenblum I. Established principles and unresolved problems in carcinogenesis. J Natl Cancer Inst. 1978;60(4):723-726
278. Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315(6015):115-122
279. Stewart TA, Bellve AR, Leder P. Transcription and promoter usage of the myc gene in normal somatic and spermatogenic cells. Science. 1984;226(4675):707-710
280. Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell. 1984;38(3):627-637
281. Ferrell SD Jr, Ahmad I, Nguyen C, Petrova SC, Wilhelm SR, Ye Y. et al. Why is cancer so common a disease in people yet so rare at a cellular level? Med Hypotheses. 2020;144:110171
282. Durgan J, Florey O. Cancer cell cannibalism: Multiple triggers emerge for entosis. Biochim Biophys Acta. 2018;1865(6):831-841
283. Krajcovic M, Overholtzer M. Mechanisms of ploidy increase in human cancers: a new role for cell cannibalism. Cancer Res. 2012;72(7):1596-1601
284. Lozupone F, Fais S. Cancer Cell Cannibalism: A Primeval Option to Survive. Curr Mol Med. 2015;15(9):836-841
285. Matarrese P, Ciarlo L, Tinari A, Piacentini M, Malorni W. Xeno-cannibalism as an exacerbation of self-cannibalism: a possible fruitful survival strategy for cancer cells. Curr Pharm Des. 2008;14(3):245-252
286. Sharma N, Dey P. Cell cannibalism and cancer. Diagn Cytopathol. 2011;39(3):229-233
287. Blagosklonny MV. NCI's provocative questions on cancer: some answers to ignite discussion. Oncotarget. 2011;2(12):1352-1367
288. Markert CL. Neoplasia: a disease of cell differentiation. Cancer Res. 1968;28(9):1908-1914
289. Raz AA, Yamashita YM. Molding immortality from a plastic germline. Curr Opin Cell Biol. 2021;73:1-8
290. Larocca D, Lee J, West MD, Labat I, Sternberg H. No Time to Age: Uncoupling Aging from Chronological Time. Genes (Basel). 2021;12(5):611-doi 10.3390/genes12050611
291. Mikula-Pietrasik J, Pakuła M, Markowska M, Uruski P, Szczepaniak-Chicheł L, Tykarski A. et al. Nontraditional systems in aging research: an update. Cell Mol Life Sci. 2021;78(4):1275-1304
292. Vaskovicova K, Zarsky V, Rosel D, Nikolic M, Buccione R, Cvrckova F. et al. Invasive cells in animals and plants: searching for LECA machineries in later eukaryotic life. Biol Direct. 2013;8:8-doi 10.1186/1745-6150-8-8
293. Yagel S, Khokha R, Denhardt DT, Kerbel RS, Parhar RS, Lala PK. Mechanisms of cellular invasiveness: a comparison of amnion invasion in vitro and metastatic behavior in vivo. J Natl Cancer Inst. 1989;81(10):768-775
294. Yagel S, Parhar RS, Jeffrey JJ, Lala PK. Normal nonmetastatic human trophoblast cells share in vitro invasive properties of malignant cells. J Cell Physiol. 1988;136(3):455-462
295. Mareel MM, Van Roy FM, De BP. The invasive phenotypes. Cancer Metastasis Rev. 1990;9(1):45-62
296. Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol. 2020;21(7):398-414
297. Gunther J, Seyfert HM. The first line of defence: insights into mechanisms and relevance of phagocytosis in epithelial cells. Semin Immunopathol. 2018;40(6):555-565
298. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558
299. Liu J. The “life code”: A theory that unifies the human life cycle and the origin of human tumors. Semin Cancer Biol. 2020;60:380-397
300. Fais S. A nonmainstream approach against cancer. J Enzyme Inhib Med Chem. 2016;31(6):882-889
301. Mazzocca A, Ferraro G, Misciagna G, Carr BI. A systemic evolutionary approach to cancer: Hepatocarcinogenesis as a paradigm. Med Hypotheses. 2016;93:132-137
302. Mazzocca A, Ferraro G, Misciagna G, Fais S. Moving the systemic evolutionary approach to cancer forward: Therapeutic implications. Med Hypotheses. 2018;121:80-87
303. Pierce GB. Relationship between differentiation and carcinogenesis. J Toxicol Environ Health. 1977;2(6):1335-1342
304. Pierce GB, Nakane PK, Martinez-Hernandez A, Ward JM. Ultrastructural comparison of differentiation of stem cells of murine adenocarcinomas of colon and breast with their normal counterparts. J Natl Cancer Inst. 1977;58(5):1329-1345
305. Pierce GB. On the boundary between development and neoplasia. An interview with Professor G. Barry Pierce. Interview by Juan Arechaga. Int J Dev Biol. 1993;37(1):5-16
306. Pierce GB. The cancer cell and its control by the embryo. Rous-Whipple Award lecture. Am J Pathol. 1983;113(1):117-124
307. Needham J. New Advances in the Chemistry and Biology of Organized Growth: (Section of Pathology). Proc R Soc Med. 1936;29(12):1577-1626
308. Needham J. SUBSTANCES PROMOTING NORMAL AND ABNORMAL GROWTH. Br Med J. 1936;2(3953):701-706
309. Witschi E. Overripeness of the egg as a cause of twinning and teratogenesis: a review. Cancer Res. 1952;12(11):763-786
310. Stevens LC. Origin of testicular teratomas from primordial germ cells in mice. J Natl Cancer Inst. 1967;38(4):549-552
311. Stevens LC. Experimental production of testicular teratomas in mice. Proc Natl Acad Sci U S A. 1964;52:654-661
312. Buta C, David R, Dressel R, Emgard M, Fuchs C, Gross U. et al. Reconsidering pluripotency tests: do we still need teratoma assays? Stem Cell Res. 2013;11(1):552-562
313. Damjanov I, Andrews PW. Teratomas produced from human pluripotent stem cells xenografted into immunodeficient mice - a histopathology atlas. Int J Dev Biol. 2016;60(10-11-12):337-419
314. Solter D, Dominis M, Damjanov I. Embryo-derived teratocarcinoma: I. The role of strain and gender in the control of teratocarcinogenesis. Int J Cancer. 1979;24(6):770-772
315. Damjanov I. Teratocarcinoma: neoplastic lessons about normal embryogenesis. Int J Dev Biol. 1993;37(1):39-46
316. Blum B, Benvenisty N. The tumorigenicity of human embryonic stem cells. Adv Cancer Res. 2008;100:133-158
317. Bustamante-Marin X, Garness JA, Capel B. Testicular teratomas: an intersection of pluripotency, differentiation and cancer biology. Int J Dev Biol. 2013;57(2-4):201-210
318. Pierce GB. Neoplasms, differentiations and mutations. Am J Pathol. 1974;77(1):103-118
319. Pierce GB. Neoplastic stem cells. Adv Pathobiol. 1977(6): 141-152.
320. Sell S. Cellular origin of cancer: dedifferentiation or stem cell maturation arrest? Environ Health Perspect. 1993;101(Suppl 5):15-26
321. Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70(1):6-22
322. Sell S. On the stem cell origin of cancer. Am J Pathol. 2010;176(6):2584-494
323. Sell S, Nicolini A, Ferrari P, Biava PM. Cancer: A Problem of Developmental Biology; Scientific Evidence for Reprogramming and Differentiation Therapy. Curr Drug Targets. 2016;17(10):1103-1110
324. Martin GR. Teratocarcinomas and mammalian embryogenesis. Science. 1980;209(4458):768-776
325. Martin GR. Teratocarcinomas as a model system for the study of embryogenesis and neoplasia. Cell. 1975;5(3):229-243
326. Arechaga J, Damjanov I. Above the borderland between normal and neoplastic development. Int J Dev Biol. 2012;56(10-12):939-948
327. Solter D. From teratocarcinomas to embryonic stem cells and beyond: a history of embryonic stem cell research. Nat Rev Genet. 2006;7(4):319-327
328. Solter D, Skreb N, Damjanov I. Extrauterine growth of mouse egg-cylinders results in malignant teratoma. Nature. 1970;227(5257):503-504
329. Stevens LC. The development of transplantable teratocarcinomas from intratesticular grafts of pre- and postimplantation mouse embryos. Dev Biol. 1970;21(3):364-382
330. Skreb N, Svajger A, Levak-Svajger B. Growth and differentiation of rat egg-cylinders under the kidney capsule. J Embryol Exp Morphol. 1971;25(1):47-56
331. Svajger A, Levak-Svajger B, Skreb N. Rat embryonic ectoderm as renal isograft. J Embryol Exp Morphol. 1986;94:1-27
332. Damjanov I. Development of teratomas from embryos transplanted into outbred and inbred adult hamsters. J Natl Cancer Inst. 1978;61(3):911-915
333. Sobis H, Verstuyf A, Vandeputte M. Endodermal origin of yolk-sac-derived teratomas. Development. 1991;111(1):75-78
334. Sobis H, van HL, Vandeputte M. Cellular events during early formation of yolk-sac-derived teratomas. J Embryol Exp Morphol. 1982;70:225-240
335. Sobis H, Vandeputte M. Development of teratomas from yolk sac of genetically sterile embryos. Dev Biol. 1982;92(2):553-556
336. Sobis H. Induction of malignant and benign tumors with embryonic tissues. Acta Zool Pathol Antverp. 1979(72): 77-82.
337. Sobis H, Vandeputte M. Teratoma induction in mice and rats in relation to the age of the visceral yolk sac. Eur J Cancer. 1979;15(2):143-151
338. Sobis H, Vandeputte M. Yolk sac derived teratomas and carcinomas in hamsters. Eur J Cancer. 1977;13(10):1175-1181
339. Vandeputte M, Sobis H. Experimental rat model for human yolk sac tumor. Eur J Cancer Clin Oncol. 1988;24(3):551-558
340. Sobis H, Verstuyf A, Vandeputte M. Visceral yolk sac-derived tumors. Int J Dev Biol. 1993;37(1):155-168
341. Sakashita S, Tsukada Y, Nakamura K, Tsuji I, Hirai H. Experimental yolk-sac tumors produced by fetectomy without virus infection in rats. Int J Cancer. 1977;20(1):83-86
342. Abad M, Mosteiro L, Pantoja C, Canamero M, Rayon T, Ors I. et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502(7471):340-345
343. Villodre ES, Felipe KB, Oyama MZ, Oliveira FH, Lopez PLDC, Solari C. et al. Silencing of the transcription factors Oct4, Sox2, Klf4, c-Myc or Nanog has different effect on teratoma growth. Biochem Biophys Res Commun. 2019;517(2):324-329
344. Zakrzewski W, Dobrzynski M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. 2019;10(1):68-doi 10.1186/s13287-019-1165-5
345. Bulic-Jakus F, Katusic BA, Juric-Lekic G, Vlahovic M, Sincic N. Teratoma: from spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip Rev Dev Biol. 2016;5(2):186-209
346. Hultman I, Bjork L, Blomberg E, Sandstedt B, Ahrlund-Richter L. Experimental teratoma: at the crossroad of fetal- and onco-development. Semin Cancer Biol. 2014;29:75-79
347. Cunningham JJ, Ulbright TM, Pera MF, Looijenga LH. Lessons from human teratomas to guide development of safe stem cell therapies. Nat Biotechnol. 2012;30(9):849-857
348. ROSE SM. Specific inhibition during differentiation. Ann N Y Acad Sci. 1955;60(7):1136-1153
349. Peterson SE, Garitaonandia I, Loring JF. The tumorigenic potential of pluripotent stem cells: What can we do to minimize it? Bioessays. 2016;38(Suppl 1):S86-S95
350. Erenpreisa J, Salmina K, Huna A, Jackson TR, Vazquez-Martin A, Cragg MS. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience. 2015;2(1):3-14
351. Salmina K, Huna A, Kalejs M, Pjanova D, Scherthan H, Cragg MS. et al. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes (Basel). 2019;10(2):doi 10.3390/genes10020083
352. Oliver CP. A reversion to wild-type associated with crossing-over in drosophila melanogaster. Proc Natl Acad Sci U S A. 1940;26(7):452-454
353. Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;40(10):721-728
354. Lai-Cheong JE, McGrath JA, Uitto J. Revertant mosaicism in skin: natural gene therapy. Trends Mol Med. 2011;17(3):140-148
355. Pasmooij AM, Jonkman MF, Uitto J. Revertant mosaicism in heritable skin diseases: mechanisms of natural gene therapy. Discov Med. 2012;14(76):167-179
356. van DE, Pretorius PJ. Point mutation instability (PIN) mutator phenotype as model for true back mutations seen in hereditary tyrosinemia type 1 - a hypothesis. J Inherit Metab Dis. 2012;35(3):407-411
357. Teotonio H, Chelo IM, Bradic M, Rose MR, Long AD. Experimental evolution reveals natural selection on standing genetic variation. Nat Genet. 2009;41(2):251-257
358. Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol. 2014;11(12):704-713
359. Fisher JPH, Tweddle DA. Neonatal neuroblastoma. Semin Fetal Neonatal Med. 2012;17(4):207-215
360. Haas D, Ablin AR, Miller C, Zoger S, Matthay KK. Complete pathologic maturation and regression of stage IVS neuroblastoma without treatment. Cancer. 1988;62(4):818-825
361. Drobyski WR, Qazi R. Spontaneous regression in non-Hodgkin's lymphoma: clinical and pathogenetic considerations. Am J Hematol. 1989;31(2):138-141
362. Cervinkova M, Kucerova P, Cizkova J. Spontaneous regression of malignant melanoma - is it based on the interplay between host immune system and melanoma antigens? Anticancer Drugs. 2017;28(8):819-830
363. Ribero S, Moscarella E, Ferrara G, Piana S, Argenziano G, Longo C. Regression in cutaneous melanoma: a comprehensive review from diagnosis to prognosis. J Eur Acad Dermatol Venereol. 2016;30(12):2030-2037
364. Askanazy M. Die Teratome nach ihrem Bau, ihrem Verlauf, ihrer Genese und im Vergleich zum exprerimentellen Teratoid. Verhandl Deutsch Pathol. 1907;11(1):39-82
365. Burgio E, Migliore L. Towards a systemic paradigm in carcinogenesis: linking epigenetics and genetics. Mol Biol Rep. 2015;42(4):777-790
366. Papaioannou VE, McBurney MW, Gardner RL, Evans MJ. Fate of teratocarcinoma cells injected into early mouse embryos. Nature. 1975;258(5530):70-73
367. Papaioannou VE. Ontogeny, pathology, oncology. Int J Dev Biol. 1993;37(1):33-37
368. Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72(9):3585-3589
369. Illmensee K, Stevens LC. Teratomas and chimeras. Sci Am. 1979;240(4):120-132
370. Illmensee K. Reversion of malignancy and normalized differentiation of teratocarcinoma cells in chimeric mice. Basic Life Sci. 1978;12:3-25
371. Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A. 1976;73(2):549-553
372. Brinster RL. The effect of cells transferred into the mouse blastocyst on subsequent development. J Exp Med. 1974;140(4):1049-1056
373. Brinster RL. Participation of teratocarcinoma cells in mouse embryo development. Cancer Res. 1976;36(9 PT 2):3412-3414
374. Brinster RL. Stem cells and transgenic mice in the study of development. Int J Dev Biol. 1993;37(1):89-99
375. Pierce GB, Lewis SH, Miller GJ, Moritz E, Miller P. Tumorigenicity of embryonal carcinoma as an assay to study control of malignancy by the murine blastocyst. Proc Natl Acad Sci U S A. 1979;76(12):6649-6651
376. Pierce GB, Pantazis CG, Caldwell JE, Wells RS. Specificity of the control of tumor formation by the blastocyst. Cancer Res. 1982;42(3):1082-1087
377. Askanazy M. Die Teratome nach ihrem Bau, ihrem Verlauf, ihrer Genese und im Vergieich zum experimentellen Teratoid. Verhandl Deutsh Pathol. 1907;11(1):39-82
378. Telerman A, Amson R. The molecular programme of tumour reversion: the steps beyond malignant transformation. Nat Rev Cancer. 2009;9(3):206-216
379. Kleinsmith LJ, Pierce Jr. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964;24:1544-1551
380. Pierce GB, Wallace C. Differentiation of malignant to benign cells. Cancer Res. 1971;31(2):127-134
381. Pierce GB, DIXON FJ Jr. Testicular teratomas. I. Demonstration of teratogenesis by metamorphosis of multipotential cells. Cancer. 1959;12(3):573-583
382. Parchment RE, Gramzinski RA, Pierce GB. Embryonic mechanisms for abrogating the malignancy of cancer cells. Prog Clin Biol Res. 1990;354A:331-344
383. PIERCE GB Jr, VERNEY EL. An in vitro and in vivo study of differentiation in teratocarcinomas. Cancer. 1961;14:1017-1029
384. Papaioannou VE, Evans EP, Gardner RL, Graham CF. Growth and differentiation of an embryonal carcinoma cell line (C145b). J Embryol Exp Morphol. 1979;54:277-295
385. Gardner RL. Extrinsic factors in cellular differentiation. Int J Dev Biol. 1993;37(1):47-50
386. Stewart TA, Mintz B. Recurrent germ-line transmission of the teratocarcinoma genome from the METT-1 culture line to progeny in vivo. J Exp Zool. 1982;224(3):465-469
387. Stewart TA, Mintz B. Successive generations of mice produced from an established culture line of euploid teratocarcinoma cells. Proc Natl Acad Sci U S A. 1981;78(10):6314-6318
388. Gootwine E, Webb CG, Sachs L. Participation of myeloid leukaemic cells injected into embryos in haematopoietic differentiation in adult mice. Nature. 1982;299(5878):63-65
389. Podesta AH, Mullins J, Pierce GB, Wells RS. The neurula stage mouse embryo in control of neuroblastoma. Proc Natl Acad Sci U S A. 1984;81(23):7608-7611
390. Livraghi T, Ceriani R, Palmisano A, Pedicini V, Pich MG, Tommasini MA. et al. Complete response in 5 out of 38 patients with advanced hepatocellular carcinoma treated with stem cell differentiation stage factors: case reports from a single centre. Curr Pharm Biotechnol. 2011;12(2):254-260
391. Proietti S, Cucina A, Pensotti A, Biava PM, Minini M, Monti N. et al. Active Fraction from Embryo Fish Extracts Induces Reversion of the Malignant Invasive Phenotype in Breast Cancer through Down-regulation of TCTP and Modulation of E-cadherin/ß-catenin Pathway. Int J Mol Sci. 2019;20(9):2151.-doi 10.3390/ijms20092151
392. Franchi F, Ielapi T, Bizzarri M, Seminara P. Embryo extracts opotherapy reduces alpha-fetoprotein levels in hepatocellular carcinoma patients. J Gastroenterol Hepatol. 2005;20(9):1467-1468
393. Proietti S, Cucina A, Pensotti A, Fuso A, Marchese C, Nicolini A. et al. Tumor reversion and embryo morphogenetic factors. Semin Cancer Biol. 2020; S1044-579X(20)30194-2.-doi: 10.1016/j.semcancer. 2020 09.005
394. Proietti S, Cucina A, Catizone A, Ricci G, Pensotti A, Bizzarri M. Zebrafish embryo extracts enhance 5-FU anti-cancer effects upon breast cancer cells. Eur Rev Med Pharmacol Sci. 2021;25(8):3235-3245
395. Bussard KM, Smith GH. The mammary gland microenvironment directs progenitor cell fate in vivo. Int J Cell Biol. 2011;2011:451676-doi 10.1155/2011/451676
396. D'Anselmi F, Masiello MG, Cucina A, Proietti S, Dinicola S, Pasqualato A. et al. Microenvironment promotes tumor cell reprogramming in human breast cancer cell lines. PLoS One. 2013;8(12):e83770-doi 10.1371/journal.pone.0083770
397. Bussard KM, Smith GH. Human breast cancer cells are redirected to mammary epithelial cells upon interaction with the regenerating mammary gland microenvironment in-vivo. PLoS One. 2012;7(11):e49221-doi 10.1371/journal.pone.0049221
398. Bussard KM, Boulanger CA, Booth BW, Bruno RD, Smith GH. Reprogramming human cancer cells in the mouse mammary gland. Cancer Res. 2010;70(15):6336-6343
399. Lucke B. Carcinoma in the leopard frog: Its probable causation by a virus. J Exp Med. 1938;68(4):457-468
400. Lucke B. Characteristics of frog carcinoma in tissue culture. J Exp Med. 1939;70(3):269-276
401. Lucke B, Schlumberger H. Heterotransplantation of frog carcinoma; charcter of growth in the eyes of alien species. J Exp Med. 1940;72(3):311-320
402. Lucke B, Schlumberger H. The effect of temperature on the growth of frog carcinoma: I. Direct microscpic observation on living intraocular transplants. J Exp Med. 1940;72(3):321-330
403. Lust JM, Carlson DL, Kowles R, Rollins-Smith L, Williams JW, III, McKinnell RG. Allografts of tumor nuclear transplantation embryos: differentiation competence. Proc Natl Acad Sci U S A. 1991;88(15):6883-6887
404. McKinnell RG, Deggins BA, Labat DD. Transplantation of pluripotential nuclei from triploid frog tumors. Science. 1969;165(3891):394-396
405. McKinnell RG. Frog tumor controversy. J Natl Cancer Inst. 1972;49(5):1471-1474
406. McKinnell RG. Neoplastic cells. Modulation of the differentiated state. Dev Biol (N Y 1985). 1989;6:199-236
407. McKinnell RG, Lust JM, Sauerbier W, Rollins-Smith LA, Williams JW, III, Williams CS. et al. Genomic plasticity of the Lucke renal carcinoma: a review. Int J Dev Biol. 1993;37(1):213-219
408. McKinnell RG. Reduced oncogenic potential associated with differentiation of the Lucke renal adenocarcinoma. In vivo. 1994;8(1):65-69
409. McKinnell RG, Carlson DL. Lucke renal adenocarcinoma, an anuran neoplasm: studies at the interface of pathology, virology, and differentiation competence. J Cell Physiol. 1997;173(2):115-118
410. Seppanen ED, McKinnell RG, Tarin D, Rollins-Smith LA, Hanson W. Temperature-dependent dissociation of Lucke renal adenocarcinoma cells. Differentiation. 1984;26(3):227-230
411. DiBerardino MA. Genomic multipotentiality of differentiated somatic cells. Cell Differ Dev. 1988;25(Suppl):129-136
412. DiBerardino MA. Genomic activation in differentiated somatic cells. Dev Biol (N Y 1985). 1989;6:175-198
413. DiBerardino MA, Orr NH, McKinnell RG. Feeding tadpoles cloned from Rana erythrocyte nuclei. Proc Natl Acad Sci U S A. 1986;83(21):8231-8234
414. Li L, Connelly MC, Wetmore C, Curran T, Morgan JI. Mouse embryos cloned from brain tumors. Cancer Res. 2003;63(11):2733-2736
415. Harris H. The role of differentiation in the suppression of malignancy. J Cell Sci. 1990;97(Pt 1):5-10
416. Harris H. A long view of fashions in cancer research. Bioessays. 2005;27(8):833-838
417. Evans EP, Burtenshaw MD, Brown BB, Hennion R, Harris H. The analysis of malignancy by cell fusion. IX. Re-examination and clarification of the cytogenetic problem. J Cell Sci. 1982;56:113-130
418. Harris H. Some thoughts about genetics, differentiation, and malignancy. Somatic Cell Genet. 1979;5(6):923-930
419. Jonasson J, Povey S, Harris H. The analysis of malignancy by cell fusion. VII. Cytogenetic analysis of hybrids between malignant and diploid cells and of tumours derived from them. J Cell Sci. 1977;24:217-254
420. Wiener F, Klein G, Harris H. The analysis of malignancy by cell fusion. IV. Hybrid between tumour cells and a malignant L cell derivative. J Cell Sci. 1973;12(1):253-261
421. Klein G, Bregula U, Wiener F, Harris H. The analysis of malignancy by cell fusion. I. Hybrids between tumour cells and L cell derivatives. J Cell Sci. 1971;8(3):659-672
422. Klinger HP, Shows TB. Suppression of tumorigenicity in somatic cell hybrids. II. Human chromosomes implicated as suppressors of tumorigenicity in hybrids with Chinese hamster ovary cells. J Natl Cancer Inst. 1983;71(3):559-569
423. Spira J, Wiener F, Babonits M, Gamble J, Miller J, Klein G. The role of chromosome 15 in murine leukemogenesis. I. Contrasting behavior of the tumor vs. normal parent-derived chromosomes No. 15 in somatic hybrids of varying tumorigenicity. Int J Cancer. 1981;28(6):785-798
424. Stanbridge EJ. Suppression of malignancy in human cells. Nature. 1976;260(5546):17-20
425. Barreto SG, Gardi N, Dutt S. Birth of a solid organ cancer-the cell fusion hypothesis presented with pancreatic cancer as a model: a narrative review. Chin Clin Oncol. 2021;10(5):45-doi 10.21037/cco-21-69
426. Pawelek JM, Chakraborty AK. The cancer cell-leukocyte fusion theory of metastasis. Adv Cancer Res. 2008;101:397-444
427. Pawelek JM. Fusion of bone marrow-derived cells with cancer cells: metastasis as a secondary disease in cancer. Chin J Cancer. 2014;33(3):133-139
428. Dittmar T. Generation of Cancer Stem/Initiating Cells by Cell-Cell Fusion. Int J Mol Sci. 2022;23(9):4514.-doi 10.3390/ijms23094514
429. Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8(5):377-386
430. Cheong KH, Koh JM, Jones MC. Paradoxical Survival: Examining the Parrondo Effect across Biology. Bioessays. 2019;41(6):e1900027-doi 10.1002/bies.201900027
431. Braun AC. An epigenetic model for the origin of cancer. Q Rev Biol. 1981;56(1):33-60
432. Binns A, Meins F. Habituation of tobacco pith cells for factors promoting cell division is heritable and potentially reversible. Proc Natl Acad Sci U S A. 1973;70(9):2660-2662
433. Carlson PS, Smith HH, Dearing RD. Parasexual interspecific plant hybridization. Proc Natl Acad Sci U S A. 1972;69(8):2292-2294
434. Meins F Jr, Binns A. Epigenetic variation of cultured somatic cells: evidence for gradual changes in the requirement for factors promoting cell division. Proc Natl Acad Sci U S A. 1977;74(7):2928-2932
435. Sacristan MD, Melchers G. The caryological analysis of plants regenerated from tumorous and other callus cultures of tobacco. Mol Gen Genet. 1969;105(4):317-333
436. Smith HH, Kao KN, Combatti NC. Interspecific hybridization by protoplast fusion in nicotiana. J Heredity. 1976;67:123-128
437. Braun AC, Wood HN. Suppression of the neoplastic state with the acquisition of specialized functions in cells, tissues, and organs of crown gall teratomas of tobacco. Proc Natl Acad Sci U S A. 1976;73(2):496-500
438. Turgeon R, Wood HN, Braun AC. Studies on the recovery of crown gall tumor cells. Proc Natl Acad Sci U S A. 1976;73(10):3562-3564
439. Wood HN, Binns AN, Braun AC. Differential expression of oncogenicity and nopaline synthesis in intact leaves derived from crown gall teratomas of tobacco. Differentiation. 1978;11:175-180
440. Braun AC. Studies on tumor inception in the crown-gall disease. Am J Botany. 1943;30:674-677
441. Braun AC, MANDLE RJ. Studies on the inactivation of the tumor-inducing principle in crown gall. Growth. 1948;12(4):255-269
442. Braun AC. Cellular autonomy in crown gall. Phytopathology. 1951;41:963-966
443. Braun AC. Recovery of crown-gall tumor cells. Cancer Res. 1951;11(11):839-844
444. Braun AC. Recovery of tumor cells from effects of the tumor-inducing principle in crown gall. Science. 1951;113(2945):651-653
445. Braun AC. Tissue culture as a tool for studying the development of autonomy in neoplastic plant cells. J Natl Cancer Inst. 1957;19(4):753-759
446. Braun AC. A Physiological Basis for Autonomous Growth of the Crown-Gall Tumor Cell. Proc Natl Acad Sci U S A. 1958;44(4):344-349
447. Braun AC. A Demonstration of the recovery of the grown-gall tumor cell with the use of complex tumors of single-cell origin. Proc Natl Acad Sci U S A. 1959;45(7):932-938
448. Braun AC. The reversal of tumor growth. Sci Am. 1965;213(5):75-83
449. Braun AC. On the origin of the cancer cells. Am Sci. 1970;58(3):307-320
450. Braun AC. Plant tumors. Biochim Biophys Acta. 1978;516(2):167-191
451. Braun AC. Genetic and biochemical studies on the suppression of and a recovery from the tumorous state in higher plants. In vitro. 1980;16(1):38-48
452. Riker AJ. Studies on the influence of some environmental factors on the development of crown gall. J Agric Res. 1926;32(1):83-96
453. Braun AC. Thermal studies on the factors responsible for tumor initiation in crown gall. Am J Bot. 1947;34(4):234-240
454. Akhtar M, Haider A, Rashid S, Al-Nabet ADMH. Paget's “Seed and Soil” Theory of Cancer Metastasis: An Idea Whose Time has Come. Adv Anat Pathol. 2019;26(1):69-74
455. Maehle AH. Ambiguous cells: the emergence of the stem cell concept in the nineteenth and twentieth centuries. Notes Rec R Soc Lond. 2011;65(4):359-378
456. Capp JP. Cancer Stem Cells: From Historical Roots to a New Perspective. J Oncol. 2019;2019:5189232-doi 10.1155/2019/5189232
457. Barrett JC. A preneoplastic stage in the spontaneous neoplastic transformation of syrian hamster embryo cells in culture. Cancer Res. 1980;40(1):91-94
458. Wells RS, Campbell EW, Swartzendruber DE, Holland LM, Kraemer PM. Role of anchorage in the expression of tumorigenicity of untransformed mouse cell lines. J Natl Cancer Inst. 1982;69(2):415-423
459. Bouillant AM, Bundza A, Genest P, Greig AS. Multisequential transformation of a pig cell line (PFT): correlations between tumorigenicity and chromosome and ultrastructural markers. J Natl Cancer Inst. 1980;64(4):783-790
460. Gorman SD, Hoffman E, Nichols WW, Cristofalo VJ. Spontaneous transformation of a cloned cell line of normal diploid bovine vascular endothelial cells. In vitro. 1984;20(4):339-345
461. Rhim JS. Neoplastic transformation of human cells in vitro. Crit Rev Oncog. 1993;4(3):313-335
462. Newbold RF, Overell RW. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature. 1983;304(5927):648-651
463. Newbold RF. Multistep malignant transformation of mammalian cells by carcinogens: induction of immortality as a key event. Carcinog Compr Surv. 1985;9:17-28
464. Newbold RF. Malignant transformation of mammalian cells in culture: delineation of stages and role of cellular oncogene activation. IARC Sci Publ. 1985;67:31-53
465. Newbold RF, Cuthbert AP, Themis M, Trott DA, Blair AL, Li W. Cell immortalization as a key, rate-limiting event in malignant transformation: approaches toward a molecular genetic analysis. Toxicol Lett. 1993;67(1-3):211-230
466. Reddel RR. The role of senescence and immortalization in carcinogenesis. Carcinogenesis. 2000;21(3):477-484
467. Prasad KN, Hovland AR, Nahreini P, Cole WC, Hovland P, Kumar B. et al. Differentiation genes: are they primary targets for human carcinogenesis? Exp Biol Med (Maywood). 2001;226(9):805-813
468. Prasad KN, Cole WC, Yan XD, Nahreini P, Kumar B, Hanson A. et al. Defects in cAMP-pathway may initiate carcinogenesis in dividing nerve cells: a review. Apoptosis. 2003;8(6):579-586
469. Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304(5927):596-602
470. Krelin Y, Zhang L, Kang TB, Appel E, Kovalenko A, Wallach D. Caspase-8 deficiency facilitates cellular transformation in vitro. Cell Death Differ. 2008;15(9):1350-1355
471. Knight RA, Vaux DL. A tumour suppressor function of caspase-8? Cell Death Differ. 2008;15(9):1337-1338
472. Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q. et al. CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med. 2002;8(10):1105-1114
473. Gordon K, Clouaire T, Bao XX, Kemp SE, Xenophontos M, de Las Heras JI. et al. Immortality, but not oncogenic transformation, of primary human cells leads to epigenetic reprogramming of DNA methylation and gene expression. Nucleic Acids Res. 2014;42(6):3529-3541
474. Sack GH Jr. Human cell transformation by simian virus 40-a review. In vitro. 1981;17(1):1-19
475. Blagosklonny MV. Cell immortality and hallmarks of cancer. Cell Cycle. 2003;2(4):296-299
476. Chang S, DePinho RA. Telomerase extracurricular activities. Proc Natl Acad Sci U S A. 2002;99(20):12520-12522
477. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400(6743):464-468
478. Stewart SA, Hahn WC, O'Connor BF, Banner EN, Lundberg AS, Modha P. et al. Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc Natl Acad Sci U S A. 2002;99(20):12606-12611
479. Belair CD, Yeager TR, Lopez PM, Reznikoff CA. Telomerase activity: a biomarker of cell proliferation, not malignant transformation. Proc Natl Acad Sci U S A. 1997;94(25):13677-13682
480. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1-2):349-361
481. Deryugina EI, Kiosses WB. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017;19(3):601-616
482. Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321(5897):1841-1844
483. Weinberg RA. Leaving home early: reexamination of the canonical models of tumor progression. Cancer Cell. 2008;14(4):283-284
484. Strakova A, Murchison EP. The cancer which survived: insights from the genome of an 11000 year-old cancer. Curr Opin Genet Dev. 2015;30:49-55
485. SCHERER WF, SYVERTON JT, GEY GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97(5):695-710
486. Coleman WB, Wennerberg AE, Smith GJ, Grisham JW. Regulation of the differentiation of diploid and some aneuploid rat liver epithelial (stemlike) cells by the hepatic microenvironment. Am J Pathol. 1993;142(5):1373-1382
487. Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7(4):246-255
488. McCullough KD, Coleman WB, Ricketts SL, Wilson JW, Smith GJ, Grisham JW. Plasticity of the neoplastic phenotype in vivo is regulated by epigenetic factors. Proc Natl Acad Sci U S A. 1998;95(26):15333-15338
489. Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW. et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci U S A. 2008;105(11):4329-4334
490. Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C. et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231-245
491. Webb CG, Gootwine E, Sachs L. Developmental potential of myeloid leukemia cells injected into midgestation embryos. Dev Biol. 1984;101(1):221-224
492. Novaro V, Radisky DC, Ramos Castro NE, Weisz A, Bissell MJ. Malignant mammary cells acquire independence from extracellular context for regulation of estrogen receptor alpha. Clin Cancer Res. 2004;10(1 Pt 2):402S-409S
493. Radisky DC, Bissell MJ. Cancer. Respect thy neighbor!. Science. 2004;303(5659):775-777
494. Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70(9-10):537-546
495. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1(1):46-54
496. Kenny PA, Bissell MJ. Tumor reversion: correction of malignant behavior by microenvironmental cues. Int J Cancer. 2003;107(5):688-695
497. Felsher DW. Reversibility of oncogene-induced cancer. Curr Opin Genet Dev. 2004;14(1):37-42
498. Kulesa PM, Kasemeier-Kulesa JC, Teddy JM, Margaryan NV, Seftor EA, Seftor RE. et al. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc Natl Acad Sci U S A. 2006;103(10):3752-3757
499. Diez-Torre A, Andrade R, Eguizabal C, Lopez E, Arluzea J, Silio M. et al. Reprogramming of melanoma cells by embryonic microenvironments. Int J Dev Biol. 2009;53(8-10):1563-1568
500. Gerschenson M, Graves K, Carson SD, Wells RS, Pierce GB. Regulation of melanoma by the embryonic skin. Proc Natl Acad Sci U S A. 1986;83(19):7307-7310
501. Kasemeier-Kulesa JC, Teddy JM, Postovit LM, Seftor EA, Seftor RE, Hendrix MJ. et al. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev Dyn. 2008;237(10):2657-2666
502. Tato F, Alemà S, Dlugosz A, Boettiger D, Holtzer H, Cossu G. et al. Development of 'revertant' myotubes in cultures of Rous sarcoma virus transformed avian myogenic cells. Differentiation. 1983;24(2):131-139
503. Boettiger D, Soltesz R, Holtzer H, Pacifici M. Infection of chick limb bud presumptive chondroblasts by a temperature-sensitive mutant of Rous sarcoma virus and the reversible inhibition of their terminal differentiation in culture. Mol Cell Biol. 1983;3(8):1518-1526
504. Moss PS, Honeycutt N, Pawson T, Martin GS. Viral transformation of chick myogenic cells. The relationship between differentiation and the expression of the SRC gene. Exp Cell Res. 1979;123(1):95-105
505. Boettiger D. Reversion and induction of Rous sarcoma virus expression in virus-transformed baby hamster kidney cells. Virology. 1974;62(2):522-529
506. McCullough KD, Coleman WB, Smith GJ, Grishan JW. Age-dependent regulation of the tumorigenic potential of neoplastically transformed rat liver epithelial cells by the liver microenvironment. Cancer Res. 1994;54(14):3668-3671
507. Rose SM, Wallingford HM. Transformation of renal tumors of frogs to normal tissues in regenerating limbs of salamanders. Science. 1948;107(2784):457
508. Rose SM. Transformed cells. Sci Am. 1949;181(6):22-24
509. Greene HS. On the development of cancer. Sci Am. 1948;179(6):40-43
510. Greene HS. Heterologous transplantation of the Brown-Pearce tumors. Cancer Res. 1949;9(12):728-735
511. Greene HS. Pathology in fields collateral to tissue culture. J Natl Cancer Inst. 1957;19(4):711-721
512. Greene HS. Heterotransplantation of tumors. Ann N Y Acad Sci. 1957;69(4):818-829
513. Greene HS, Harvey EK. Metastasis of heterologously transplanted tumors. Cancer Res. 1964;24:1678-1687
514. Stoker M. Regulation of growth and orientation in hamster cells transformed by polyoma virus. Virology. 1964;24:165-174
515. Rubin H. The suppression of morphological alterations in cells infected with Rous sarcoma virus. Virology. 1960;12:14-31
516. Stoker MG, Shearer M, O'Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci. 1966;1(3):297-310
517. Friend C, Scher W, Holland JG, Sato T. Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci U S A. 1971;68(2):378-382
518. Andersson LC, Jokinen M, Gahmberg CG. Induction of erythroid differentiation in the human leukaemia cell line K562. Nature. 1979;278(5702):364-365
519. Lozzio CB, Lozzio BB, Machado EA, Fuhr JE, Lair SV, Bamberger EG. Effects of sodium butyrate on human chronic myelogenous leukaemia cell line K562. Nature. 1979;281(5733):709-710
520. Andersson LC, Nilsson K, Gahmberg CG. K562-a human erythroleukemic cell line. Int J Cancer. 1979;23(2):143-147
521. Rutherford TR, Clegg JB, Weatherall DJ. K562 human leukaemic cells synthesise embryonic haemoglobin in response to haemin. Nature. 1979;280(5718):164-165
522. Metcalf D. The granulocyte-macrophage colony-stimulating factors. Science. 1985;229(4708):16-22
523. Sachs L. Cell differentiation and malignancy. Cell Biophys. 1986;9(1-2):225-242
524. Lotem J, Sachs L. Control of in vivo differentiation of myeloid leukemic cells. Leukemia. 1988;2(12 Suppl):24S-37S
525. Reitsma PH, Rothberg PG, Astrin SM, Trial J, Bar-Shavit Z, Hall A. et al. Regulation of myc gene expression in HL-60 leukaemia cells by a vitamin D metabolite. Nature. 1983;306(5942):492-494
526. Uriel J. Cell injury, retrodifferentiation and the cancer treatment paradox. Tumour Biol. 2015;36(10):7365-7374
527. Biava PM, Burigana F, Germano R, Kurian P, Verzegnassi C, Vitiello G. Stem Cell Differentiation Stage Factors and their Role in Triggering Symmetry Breaking Processes during Cancer Development: A Quantum Field Theory Model for Reprogramming Cancer Cells to Healthy Phenotypes. Curr Med Chem. 2019;26(6):988-1001
528. Martelli C, King A, Simon T, Giamas G. Graphene-Induced Transdifferentiation of Cancer Stem Cells as a Therapeutic Strategy against Glioblastoma. ACS Biomater Sci Eng. 2020;6(6):3258-3269
529. Sachs L. Cell differentiation and bypassing of genetic defects in the suppression of malignancy. Cancer Res. 1987;47(8):1981-1986
530. Rubin H. Ordered heterogeneity and its decline in cancer and aging. Adv Cancer Res. 2007;98:117-147
531. Deng CT, Boettiger D, Macpherson I, Varmus HE. The persistence and expression of virus-specific DNA in revertants of Rous sarcoma virus-transformed BHK-21 cells. Virology. 1974;62(2):512-521
532. Macpherson I. Reversion in Hamster Cells Transformed by Rous Sarcoma Virus. Science. 1965;148(3678):1731-1733
533. Visfeldt J. Transformation of sympathicoblastoma into ganglioneuroma, with a case report. Acta Pathol Microbiol Scand. 1963;58:414-428
534. Kimhi Y, Palfrey C, Spector I, Barak Y, Littauer UZ. Maturation of neuroblastoma cells in the presence of dimethylsulfoxide. Proc Natl Acad Sci U S A. 1976;73(2):462-466
535. Enane FO, Saunthararajah Y, Korc M. Differentiation therapy and the mechanisms that terminate cancer cell proliferation without harming normal cells. Cell Death Dis. 2018;9(9):912-doi 10.1038/s41419-018-0919-9
536. Murray KN, Wolf JC, Spagnoli ST, Lains D, Budrow N, Kent ML. Reversibility of Proliferative Thyroid Lesions Induced by Iodine Deficiency in a Laboratory Zebrafish Colony. Zebrafish. 2018;15(6):558-565
537. Wiatrak B, Kubis-Kubiak A, Piwowar A, Barg E. PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions. Cells. 2020;9(4):958-doi 10.3390/cells9040958
538. Falsafi N, Soleimani T, Fallahi H, Azadbakht M. Regulatory networks upon neurogenesis induction in PC12 cell line by small molecules. J Cell Physiol. 2019;234(10):18813-18824
539. Moghadam FH, Mesbah-Ardakani M, Nasr-Esfahani MH. Ferulic Acid exerts concentration-dependent anti-apoptotic and neuronal differentiation-inducing effects in PC12 and mouse neural stem cells. Eur J Pharmacol. 2018;841:104-112
540. Butawan M, Benjamin RL, Bloomer RJ. Methylsulfonylmethane: Applications and Safety of a Novel Dietary Supplement. Nutrients. 2017;9(3):290-doi 10.3390/nu9030290
541. Muizzuddin N, Benjamin R. Beauty from within: Oral administration of a sulfur-containing supplement methylsulfonylmethane improves signs of skin ageing. Int J Vitam Nutr Res. 2020:1-10
542. Caron JM, Bannon M, Rosshirt L, Luis J, Monteagudo L, Caron JM. et al. Methyl sulfone induces loss of metastatic properties and reemergence of normal phenotypes in a metastatic cloudman S-91 (M3) murine melanoma cell line. PLoS One. 2010;5(8):e11788-doi 10.1371/journal.pone.0011788
543. Caron JM, Monteagudo L, Sanders M, Bannon M, Deckers PJ. Methyl sulfone manifests anticancer activity in a metastatic murine breast cancer cell line and in human breast cancer tissue-part 2: human breast cancer tissue. Chemotherapy. 2013;59(1):24-34
544. Caron JM, Bannon M, Rosshirt L, O'Donovan L. Methyl sulfone manifests anticancer activity in a metastatic murine breast cancer cell line and in human breast cancer tissue-part I: murine 4T1 (66cl-4) cell line. Chemotherapy. 2013;59(1):14-23
545. Kowalska K, Habrowska-Górczyńska DE, Domińska K, Urbanek KA, Piastowska-Ciesielska AW. Methylsulfonylmethane (organic sulfur) induces apoptosis and decreases invasiveness of prostate cancer cells. Environ Toxicol Pharmacol. 2018;64:101-111
546. Nipin SP, Darvin P, Yoo YB, Joung YH, Kang DY, Kim DN. et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer. 2015;15:474-doi 10.1186/s12885-015-1445-0
547. Tallman MS, Nabhan C, Feusner JH, Rowe JM. Acute promyelocytic leukemia: evolving therapeutic strategies. Blood. 2002;99(3):759-767
548. Douer D, Tallman MS. Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol. 2005;23(10):2396-2410
549. Prasad S, Gupta SC, Aggarwal BB. Serendipity in Cancer Drug Discovery: Rational or Coincidence? Trends Pharmacol Sci. 2016;37(6):435-450
550. Gordeeva O. TGF-beta Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells' Fate Decisions: Balancing Between Self-Renewal, Differentiation, and Cancer. Cells. 2019;8(12):1500-doi 10.3390/cells8121500
551. Kelly GM, Gatie MI. Mechanisms Regulating Stemness and Differentiation in Embryonal Carcinoma Cells. Stem Cells Int. 2017;2017:3684178-doi 10.1155/2017/3684178
552. Amson R, Karp JE, Telerman A. Lessons from tumor reversion for cancer treatment. Curr Opin Oncol. 2013;25(1):59-65
553. Telerman A, Amson R, Hendrix MJ. Tumor reversion holds promise. Oncotarget. 2010;1(4):233-234
554. Tuynder M, Susini L, Prieur S, Besse S, Fiucci G, Amson R. et al. Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc Natl Acad Sci U S A. 2002;99(23):14976-14981
555. Asashima M, Komazaki S, Satou C, Oinuma T. Seasonal and geographical changes of spontaneous skin papillomas in the Japanese newt Cynops pyrrhogaster. Cancer Res. 1982;42(9):3741-3746
556. Asashima M, Oinuma T, Matsuyama H, Nagano M. Effects of temperature on papilloma growth in the newt, Cynops pyrrhogaster. Cancer Res. 1985;45(3):1198-1205
557. Asashima M, Koyama H, Shimada K, Pfeiffer CJ. Temperature-induced alterations in protein composition of newt papilloma cells. Cell Mol Biol. 1989;35(6):669-677
558. Getchell RG, Gasey JW, Bowser PR. Seasonal occurence of virally induced skin tumors in wild fish. J Aquat Anim Health. 1998;10:191-201
559. Holzschu D, Lapierre LA, Lairmore MD. Comparative pathogenesis of epsilonretroviruses. J Virol. 2003;77(23):12385-12391
560. Peters G, Peters N. Temperature-dependent growth and regression of epidermal tumors in the european eel (Anguilla anguilla L.). Ann N Y Acad Sci. 1978;298:245-260
561. Pfeiffer CJ, Nagai T, Fujimura M, Tobe T. Spontaneous regressive epitheliomas in the Japanese newt, Cynops pyrrhogaster. Cancer Res. 1979;39(6 Pt 1):1904-1910
562. Wojciechowska S, Zeng Z, Lister JA, Ceol CJ, Patton EE. Melanoma Regression and Recurrence in Zebrafish. Methods Mol Biol. 2016;1451:143-153
563. Coffee LL, Casey JW, Bowser PR. Pathology of tumors in fish associated with retroviruses: a review. Vet Pathol. 2013;50(3):390-403
564. Mizuno S, Fujinaga T, Hagio M. Role of lymphocytes in spontaneous regression of experimentally transplanted canine transmissible venereal sarcoma. J Vet Med Sci. 1994;56(1):15-20
565. Yang TJ. Immunobiology of a spontaneously regressive tumor, the canine transmissible venereal sarcoma (review). Anticancer Res. 1988;8(1):93-95
566. Prehn RT. Cancers beget mutations versus mutations beget cancers. Cancer Res. 1994;54(20):5296-5300
567. Hu T, Kumar Y, Shazia I, Duan SJ, Li Y, Chen L. et al. Forward and reverse mutations in stages of cancer development. Hum Genomics. 2018;12(1):40. doi: 10.1186/s40246-018-0170-6
568. Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol. 2016;8(9):pii a019505. doi: 10.1101/cshperspect.a019505
569. Jinesh GG, Brohl AS. The genetic script of metastasis. Biol Rev Camb Philos Soc. 2020;95(2):244-266
570. Persi E, Wolf YI, Leiserson MDM, Koonin EV, Ruppin E. Criticality in tumor evolution and clinical outcome. Proc Natl Acad Sci U S A. 2018;115(47):E11101-E11110
571. Lotem J, Sachs L. Epigenetics wins over genetics: induction of differentiation in tumor cells. Semin Cancer Biol. 2002;12(5):339-346
572. Cruz FD, Matushansky I. Solid tumor differentiation therapy - is it possible? Oncotarget. 2012;3(5):559-567
573. Nowak D, Stewart D, Koeffler HP. Differentiation therapy of leukemia: 3 decades of development. Blood. 2009;113(16):3655-3665
574. Sachs L. Origin and reversibility of malignancy. Carcinog Compr Surv. 1985;10:23-33
575. Papaioannou VE, Gardner RL, McBurney MW, Babinet C, Evans MJ. Participation of cultured teratocarcinoma cells in mouse embryogenesis. J Embryol Exp Morphol. 1978;44:93-104
576. Liu B, Xu N, Man Y, Shen H, Avital I, Stojadinovic A. et al. Apoptosis in Living Animals Is Assisted by Scavenger Cells and Thus May Not Mainly Go through the Cytochrome C-Caspase Pathway. J Cancer. 2013;4(9):716-723
577. Liu X, Yang W, Guan Z, Yu W, Fan B, Xu N. et al. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell Biosci. 2018;8:6-doi 10.1186/s13578-018-0206-6
578. Ma Y, Jia Y, Chen L, Ezeogu L, Yu B, Xu N. et al. Weaknesses and Pitfalls of Using Mice and Rats in Cancer Chemoprevention Studies. J Cancer. 2015;6(10):1058-1065
579. Choi PS, Li Y, Felsher DW. Addiction to multiple oncogenes can be exploited to prevent the emergence of therapeutic resistance. Proc Natl Acad Sci U S A. 2014;111(32):E3316-E3324
580. Li Y, Casey SC, Felsher DW. Inactivation of MYC reverses tumorigenesis. J Intern Med. 2014;276(1):52-60
581. Ewald D, Li M, Efrat S, Auer G, Wall RJ, Furth PA. et al. Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science. 1996;273(5280):1384-1386
582. Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24(1):57-60
583. Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M. et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297(5578):102-104
584. He Y, Yuan C, Chen L, Liu Y, Zhou H, Xu N. et al. While it is not deliberate, much of today's biomedical research contains logical and technical flaws, showing a need for corrective action. Int J Med Sci. 2018;15(4):309-322
585. Fischer B. Die experimentelle erzeugung atypischer epithelwucherungen und die entstehung bosartiger geschwulste. Munch Med Woschschr. 1906;53:2041-2047
586. Marin G, Macpherson I. Reversion in polyoma-transformed cells: retransformation, induced antigens and tumorigenicity. J Virol. 1969;3(2):146-149
587. Macpherson I. Reversion in virus-transformed cells. Biochem Pharmacol. 1971;20(5):1005-1008
588. Wiblin CN, Macpherson I. Reversion in hybrids between SV40-transformed hamster and mouse cells. Int J Cancer. 1973;12(1):148-161
589. Macera-Bloch L, Houghton J, Lenahan M, Jha KK, Ozer HL. Termination of lifespan of SV40-transformed human fibroblasts in crisis is due to apoptosis. J Cell Physiol. 2002;190(3):332-344
590. Liao DJ, Dickson RB. Cell death in MMTV-c-myc transgenic mouse mammary tumors may not be typical apoptosis. Lab Invest. 2003;83(10):1437-1449
591. Liao DJ. The scavenger cell hypothesis of apoptosis: apoptosis redefined as a process by which a cell in living tissue is destroyed by phagocytosis. Med Hypotheses. 2005;65(1):23-28
592. Liu XD, Yang WX, Guan ZZ, Yu WF, Fan B, Xu NZ. et al. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell & Bioscience. 2018;8(6):doi.org /10.1186/s13578-018-0206-6
593. Zellmer L, Han YP, Chen LC, Xu NZ, Liao DJ. Does the cytochrome c-caspase pathway of cell death occur physiologically in animals? Journal Tumor Med Prev. 2017;1(2):JTMP.MS.ID.555557.pdf
594. LeGrand EK. Implications of early apoptosis of infected cells as an important host defense. Med Hypotheses. 2000;54(4):591-596
595. LeGrand EK. Genetic conflict and apoptosis. Perspect Biol Med. 2001;44(4):509-521
596. Anso E, Mullen AR, Felsher DW, Matѐs JM, DeBerardinis RJ, Chandel NS. Metabolic changes in cancer cells upon suppression of MYC. Cancer Metab. 2013;1(1):7-doi 10.1186/2049-3002-1-7
597. Casey SC, Li Y, Fan AC, Felsher DW. Oncogene withdrawal engages the immune system to induce sustained cancer regression. J Immunother Cancer. 2014;2:24-doi 10.1186/2051-1426-2-24
598. Casey SC, Li Y, Felsher DW. An essential role for the immune system in the mechanism of tumor regression following targeted oncogene inactivation. Immunol Res. 2014;58(2-3):282-291
599. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-257
600. Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A. et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12(2):439-451
601. Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest. 2010;120(7):2619-2626
602. Sacco A, Kawano Y, Moschetta M, Zavidij O, Huynh D, Reagan M. et al. A novel in vivo model for studying conditional dual loss of BLIMP-1 and p53 in B-cells, leading to tumor transformation. Am J Hematol. 2017;92(8):E138-E145
603. Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B. et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180(2):599-607
604. Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X. et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A. 2012;109(49):20035-20040
605. Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25(6):681-686
606. Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L. et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88(12):5096-5100
607. Sakairi T, Abe Y, Kajiyama H, Bartlett LD, Howard LV, Jat PS. et al. Conditionally immortalized human podocyte cell lines established from urine. Am J Physiol Renal Physiol. 2010;298(3):F557-F567
608. Kawata S, Suzuki J, Maruoka M, Mizutamari M, Ishida-Kitagawa N, Yogo K. et al. Retrovirus-mediated conditional immortalization and analysis of established cell lines of osteoclast precursor cells. Biochem Biophys Res Commun. 2006;350(1):97-104
609. Lidington EA, Rao RM, Marelli-Berg FM, Jat PS, Haskard DO, Mason JC. Conditional immortalization of growth factor-responsive cardiac endothelial cells from H-2K(b)-tsA58 mice. Am J Physiol Cell Physiol. 2002;282(1):C67-C74
610. O'Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C. et al. Conditional immortalization of freshly isolated human mammary fibroblasts and endothelial cells. Proc Natl Acad Sci U S A. 2001;98(2):646-651
611. Noble M, Groves AK, Ataliotis P, Ikram Z, Jat PS. The H-2KbtsA58 transgenic mouse: a new tool for the rapid generation of novel cell lines. Transgenic Res. 1995;4(4):215-225
612. Tegtmeyer P. Function of simian virus 40 gene A in transforming infection. J Virol. 1975;15(3):613-618
613. Petit CA, Gardes M, Feunteun J. Immortalization of rodent embryo fibroblasts by SV40 is maintained by the A gene. Virology. 1983;127(1):74-82
614. Zaret KS, DiPersio CM, Jackson DA, Montigny WJ, Weinstat DL. Conditional enhancement of liver-specific gene transcription. Proc Natl Acad Sci U S A. 1988;85(23):9076-9080
615. Radna RL, Caton Y, Jha KK, Kaplan P, Li G, Traganos F. et al. Growth of immortal simian virus 40 tsA-transformed human fibroblasts is temperature dependent. Mol Cell Biol. 1989;9(7):3093-3096
616. Jat PS, Sharp PA. Cell lines established by a temperature-sensitive simian virus 40 large-T-antigen gene are growth restricted at the nonpermissive temperature. Mol Cell Biol. 1989;9(4):1672-1681
617. Wojcik BE, Dermody JJ, Ozer HL, Mun B, Mathews CK. Temperature-sensitive DNA mutant of Chinese hamster ovary cells with a thermolabile ribonucleotide reductase activity. Mol Cell Biol. 1990;10(11):5688-5699
618. Liao DJ, Wang Y, Wu J, Adsay NV, Grignon D, Khanani F. et al. Characterization of pancreatic lesions from MT-tgfalpha, Ela-myc and MT-tgfalpha/Ela-myc single and double transgenic mice. J Carcinog. 2006;5:DOI 10.1186/1477-3163-5-19
619. Liao JD, Adsay NV, Khannani F, Grignon D, Thakur A, Sarkar FH. Histological complexities of pancreatic lesions from transgenic mouse models are consistent with biological and morphological heterogeneity of human pancreatic cancer. Histol Histopathol. 2007;22(6):661-676
620. Abascal F, Harvey LMR, Mitchell E, Lawson ARJ, Lensing SV, Ellis P. et al. Somatic mutation landscapes at single-molecule resolution. Nature. 2021;593(7859):405-410
621. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y. et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94-101
622. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495-501
623. Soussi T, Hamroun D, Hjortsberg L, Rubio-Nevado JM, Fournier JL, Beroud C. MUT-TP53 2.0: a novel versatile matrix for statistical analysis of TP53 mutations in human cancer. Hum Mutat. 2010;31(9):1020-1025
624. Wotherspoon AC, Doglioni C, Diss TC, Pan L, Moschini A, de BM. et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993;342(8871):575-577
625. Wundisch T, Thiede C, Morgner A, Dempfle A, Gunther A, Liu H. et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23(31):8018-8024
626. Morgner A, Thiede C, Bayerdorffer E, Alpen B, Wundisch T, Neubauer A. et al. Long-term follow-up of gastric MALT lymphoma after H. pylori eradication. Curr Gastroenterol Rep. 2001;3(6):516-522
627. Morgner A, Miehlke S, Fischbach W, Schmitt W, Muller-Hermelink H, Greiner A. et al. Complete remission of primary high-grade B-cell gastric lymphoma after cure of Helicobacter pylori infection. J Clin Oncol. 2001;19(7):2041-2048
628. Wundisch T, Dieckhoff P, Greene B, Thiede C, Wilhelm C, Stolte M. et al. Second cancers and residual disease in patients treated for gastric mucosa-associated lymphoid tissue lymphoma by Helicobacter pylori eradication and followed for 10 years. Gastroenterology. 2012;143(4):936-942
629. Bertoni F, Conconi A, Capella C, Motta T, Giardini R, Ponzoni M. et al. Molecular follow-up in gastric mucosa-associated lymphoid tissue lymphomas: early analysis of the LY03 cooperative trial. Blood. 2002;99(7):2541-2544
630. Park JB, Koo JS. Helicobacter pylori infection in gastric mucosa-associated lymphoid tissue lymphoma. World J Gastroenterol. 2014;20(11):2751-2759
631. El HH, El-Sabban M, Hasegawa H, Zaatari G, Ablain J, Saab ST. et al. Therapy-induced selective loss of leukemia-initiating activity in murine adult T cell leukemia. J Exp Med. 2010;207(13):2785-2792
632. Gill PS, Harrington W Jr, Kaplan MH, Ribeiro RC, Bennett JM, Liebman HA. et al. Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med. 1995;332(26):1744-1748
633. Hermine O, Bouscary D, Gessain A, Turlure P, Leblond V, Franck N. et al. Brief report: treatment of adult T-cell leukemia-lymphoma with zidovudine and interferon alfa. N Engl J Med. 1995;332(26):1749-1751
634. Fernandes R, Ferreira S, Botelho MC. Commentary: Theileria Parasites Secrete a Prolyl Isomerase to Maintain Host Leukocyte Transformation. Front Med (Lausanne). 2018;5:120-doi 10.3389/fmed.2018.00120
635. Marsolier J, Perichon M, DeBarry JD, Villoutreix BO, Chluba J, Lopez T. et al. Theileria parasites secrete a prolyl isomerase to maintain host leukocyte transformation. Nature. 2015;520(7547):378-382
636. Rchiad Z, Haidar M, Ansari HR, Tajeri S, Mfarrej S, Ben RF. et al. Novel tumour suppressor roles for GZMA and RASGRP1 in Theileria annulata-transformed macrophages and human B lymphoma cells. Cell Microbiol. 2020;22(12):e13255-doi 10.1111/cmi.13255
637. Baum JK, Bookstein JJ, Holtz F, Klein EW. Possible association between benign hepatomas and oral contraceptives. Lancet. 1973;2(7835):926-929
638. Horvath E, Kovacs K, Ross RC. Letter: Benign hepatoma in a young woman on contraceptive steroids. Lancet. 1974;1(7853):357-358
639. Knapp WA, Ruebner BH. Letter: Hepatomas and oral contraceptives. Lancet. 1974;1(7851):270-271
640. Lingeman CH. Letter: Liver-cell neoplasms and oral contraceptives. Lancet. 1974;1(7846):64. doi: 10.1016/s0140-6736(74)93063-3
641. Oral contraceptives and cancer. Lancet. 1972; 2(7783): 911.
642. Thalassinos NC, Lymberatos C, Hadjioannou J, Gardikas C. Letter: Liver-cell carcinoma after long-term oestrogen-like drugs. Lancet. 1974;1(7851):270
643. Wendel HA. Oral contraceptives and cancer. Lancet. 1972;2(7787):1139
644. Potter VR. Blocked ontogeny. Science. 1987;237(4818):964
645. Potter VR. Phenotypic diversity in experimental hepatomas: the concept of partially blocked ontogeny. The 10th Walter Hubert Lecture. Br J Cancer. 1978;38(1):1-23
646. Mills J. Paligenosis: A conserved program differentiated cells use in regeneration and misuse in cancer. FASEB J. 2022; 36 Suppl 1. doi: 10.1096/fasebj. 2022 36.S1.0I659
647. Brown JW, Cho CJ, Mills JC. Paligenosis: Cellular Remodeling During Tissue Repair. Annu Rev Physiol. 2022;84:461-483
648. Bignold LP. Embryonic reversions and lineage infidelities in tumour cells: genome-based models and role of genetic instability. Int J Exp Pathol. 2005;86(2):67-79
649. Liao DZ, Porsch-Hallstrom I, Gustafsson JA, Blanck A. Persistent sex differences in growth control of early rat liver lesions are programmed during promotion in the resistant hepatocyte model. Hepatology. 1996;23(4):835-839
650. Katase N, Tamamura R, Gunduz M, Murakami J, Asaumi J, Tsukamoto G. et al. A spindle cell carcinoma presenting with osseous metaplasia in the gingiva: a case report with immunohistochemical analysis. Head Face Med. 2008;4:28-doi 10.1186/1746-160X-4-28
651. Kazakov DV, Belousova IE, Bisceglia M, Calonje E, Emberger M, Grayson W. et al. Apocrine mixed tumor of the skin ("mixed tumor of the folliculosebaceous-apocrine complex"). Spectrum of differentiations and metaplastic changes in the epithelial, myoepithelial, and stromal components based on a histopathologic study of 244 cases. J Am Acad Dermatol. 2007;57(3):467-483
652. Navani SS, Alvarado-Cabrero I, Young RH, Scully RE. Endometrioid carcinoma of the fallopian tube: a clinicopathologic analysis of 26 cases. Gynecol Oncol. 1996;63(3):371-378
653. Pai T, Shet T, Desai S, Patil A, Nair N, Parmar V. et al. Impact of Squamous Differentiation in Breast Carcinoma. Int J Surg Pathol. 2016;24(6):483-489
654. Willis GW. Metastatic metaplastic carcinoma from a pseudosarcoma (Lane tumor) of the mouth. South Med J. 1977;70(12):1467-1468
655. Barnes PJ, Boutilier R, Chiasson D, Rayson D. Metaplastic breast carcinoma: clinical-pathologic characteristics and HER2/neu expression. Breast Cancer Res Treat. 2005;91(2):173-178
656. Catroppo JF, Lara JF. Metastatic metaplastic carcinoma of the breast (MCB): an uncharacteristic pattern of presentation with clinicopathologic correlation. Diagn Cytopathol. 2001;25(5):285-291
657. Eble JN, Young RH. Carcinoma of the urinary bladder: a review of its diverse morphology. Semin Diagn Pathol. 1997;14(2):98-108
658. Hanada M, Nakano K, Ii Y, Yamashita H. Carcinosarcoma of the esophagus with osseous and cartilagenous production. A combined study of keratin immunohistochemistry and electron microscopy. Acta Pathol Jpn. 1984;34(3):669-678
659. Elsensohn A, Mo JH, Maly TJ, Lee PK, de FS. Myoepithelioma of Soft Tissue with both Squamous and Adipocytic Metaplasia. Am J Dermatopathol. 2018;40(2):142-144
660. McKenney JK. Precursor lesions of the urinary bladder. Histopathology. 2019;74(1):68-76
661. Yorita K, Nakagawa H, Miyazaki K, Fukuda J, Ito S, Kosai M. Infarcted Warthin tumor with mucoepidermoid carcinoma-like metaplasia: a case report and review of the literature. J Med Case Rep. 2019;13(1):12-doi 10.1186/s13256-018-1941-3
662. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674
663. Weinberg RA. Coming full circle-from endless complexity to simplicity and back again. Cell. 2014;157(1):267-271
664. Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21(1):12-20
665. Lazebnik Y. What are the hallmarks of cancer? Nat Rev Cancer. 2010;10(4):232-233
666. Srivastava S, Koay EJ, Borowsky AD, De Marzo AM, Ghosh S, Wagner PD. et al. Cancer overdiagnosis: a biological challenge and clinical dilemma. Nat Rev Cancer. 2019;19(6):349-358
667. Welch HG. Cancer Screening, Overdiagnosis, and Regulatory Capture. JAMA Intern Med. 2017;177(7):915-916
668. Jegerlehner S, Bulliard JL, Aujesky D, Rodondi N, Germann S, Konzelmann I. et al. Overdiagnosis and overtreatment of thyroid cancer: A population-based temporal trend study. PLoS One. 2017;12(6):e0179387
669. Makarewich CA, Olson EN. Mining for Micropeptides. Trends Cell Biol. 2017;27(9):685-696
670. Takano T. Natural history of thyroid cancer [Review]. Endocr J. 2017;64(3):237-244
671. Kakudo K, Bychkov A, Abelardo A, Keelawat S, Kumarasinghe P. Malpractice Climate Is a Key Difference in Thyroid Pathology Practice Between North America and the Rest of the World. Arch Pathol Lab Med. 2019;143(10):1171-doi 10.5858/arpa.2019-0228-LE
672. Heng HH, Stevens JB, Bremer SW, Ye KJ, Liu G, Ye CJ. The evolutionary mechanism of cancer. J Cell Biochem. 2010;109(6):1072-1084
673. Kadota M, Yang HH, Gomez B, Sato M, Clifford RJ, Meerzaman D. et al. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PLoS One. 2010;5(2):e9201. doi: 10.1371/journal.pone.0009201
674. Miller FR. Xenograft models of premalignant breast disease. J Mammary Gland Biol Neoplasia. 2000;5(4):379-391
675. Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3(4):355-359
676. Wang Y, Thakur A, Sun Y, Wu J, Biliran H, Bollig A. et al. Synergistic effect of cyclin D1 and c-Myc leads to more aggressive and invasive mammary tumors in severe combined immunodeficient mice. Cancer Res. 2007;67(8):3698-3707
677. Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197(4302):461-463
678. Cowley G, Gusterson B, Knight J. Growth in agar and tumor formation in immunologically incompetent mice as criteria for keratinocyte transformation. Cancer Lett. 1983;21(1):95-104
679. Hamburger AW. The human tumor clonogenic assay as a model system in cell biology. Int J Cell Cloning. 1987;5(2):89-107
680. Stanbridge EJ, Wilkinson J. Analysis of malignancy in human cells: malignant and transformed phenotypes are under separate genetic control. Proc Natl Acad Sci U S A. 1978;75(3):1466-1469
681. Doyle LA, Fletcher CD. Metastasizing "benign" cutaneous fibrous histiocytoma: a clinicopathologic analysis of 16 cases. Am J Surg Pathol. 2013;37(4):484-495
682. Soufi M, Lupinacci RM, Godiris-Petit G, Vignot S, Genestie C, Menegaux F. et al. Growing teratoma syndrome of the ovary presenting with liver metastasis: report of a case. Eur J Gynaecol Oncol. 2015;36(4):473-476
683. Kataria SP, Varshney AN, Nagar M, Mandal AK, Jha V. Growing Teratoma Syndrome. Indian J Surg Oncol. 2017;8(1):46-50
684. Pienta KJ, Hammarlund EU, Axelrod R, Amend SR, Brown JS. Convergent Evolution, Evolving Evolvability, and the Origins of Lethal Cancer. Mol Cancer Res. 2020;18(6):801-810
685. Porta-Pardo E, Valencia A, Godzik A. Understanding oncogenicity of cancer driver genes and mutations in the cancer genomics era. FEBS Lett. 2020: 10.1002/1873-3468.13781.-doi: 10.1002/1873-3468.13781.
686. Priestley P, Baber J, Lolkema MP, Steeghs N, de BE, Shale C. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575(7781):210-216
687. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789-799
688. Giunta S. Decoding human cancer with whole genome sequencing: a review of PCAWG Project studies published in February 2020. Cancer Metastasis Rev. 2021:1-16
689. Dentro SC, Leshchiner I, Haase K, Tarabichi M, Wintersinger J, Deshwar AG. et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell. 2021;184(8):2239-2254
690. Sinha S, Gajra A. Nasopharyngeal Cancer: StatPearls Publishing. 2021. https://www.ncbi.nlm.nih.gov/books/NBK459256/.
691. Stepan KO, Mazul AL, Skillington SA, Paniello RC, Rich JT, Zevallos JP. et al. The prognostic significance of race in nasopharyngeal carcinoma by histological subtype. Head Neck. 2021;43(6):1797-1811
692. Guo R, Wu H, Wang J, Lian CL, He ZY, Zhang WW. et al. Lymph Node Status and Outcomes for Nasopharyngeal Carcinoma According to Histological Subtypes: A SEER Population-Based Retrospective Analysis. Adv Ther. 2019;36(11):3123-3133
693. Richardson LC, Wingo PA, Zack MM, Zahran HS, King JB. Health-related quality of life in cancer survivors between ages 20 and 64 years: population-based estimates from the Behavioral Risk Factor Surveillance System. Cancer. 2008;112(6):1380-1389
694. Marcotte EL, Spector LG, Mendes-de-Almeida DP, Nelson HH. The Prenatal Origin of Childhood Leukemia: Potential Applications for Epidemiology and Newborn Screening. Front Pediatr. 2021;9:639479-doi 10.3389/fped.2021.639479. eCollection 2021
695. Shimizu N. Gene Amplification and the Extrachromosomal Circular DNA. Genes (Basel). 2021;12(10):1533.-doi 10.3390/genes12101533
696. Wu S, Bafna V, Chang HY, Mischel PS. Extrachromosomal DNA: An Emerging Hallmark in Human Cancer. Annu Rev Pathol. 2021: -doi: 10.1146/annurev-pathmechdis-051821-114223.
697. Abascal F, Juan D, Jungreis I, Kellis M, Martinez L, Rigau M. et al. Corrigendum: Loose ends: almost one in five human genes still have unresolved coding status. Nucleic Acids Res. 2018;46(22):12194. doi: 10.1093/nar/gky1146
698. Abascal F, Juan D, Jungreis I, Kellis M, Martinez L, Rigau M. et al. Loose ends: almost one in five human genes still have unresolved coding status. Nucleic Acids Res. 2018;46(14):7070-7084
699. Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer. 2003;3(12):952-959
700. Dujon AM, Aktipis A, Alix-Panabieres C, Amend SR, Boddy AM, Brown JS. et al. Identifying key questions in the ecology and evolution of cancer. Evol Appl. 2021;14(4):877-892
701. Boutry J, Tissot S, Ujvari B, Capp JP, Giraudeau M, Nedelcu AM. et al. The evolution and ecology of benign tumors. Biochim Biophys Acta Rev Cancer. 2021;1877(1):188643. doi: 10.1016/j.bbcan.2021.188643
702. Marino-Enriquez A, Fletcher CD. Shouldn't we care about the biology of benign tumours? Nat Rev Cancer. 2014;14(11):701-702
703. Zhu S, Wang J, Zhou H, Zhao Y, He Y, Zellmer L. et al. Conflicting Cancer Theories by Recognizing the Roles of Epigenetic and Genetic Alterations in the Immediate-Cancer-Causing Genes that Establish Cellular Immortality and Autonomy. Preprints 2020:202011.0708.v1-https://www.preprints.org/manuscript/202011.0708/v1.
Author contact
![]() Corresponding authors: Hong Ma, Department of Oral and Maxillofacial Surgery, School of Stomatology, Guizhou Medical University, 9 Beijing Road, Guiyang 550004, Guizhou Province, P.R. China. E-mail: mahongedu.cn; Fei Deng, Department of Pathology, The Third Affiliated Hospital of Zunyi Medical University, Gun-Yi City 563000, Guizhou Province, P.R. China. E-mail: 1296223120com; Wenxiu Yang, Department of Pathology, The Affiliated Hospital, Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China. E-mail: ypq1964com; Joshua Liao, Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou Province 550004, P. R. China. E-mail: djliaoedu.cn.
Corresponding authors: Hong Ma, Department of Oral and Maxillofacial Surgery, School of Stomatology, Guizhou Medical University, 9 Beijing Road, Guiyang 550004, Guizhou Province, P.R. China. E-mail: mahongedu.cn; Fei Deng, Department of Pathology, The Third Affiliated Hospital of Zunyi Medical University, Gun-Yi City 563000, Guizhou Province, P.R. China. E-mail: 1296223120com; Wenxiu Yang, Department of Pathology, The Affiliated Hospital, Guizhou Medical University, Guiyang 550004, Guizhou Province, P.R. China. E-mail: ypq1964com; Joshua Liao, Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China in Guizhou Medical University, Guiyang, Guizhou Province 550004, P. R. China. E-mail: djliaoedu.cn.