Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(11):3209-3220. doi:10.7150/jca.76695 This issue Cite
Review
KRAS as a Key Oncogene in the Clinical Precision Diagnosis and Treatment of Pancreatic Cancer
Manxiong Dai1,3, Shaofeng Chen1,3, Xiong Teng1,3, Kang Chen1,3 ![]() , Wei Cheng1,2 3
, Wei Cheng1,2 3 ![]()
1. Department of Hepatobiliary Surgery, Hunan Provincial People's Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, 410005 Hunan Province, China.
2. Xiangyue Hospital Affiliated to Hunan Institute of Parasitic Diseases, National Clinical Center for Schistosomiasis Treatment, Yueyang 414000, Hunan Province, China.
3. Translational Medicine Laboratory of Pancreas Disease of Hunan Normal University, Changsha 410005, China.
Received 2022-6-30; Accepted 2022-8-19; Published 2022-8-31
Abstract

Pancreatic ductal adenocarcinoma (PDAC) is one of the most malignant tumors, with a 5-year survival rate of less than 10%. At present, the comprehensive treatment based on surgery, radiotherapy and chemotherapy has encountered a bottleneck, and targeted immunotherapy turns to be the direction of future development. About 90% of PDAC patients have KRAS mutations, and KRAS has been widely used in the diagnosis, treatment, and prognosis of PDAC in recent years. With the development of liquid biopsy and gene testing, KRAS is expected to become a new biomarker to assist the stratification and prognosis of PDAC patients. An increasing number of small molecule inhibitors acting on the KRAS pathway are being developed and put into the clinic, providing more options for PDAC patients.
Keywords: Pancreatic neoplasms, KRAS gene, Targeted therapy, Diagnosis, Review
Introduction
In 2020, there were 490,000 new cases of pancreatic cancer and more than 460,000 deaths worldwide [1], with a 5-year survival rate less than 10% [2]. Pancreatic cancer, a serious disease threatening the life and health of people, is projected to become the second leading cause of cancer death worldwide by 2030 [3]. Despite the continuous development of surgical techniques and the emergence of various chemotherapies for PDAC, the improvement in patient survival is still rather limited.
In 2008, Jones et al. [4] conducted the first full exon sequencing of pancreatic cancer, with different gene mutations in 12 tumor-related pathways observed. Subsequent studies confirmed these findings and identified four major driver mutations including KRAS, tumor protein p53 (TP53), SMAD family member 4 (SMAD4) and cyclin dependent kinase inhibitor 2A (CDKN2A) [5], among which, KRAS mutation is the most common one, with a mutation rate up to 90%. However, how KARS mutation plays its clinical value in the clinical diagnosis and treatment of PDAC and whether it has the possibility of becoming a molecular targeted therapy site to improve the diagnosis and treatment of PDAC are all questions being frequently explored in the field of precision diagnosis and treatment of pancreatic tumors. Against such a context, the focus of this paper will be placed on the occurrence of KRAS mutations in PDAC, types of KRAS mutations and their correlation with the clinical prognosis of PDAC, and the research progress in precision diagnosis and treatment related to KRAS mutations.
1. The Oncogenic Mechanism of KRAS Gene Mutation and Its Relationship with Pancreatic Cancer
KRAS mutations are the most common mutations in human solid tumors [6], and can be found in 90% of pancreatic cancer patients [7]. Similar alternative mutations activating RAS downstream pathways are detected in another 10% of KRAS wild-type patients [8]. KRAS is a subtype of the RAS gene family, which also includes HRAS and NRAS. KRAS gene is located on human chromosome 12, encoding KRAS4A and KRAS4B proteins, also known as P12 protein due to its molecular weight of 12KDa[9]. KRAS protein is essentially a GTP enzyme, activated when binding to GTP and activating more than 80 downstream effector proteins and multiple pathways including rapidly accelerated fibrosarcoma (RAF)- mitogen activated protein kinase (MEK)- extracellular signal regulated kinase (ERK), phosphatidylinositol 3 kinase (PI3K)- protein kinase B (AKT)- mammalian target of rapamycin (mTOR), Hippo-yes associated protein (YAP), etc. KRAS protein is inactivated when binding to GDP. The conversion process of KRAS protein between its activated and inactivated states is regulated by two types of factors, i.e., guanine nucleotide exchange factor (GEF) and GTP-activated protein (GAP). The former promotes the binding of KRAS and GTP to activate KRAS, while the latter hydrolyzes GTP for the inactivation of KRAS [10]. Epidermal growth factor receptor (EGFR) upstream of RAS protein is stimulated by growth factors and mitogen, thereby causing GEF aggregation in RAS protein, promoting RAS protein activation and activating downstream pathways. KRAS gene does matter considerably in regulating cell growth, proliferation, differentiation and apoptosis, the mutation of which inhibits the hydrolysis of RAS-GTP to RAS-GDP, and leads to the continuous activation of downstream pathways, thereby ultimately bringing about tumorigenesis [11].
Based on the degree of tissue structural disorder and nuclear abnormality, intraepithelial neoplasia of the pancreas (PanIN), a precancerous lesion of PDAC, is divided into three stages (I-III) from low to high [12]. KRAS gene mutation is the initiating factor for the occurrence of PDAC, and over 90% of low-grade PanIN is coupled with KRAS gene mutation and shortened telomere corrosion [13, 14]. In the PDAC gene engineered mouse model (GEMM), KRAS mutation and TP53 allele deletion jointly lead to the dedifferentiation and PanIN of pancreatic epithelial cells, while the loss of KRAS mutation results in the regression of lesions, indicating the decisive role of KRAS mutation in the development of PDAC [15]. Besides, studies have also shown that a single KRAS gene mutation is not enough to maintain the concentration of activated KRAS protein that brings about PDAC. TP53 mutation can inhibit GAP activity and have more KRAS protein activated, thereby causing PDAC [16]. At the same time, some studies have indicated that the larger the copy number of KRAS gene mutation is, the higher the malignant degree of the tumor becomes, which plays an important role in the substitution of low-dose KRAS mutated tumor MYC proto-oncogene (MYC), YAP and nuclear factor of kappa light polypeptide gene enhancer in B cells 2 (NFKB2), and promotes the invasion and metastasis of the tumor [17]. To this end, the conclusion can be drawn that KRAS mutation is the initiating factor of PDAC, and that TP53, SMAD4 and other genes play an important synergistic role in the occurrence and development of PDAC.
2. Mutation Types of KRAS Gene and Their Relationship with PDAC Diagnosis and Prognosis
KRAS mutations in PDAC are mainly missense mutations, most observed in codon 12. According to the different bases of missense mutations, KRAS mutations can be divided into G12D, G12V, G12R, G12C, G12A and G12S, among which, G12D mutation is the most common one. G12D patients account for 51% of all mutations, while G12V, 32%; G12R, 12%, and all other phenotypes, less than 5% [13, 18].
Other mutations with a lower frequency may also occur on codons 11, 13, 61, and 146[19]. The prognosis of patients with KRAS gene mutation has been studied for a long time, and most study results show that compared with patients with KRAS wild-type, those with KRAS mutation are exposed to a worse prognosis [20]. For patients with resectable or unresectable PDAC, the prognosis is different for those with different KRAS mutation subtypes, among which, the overall survival (OS) period of patients with G12D mutation is 6 months, significantly shorter than that of those with other KRAS mutation types (KRAS wild type 9 months, G12V 9 months, G12R 14 months, P = 0.003)[21]. For patients with unresectable PDAC, G12D mutation is also an independent risk factor for poor prognosis, while KRAS mutation Q61 subtype predicts a better survival [22], which may be mainly attributed to the subtle differences in downstream effector proteins and downstream pathways of KRAS mutations of different subtypes [23, 24].
KRAS pathway and its targeted inhibitors. EGFR transmits signals to KRAS and activates KRAS; GRB2 promotes the activation of KRAS; and SHP2 is a scaffold protein that connects KRAS, GRB2 and SOS1. The red text represents the targeted inhibitor for this target.
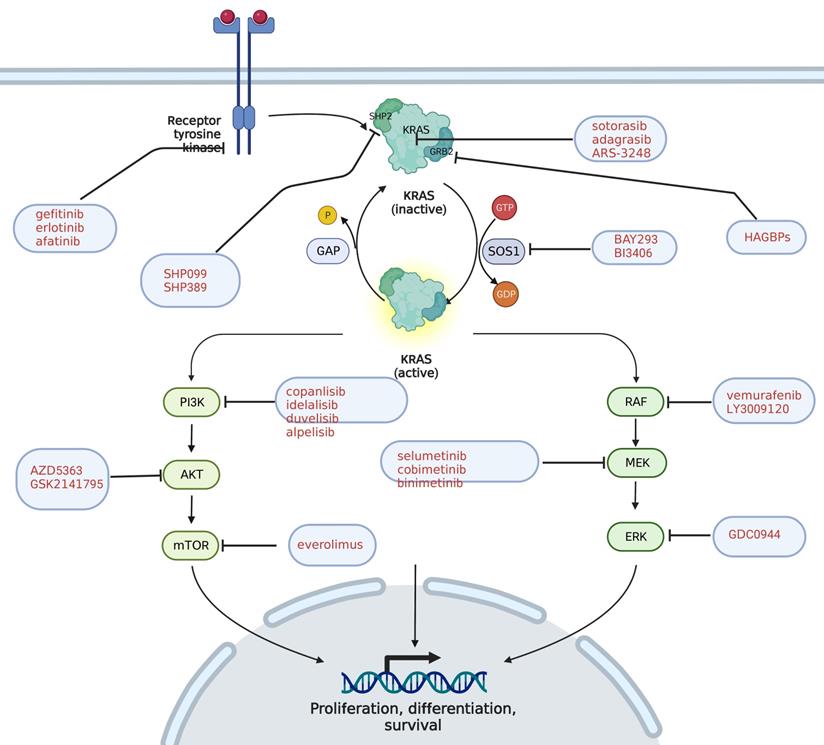
Liquid biopsy technology is an emerging non-invasive pathological detection technology, and obtains tumor gene or protein expression in patients mainly by detecting the marker information released by tumor cells into body fluids. Circulating tumor DNA (ctDNA) is a portion of DNA released by tumor cells into the peripheral blood, with a short half-life, from 15 minutes to less than 2 hours, and is cleared quickly in the peripheral blood, making it a marker for monitoring tumor dynamics in real time. ctDNA is used to monitor efficacy, drug resistance and recurrence. Previous studies have shown that the ctDNA detection results for patients after PDAC are closely related to their prognosis, which can effectively predict tumor recurrence and metastasis, and stratify the risk of these patients. On the one hand, the specificity of ctDNA KRAS mutation is as high as 96% in the early diagnosis of PDAC, while the sensitivity remains only 30%. In combination with other protein biomarkers carbohydrate antigen 199 (CA199), carcinoembryonic antigen (CEA) and osteopontin can be increased to 64% [25]. Therefore, ctDNA can assist the clinical more sensitive detection of abnormalities in the early stage of PDAC. On the other, KRAS mutations in ctDNA have also been used for predicting prognosis. Lee et al. [26] detected preoperative and postoperative ctDNA KRAS mutations in 38 patients with surgically resectable PDAC, and the results show that patients with KRAS positive ctDNA preoperatively experience a shorter median recurrence-free survival (RFS) (10.3 months, P=0.002) and a shorter median OS (13.6 months, P=0.015) compared with those without KRAS. Postoperative ctDNA-KRAS positive patients are exposed to a faster rate of recurrence, a shorter median RFS (5.4 months versus 17.1 months, P < 0.001), and a shorter median OS (10.6 months, P =0.003) than negative patients, indicating that positive ctDNA KRAS is closely related to poor prognosis. CtDNA variation can be detected by blood during the perioperative period, which thereby assists the judgment of clinical prognosis.
3. Advances in KRAS Targeting Drugs for Pancreatic Cancer
Currently, no KRAS inhibitor for PDAC has been approved, and KRAS was once considered drug-resistant. In turn, the KRAS gene is indirectly inhibited by inhibiting its upstream and downstream targets. With recent advances in research, direct inhibitors of KRAS gene in lung cancer have been approved for marketing. According to the location of inhibition targets, drugs acting on KRAS pathway can be divided into three categories, i.e., upstream KRAS molecular targeting drugs, KRAS targeted drugs and KRAS downstream molecular targeting drugs.
3.1 KRAS upstream molecular targeting agents
First-generation EGFR inhibitors, such as gefitinib and erlotinib, improve little in patients with PDAC either in combination with gemcitabine or with tigio, and the overall survival time (OS) is slightly lower in the gemcitabine plus erlotinib group than in the gemcitabine monotherapy group in Phase III clinical trial (LAP07) in advanced pancreatic cancer [27]. A Phase II clinical study in Korea showed that the OS of the gemcitabine and tigio combined with erlotinib group was lower than that of the gemcitabine and tigio group [28], which is attributed to the compensatory expression of three other members of the human epidermal growth factor receptor family (HER), including four members, i.e., EGFR/HER1, ERBB-2 /HER-2, ERBB-3 /HER-3 and ERBB-4 /HER-4[29]. Inhibitors of both EGFR and ERBB, such as afatinib, have been frequently studied in lung cancer and been proven effective [30]. A phase II clinical study of afatinib in patients with PDAC (NCT02451553) is currently underway.
Other upstream targets include son of sevenless 1 (SOS1), a GEF member that inhibits KRAS activation by preventing its binding to KRAS. Previous studies mainly focused on synthetic drugs directly binding SOS1 to prevent it from binding KRAS. Although these compounds can effectively bind SOS1, the blocking efficiency remains low [31, 32]. New SOS1 inhibitors BAY293 [33] and BI3406 [34] have achieved a good inhibition effect on tumor cells, and further animal and human tests are needed for verification.
Protein tyrosine phosphatase (SHP2) is both a non-receptor protein tyrosinase and a scaffold adaptor binding to effector proteins[35], which connects SOS1, growth factor receptor bound protein 2 (GRB2) and KRAS in the KRAS pathway, thereby mediating the activation of KRAS[36]. In 2016, the first selective oral SHP2 inhibitor SHP099 appeared [37]. With the discovery of drug binding sites, an increasing number of SHP2 inhibitors, such as SHP389, BBP398, HBI2376, etc., have come out [38-40]. No targeted inhibitors for SHP2 are currently approved for marketing, and most of the drugs are in Phase I and II clinical trials in patients with advanced solid tumors, including PDAC.
GRB2 is a 25kDa adaptor protein composed of one SH2 and two SH3 domains, which can bind to SOS1 for promoting the activation of KRAS, known as a potential therapeutic target for KRAS-related tumors [41]. The high-affinity GRB2-binding peptides HAGBPs and PTD-GRB2-SH2 [42] can block the connection between SH3 domain and SOS1, thus blocking the conduction of carcinogenic signal. Current research on GRB2 mainly focuses on hematologic tumor and breast cancer, and a Phase I clinical trial (NCT04196257) is underway for solid tumors such as PDAC.
3.2 KRAS molecular targeting agents
KRAS is the first discovered human tumor gene, and KRAS protein is equipped with a nearly spherical structure with no obvious binding site, which makes it difficult to synthesize a compound that can target binding and inhibit its activity. It was not until 2013 that Shokat and his team [43] discovered an allosteric binding pocket behind the KRAS-G12C protein switch-II. It was exactly the very discovery that led to the development of several small molecule covalent inhibitors targeting KRAS G12C mutations. Among them, Amgen's sotorasib(AMG510) was approved by the US Food and Drug Administration(FDA) in May 2021 for non-small cell lung cancer patients with KRAS G12C mutation, becoming the first KRAS-targeted drug hitting the world market. Sotorasib is a highly selective small-molecule inhibitor that targets KRAS-G12C and covalently binds to the switch-II allosteric binding pocket, which keeps KRAS-G12C inactive and inhibits the downward transmission of KRAS-oncogenic signals. It has shown promising results in Phase I and II trials in advanced solid tumors, with 8 of the 12 enrolled patients with advanced PDAC suffering from stable disease (SD) and 1 having a partial remission [44]. Meanwhile, several KRAS G12C inhibitors are in Phase I/II clinical trials. Mirati Therapeutics' adagrasib is attracting extensive attention. MRTX1257, another KRAS G12C inhibitor of the company, is still in preclinical development. FDA has approved a clinical trial application developed by Wellspring Biosciences for ARS-3248, and Janssen Biotech will take over the Phase I clinical trial and subsequent development. Other KRAS-G12C targeted inhibitors for PDAC, such as GFH925 and JAB21822, are currently in Phase I or II clinical trials.
In addition to small molecule drugs directly targeting KRAS, other innovative therapeutic modalities for KRAS, such as RNA Interference (RNAi) therapies and KRAS-associated tumor vaccines, are also being extensively explored.
RNAi has been a hot research topic since it was first reported in 1998. In recent years, small interfering RNA (siRNA) has been frequently used for targeted therapy of PDAC patients, with an efficient delivery platform acting as the key to its clinical efficacy. Early delivery platforms were divided into viral vectors (adenoviruses and retroviruses) and non-viral vectors (cationic liposomes, etc.) [45]. The extracellular matrix of pancreatic cancer is composed of dense connective tissue. Exosomes are extracellular vesicles containing mRNA and proteins involved in the transmission of information between cells [46]. The CD47 transmembrane protein of exosomes helps to evade host immune clearance, making it an efficient delivery vector for siRNA [47]. Exosome delivery siRNA targeting KRAS-G12D has presented superior efficacy in animal studies [48], and a Phase I clinical trial (NCT03608631) is currently underway.
A KRAS-associated cancer vaccine has emerged more than once. Currently, the most promising pancreatic cancer vaccine is GI4000, which expresses KRAS peptides of various mutant subtypes. In the Phase II clinical trial of KRAS-mutated lung cancer patients, 50% of them exhibited specific antigen-antibody reactivity [49]. KRAS peptide vaccine is currently in Phase I/II clinical trials for patients with PDAC (NCT03329248), and Mdc3/8 dendritic cell vaccine targeting KRAS mutations is also currently undergoing Phase I clinical trials (NCT03592888) [50].
3.3 KRAS downstream molecular targeting agents
Downstream of KRAS involves multiple signal transduction pathways and more than 80 effector proteins, among which, the most studied and understood are RAF-MEK-ERK, MAPK cascade signaling, PI3K-AkT and Hipoo-YAP pathway. Specific inhibitors targeting these targets have been or are being tested in clinical trials.
RAF proteins, including ARAF, BRAF and CRAF, are key components of MAPK. RAF, the first kinase of this pathway, is considered an ideal target for the development of anticancer drugs [54]. First-generation RAF inhibitors have been developed and used for BRAF (V600E) - carrying therapies, such as Vemurafenib [55], Dabrafenib [56], and Encorafenib [57]. Good results have been obtained at the initial treatment stage, but tumor drug resistance remains the main problem existing at present. The main mechanisms are: (i) upregulation of KRAS activity, which brings about abnormal activation of ERK; and (ii) BRAF producing N-terminal truncated variants, and reducing drug affinity [58]. The second generation of pan-RAF inhibitors, including LY3009120 [59], TAK632 [60], TAK580 [61], CCT3833 [62] and other drugs, can effectively avoid the above-mentioned problem. However, there are limited clinical data of both first-generation and second-generation RAF inhibitors in advanced solid tumors such as PDAC. A Phase II clinical trial (NCT04390243) is currently underway in PDAC patients in combination with the RAF inhibitor Encorafenib and the MEK inhibitor Binimetinib.
Single-target clinical trials
| Target | Disease | Agent | Phase | Clinical Trial Number | Status/Results |
|---|---|---|---|---|---|
| EGFR | PDAC | Erlotinib | II | NCT01608841 | [51] |
| EGFR | PDAC | Afatnib | II | NCT02451553 | Active |
| GRB2 | Solid tumor | BP1001 | I | NCT04196257 | Recruiting |
| SHP2 | Solid tumor | HBI2376 | I | NCT05163028 | Recruiting |
| KRAS-G12C | Solid tumor | sotorasib | III | NCT03600883 | [44] |
| KRAS | PDAC | Rat sarcoma KRAS vaccine | I | NCT05013216 | Active |
| KRAS | PDAC | mDC318 KRAS vaccine | I | NCT03608631 | Recruiting |
| KRAS-G12D | PDAC | exosome-KRAS(G12D)-siRNA | I | NCT03608631 | Recruiting |
| KRAS | Solid tumor | MRTX849 | I、II | NCT03785249 | Recruiting |
| BRAF | PDAC | Vemurafenib | II | NCT05068757 | Recruiting |
| MEK | Solid tumor | Pimasertib | I、II | NCT01016483 | [52] |
| MEK | Solid tumor | GSK1120212 | II | NCT01231581 | [53] |
| ERK | Solid tumor | Ulixertinib | I | NCT04566393 | Active |
| mTOR | PDAC | Sirolimus | I、II | NCT03662412 | Recruiting |
The most important members in the MEK family are MEK1 and MEK2, two key nodes in the MAPK level pathway. Currently, most of the inhibitors targeting MEK are non-ATP competitive inhibitors, which function by combining themselves with MEK hydrophobic region to inhibit phosphorylation [63]. Currently, there are four MEK1/2 inhibitors on the market, i.e., Selumetinib [64], Binimetinib [65], Cobimetinib [66] and Trametinib [67]. Other MEK inhibitors include Pimasertib. In the Phase I/II clinical trial (NCT01016483) of pimasertib combined with gemcitabine in patients with advanced PDAC, the OS and Progression Free Survival (PFS) of patients in the Pimasertib combined with gemcitabine group were found 0.33 and 0.2 months longer than those in the gemcitabine group alone, not statistically significant, which increased the incidence of adverse reactions [52]. A similar Phase II trial of Trametinib in combination with gemcitabine (NCT01231581) also presented limited efficacy [53]. The poor efficacy of MEK single drug also results from tumor drug resistance, and the current drug resistance mechanism includes reactivation of MAPK level pathway; up-regulation of other parallel pathways, such as PI3K, signal transducer and activator of transcription (STAT) and Hippo signaling pathway [68-70]; and transformation of cell phenotypic [71]. Attempts have been made on several combinations to address these resistance mechanisms, among which, MEK inhibitors combined with RAF inhibitors are approved by the FDA for the treatment of melanoma [72, 73]. MEK inhibitors combined with PI3K inhibitors, AKI inhibitors, PI3K/mTOR dual inhibitors, YAP inhibitors and STAT3 inhibitors have all achieved good preclinical efficacy [69, 74-76]. Although pancreatic cancer has not yet benefited from immunotherapy, MEK inhibitors have been proven to increase T cell infiltration in the tumor microenvironment and present significant synergies with immunotherapy such as programmed death-receptor 1 (PD-1) and programmed cell death-ligand 1 (PD-L1), compared with MEK inhibitors alone [77].
ERK is another target of MAPK-class pathway, with ERK1 and ERK2 included in the ERK family [78]. Currently, there are no approved ERK1/2 inhibitors on the market, but some small molecule ERK inhibitors, such as GDC0944 [79], Ulixertinib [80], CC90003 [81] and other drugs, have already been under preclinical or clinical studies. ERK1/2 inhibitors can inhibit epithelial mesenchymal transition (EMT), up-regulate cellular markers of aging, and activate pancreatic stellate cell autophagy in pancreatic cancer.
Another important pathway for the occurrence and development of PDAC is the PI3K-Akt-mTOR pathway. PI3K/Akt/mTOR signal transduction pathway widely exists in tissue cells and is involved in the regulation of cell growth, proliferation, and differentiation. PI3K is the most important component of THE PI3K/AKt/mTOR signaling pathway. Activation can further phosphorylate phosphatidylinositol 4, 5-diphosphate (PIP2) into phosphatidylinositol 3,4, 5-triphosphate (PIP3). PIP3 acts as a second messenger to translocation AKt from the cell membrane to the cytoplasm, and activate configurational changes. Thus, downstream mTOR and other targets can be activated to regulate protein translation and cell growth. Abnormal activation of any of these targets is strongly associated with malignancy [82]. Up to now, five PI3K inhibitors, including Idelalisib [83], Copanlisib [84], Duvelisib [85], Alpelisib [86] and Umbralisib [87], have been approved for marketing worldwide, which are mostly used for the treatment of breast cancer and hematological tumors, but have not been used for PDAC treatment. Preliminary studies have proved that PI3K inhibitors alone are not safe for PDAC patients, and their efficacy is limited. A Phase II/III trial of Rigosertib (a PI3K targeted inhibitor) in combination with gemcitabine in patients with advanced PDAC showed that Rigosertib failed to improve patient outcomes [88]. Currently, no AKI targeted inhibitors have been approved for marketing, but inhibitors including AZD5363 [89], GSK2141795 [90] and other drugs have been or are undergoing clinical trials, with preliminary data showing limited efficacy of single agents. mTOR includes mTORC1 and mTORC2. The former regulates cell growth while the latter is related to cell survival and proliferation [91, 92]. Already approved drugs for the treatment of renal cell carcinoma, lymphoma and other diseases include sirolimus, everolimus, tesirolimus, etc., and clinical trials are underway for patients with PDAC alone or in combination.
Hippo-YAP pathway is another important downstream effector pathway of KRAS, involved in cell proliferation, apoptosis, and control of organ size [93]. The core of Hippo pathway is a kinase cascade reaction, in which MST1/2 kinase and SAV1 form a complex that phosphorylates and activates LATs1/2. LATs1/2 can further phosphorylate YAP/TAZ, and the phosphorylated YAP/TAZ cannot enter the nucleus and will be ubiquitinated and degraded in the cytoplasm, which cannot play its proliferation-promoting and anti-programmed cell death activities, either. If the MST1/2-YAP/TAZ pathway is blocked or inactivated, the unphosphorylated YAP/TAZ enters the nucleus and binds with downstream transcription factors such as transcriptional enhanced associate domain (TEAD) to promote the expression of target genes [94]. Yiet al. [95] found that YAP plays a limited role in the first step of PDAC transformation, acino-ductal metaplasia (ADM), but a considerable role in the transformation of PanIN to PDAC. In addition, YAP can disrupt tumor-matrix interactions that promote PDAC invasion and metastasis [96]. High expression of YAP is associated with poor prognosis of PDAC [97-99]. At the same time, studies have shown that Hippo-YAP pathway is related to drug resistance of tumor cells to chemotherapy and targeted therapy [100, 101]. Targeted blocking of the MAPK cascade signaling pathway will lead to the upregulation of Hippo pathway, thereby mediating the drug resistance of targeted drugs. Albumin-paclitaxel combined with gemcitabine is a first-line chemotherapy regimen widely applied to pancreatic cancer, but most patients rapidly develop drug resistance after several courses of treatment. Albumin paclitaxel is an anti-tubulin drug, and its action mechanism depends on the activation of cell cycle kinase CDK1 to induce apoptosis of cancer cells, while the mutation and upregulation of YAP targets can reduce the apoptosis inducing ability of anti-tubulin drugs [102, 103]. The combination of PD-1 and PD-L1 can down-regulate the response of the immune system to human cells, regulate the immune system, and promote the tolerance by inhibiting the inflammatory activity of T cells. The expression of PD-1 and PD-L1 in tumor cells can lead to immune evasion, while the activation of YAP or TAZ can up-regulate the expression of PD-1 and PD-L1 in tumor cells, thereby mediating the immune escape of tumor cells [104-106].
The pathogenesis of the Hippo pathway mentioned above also provides a new treatment vision, i.e., YAP targeted inhibitors combined with chemotherapy, targeted and immunotherapy. Current YAP targeting inhibitors mainly include two types, i.e., inhibitors directly targeting YAP and those targeting YAP downstream transcription factors. Verteporfin is the only existing small molecule inhibitor that acts directly on YAP targets and can bind to YAP to block the interaction between YAP and TEAD [107]. Nie et al. [108] have treated patients with KRAS mutation-positive pancreatic cancer using Verteporfin in combination with a pan-Raf targeted inhibitor (LY3009120), and the results show that the combination significantly enhances the anti-tumor efficacy of LY3009120; Roberge et al.[109] found in animal studies that compared to gemcitabine alone, Verteporfin in combination with gemcitabine improves survival in mice, which, however, is photosensitive and cytotoxic and has been proven exposed to relatively large side effects[110, 111]. To this end, the downstream TEAD transcription factor targets such as Vinylsulfonamide derivatives have been studied instead [112].
3.4 KRAS gene mutation and autophagy
Cell autophagy is mediated by lysosomes in eukaryotes, the degradation of highly conservative in the energy shortage, reactive oxygen species under various stress conditions such as accumulation, cells that can pass autophagosome formation, the intracellular substances to the soluble enzyme degradation and recycling in the body, and thus obtains the material and energy needed for powering a cell survival [113]. Compared with normal pancreatic duct cells, autophagy in PanIN and PDAC cells is up-regulated [114]. However, the activation of the Raf-MEK-ERK pathway can inhibit autophagy [115, 116]. The targeted inhibition of the Raf-MEK-ERK pathway can up-regulate autophagy and increase the PDAC cells' tolerance to autophagy. The combination of RAF-MEK-ERK pathway targeting inhibitors and autophagy inhibitors chloroquine or hydroxychloroquine can effectively inhibit the growth of PDAC cells [117-119]. Several studies have initiated clinical trials of combined MAPK pathway targeting inhibitors and autophagy inhibitors, i.e., NCT04145297, NCT03825289, and NCT04132505. Not only can the inhibition of autophagy inhibit the growth of PDAC cells, but also excessive induction of autophagy can inhibit the proliferation of PDAC cells. MTOR regulates autophagy by sensing changes in nutrient levels inside and outside cells, and inhibits autophagy when activated [120]. The mTOR targeting inhibitor Sirolimus can over-induce autophagy, thereby inhibiting the proliferation of PDAC cells [121].
3.5 KRAS gene mutation and pancreatic cancer microenvironment
The microenvironment of pancreatic cancer refers to the local environment of pancreatic cancer cells, involving cellular and non-cellular components. The former mainly include pancreatic stellate cells (PSC), cancer-associated fibroblasts (CAF), and immune cells, while the latter, also known as extracellular matrix (ECM), contain collagen, matrix proteins and a variety of soluble factors. Different from other solid tumors, pancreatic stroma accounts for more than 80% of the tumor volume and contains abundant extracellular matrix, pancreatic stellate cells and tumor-associated fibroblasts, known as one of the reasons for the poor response to chemoradiotherapy, targeted and immunotherapy[122]. Mutation of KRAS gene can alter the tumor microenvironment of pancreatic cancer by changing the composition of extracellular matrix after inducing the production of various chemokines and fibroblasts. It is widely acknowledged that Hedgehog signaling pathway matters considerably in embryonic development, stem cell regulation, etc., and is highly activated in PDAC. Besides, sonic hedgehog (SHH) is the ligand of hedgehog signaling pathway [123]. Tape and colleagues found that KRAS-G12D can activate SHH, thereby promoting the secretion of multiple ECM [124]. Targeted inhibition of SHH can promote the TME of pancreatic cancer, and thus improve the response of pancreatic cancer cells to chemotherapy. SHH inhibitors combined with chemotherapy have achieved good efficacy in animal experiments [125, 126]. However, several clinical studies in patients with advanced PDAC have found that SHH-targeting inhibitor Vismodegib combined with gemcitabine or paclitaxel fails to improve the OS and PFS in patients with PDAC [127, 128]. The combination of saridegib, another SHH inhibitor, with gemcitabine, results in a higher incidence of PDAC progression than placebo and gemcitabine monotherapy [129].
Hippo pathway and its targeted inhibitors. Mst1/2 kinase and SAV1 form a complex that phosphorylates and activates LATs1/2; Lats1/2 kinase in turn phosphorylates and inhibits the transcription coactivators YAP and TAZ; and after dephosphorylation, YAP/TAZ is transferred to the cell nucleus and activates transcription factors such as TEAD1-4, thereby inducing and promoting cell proliferation, and inhibiting apoptosis. The red text represents the targeted inhibitor for this target.
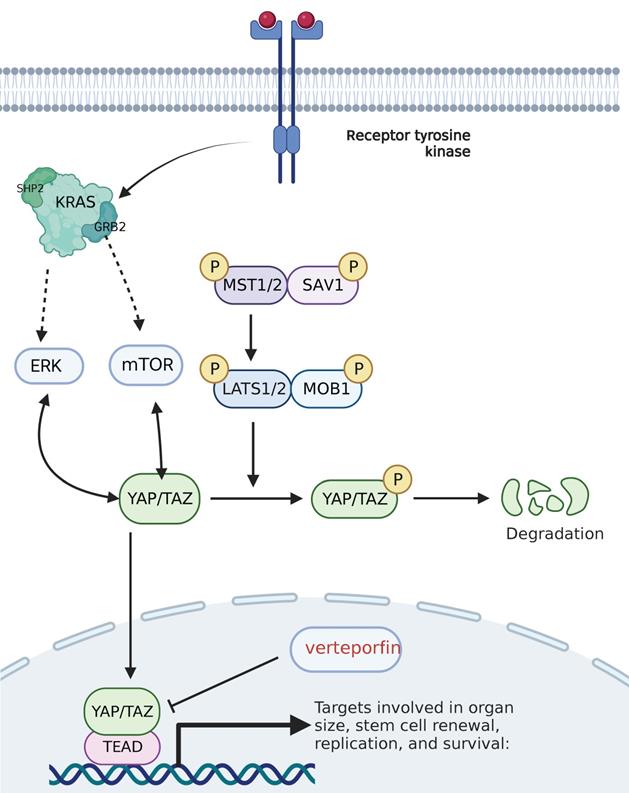
Clinical trials of drug combinations
| Target | Disease | Agent | Phase | Clinical Trial Number | Status/ Results |
|---|---|---|---|---|---|
| KRAS-G12C+SOS1 | PDAC | MRTX849+BI1701963 | I | NCT049752356 | Recruiting |
| ERK+SHP2 | PDAC | LY3214996+RMC4630 | I | NCT04916236 | Active |
| ERK+autophagy | Solid tumor | Binimetinib+ hydroxychloroquine | I | NCT04132505 | Recruiting |
| KRAS-G12C+SHP2 | PDAC | MRTX849+TN0155 | I、II | NCT04330664 | Active |
| ERK+BRAF | PDAC | Binimetinib+Encorafenib | II | NCT04390243 | Recruiting |
| MEK+autophagy | PADC | Trametinib+ hydroxychloroquine | I | NCT03825289 | Recruiting |
| CDK4/6+PI3K | PDAC | Palbociclib+Gedatolisib | I | NCT03065062 | Recruiting |
| PI3K+mTOR | Solid tumor | Alpelisib+Everolimus | I | NCT02677933 | Recruiting |
To sum up the above progress of cancer-pathway targeting drugs in upstream and downstream KRAS, it is found that whether for longitudinal administration of AKT inhibitor in combination with PI3K inhibition, or for horizontal administration of BRAF inhibitor in combination with mTOR inhibitor [130], or vertical administration of downstream targeted in combination with upstream and middle targets, the clinical efficacy is superior to that of single administration and can achieve more lasting antitumor effect. Inhibitors simultaneously inhibiting multiple target sites, such as PI103 [131], PKI587 [132] and PKI179 [133], known as dual targeting inhibitors of mTOR and PI3K, are also being developed. Studies have also shown that targeted inhibition of the PI3K-AKT-mTOR pathway can prevent cell repair of DNA damage, thereby making patients more sensitive to radiotherapy and chemotherapy [134]. This finding has been used to treat head and neck malignancies [135, 136]. Besides, inhibition of PI3K-AKT-mTOR pathway in pancreatic cancer cells can inhibit cancer-associated fibroblasts (CAF), reduce interstitial fibrosis, and alter pancreatic cancer tumor microenvironment (TME), which will thereby increase the lethality of chemotherapy drugs [137]. Therefore, combined radiotherapy and immunotherapy, not only the combination of targeted drugs with different targets, but also that of targeted drugs with chemotherapy, are both the direction for future research [138]. However, the underlying mechanisms for the combination therapy remain unclear, and avoiding the accumulation of toxic effects of drug combination on normal cells is still a tough challenge in the future [139].
4. Summary and Outlook
In summary, the clinical value of KRAS gene in the treatment of PDAC is beginning to dawn. On the one hand, the role of KRAS mutations in the occurrence and development of PDAC has been basically clear, while on the other, the medicinal properties of KRAS gene have achieved a breakthrough in solid tumors such as lung cancer and colorectal cancer. Although the mutation rate of KRAS G12C is less than 5% in Chinese lung cancer population while that in PDAC is less than 1% [140], it still brings promising treatment efficacy to the patients, which seems to open a new pattern of clinical research on KRAS mutated tumors. In addition to targeted drugs, Sotorasib is found to alter TME in mice, promote T cell infiltration, and produce pro-inflammatory TME, thereby producing long-lasting anti-tumor effects alone or in combination with immune checkpoint inhibitors. Besides, it is widely acknowledged that the immunotherapy of PDAC progresses slowly. Studies have found that PDAC lacks CD8+T cell infiltration, and the high infiltration of myeloid suppressor cells and tumor-associated macrophages results in PDAC alone not being sensitive to anti-PD-L1 therapy. Most clinical trials have presented poor immunotherapy effect, and no obvious efficacy has been observed. Therefore, PDAC is recognized as a "cold tumor". In this case, whether KRAS-targeted drugs can also bring a breakthrough in immunotherapy for PDAC remains to be seen. Finally, it is also noteworthy that the drug resistance of targeted drugs in tumor therapy should not be ignored. In June 2021, the drug resistance mechanism of Adagrasib was published on the New England Journal, which involves secondary mutation or amplification of KRAS, bypass activation, other driver mutations, and histopathological transformation, etc. [141]. Studies have been conducted to explore these mechanisms and solutions concerning the acquired resistance to KRAS inhibitors [142].
It is still expected that with the technological progress and the continuous accumulation of clinical practice, an increasing number of biomarkers, signaling pathways and targets can be found for the individualized diagnosis and treatment of PDAC, and ultimately benefit the related patients.
Acknowledgements
This work was supported by grants from the Natural Science Foundation of Hunan Province (2022JJ40216), Key Projects of Hunan Provincial Department of Education (21A0026), the Natural Science Foundation of Changsha City (kq2202447), and Youth Program of Hunan Provincial Department of Education (21B0032).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7-33
3. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
4. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801-6
5. Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A. et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology. 2019;156:2242-53.e4
6. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828-51
7. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M. et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730-3.e9
8. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017; 32: 185-203.e13.
9. Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011;2:216-31
10. Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ. et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112:779-84
11. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170:17-33
12. Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218-49
13. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91-100
14. van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL. et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541-7
15. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-70
16. Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK. et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell. 2020;38:198-211.e8
17. Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T. et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554:62-8
18. Brychta N, Krahn T, von Ahsen O. Detection of KRAS Mutations in Circulating Tumor DNA by Digital PCR in Early Stages of Pancreatic Cancer. Clin Chem. 2016;62:1482-91
19. Haigis KM. KRAS Alleles: The Devil Is in the Detail. Trends Cancer. 2017;3:686-97
20. Kim MK, Woo SM, Park B, Yoon KA, Kim YH, Joo J. et al. Prognostic Implications of Multiplex Detection of KRAS Mutations in Cell-Free DNA from Patients with Pancreatic Ductal Adenocarcinoma. Clin Chem. 2018;64:726-34
21. Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153-68
22. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J. et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744
23. Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P. et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157
24. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR. et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228-39
25. Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P. et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci U S A. 2017;114:10202-7
26. Lee B, Lipton L, Cohen J, Tie J, Javed AA, Li L. et al. Circulating tumor DNA as a potential marker of adjuvant chemotherapy benefit following surgery for localized pancreatic cancer. Ann Oncol. 2019;30:1472-8
27. Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P. et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. Jama. 2016;315:1844-53
28. Han B, Kim BJ, Kim HS, Choi DR, Shim BY, Lee KH. et al. A phase II study of gemcitabine, erlotinib and S-1 in patients with advanced pancreatic cancer. J Cancer. 2021;12:912-7
29. Jacobsen HJ, Poulsen TT, Dahlman A, Kjær I, Koefoed K, Sen JW. et al. Pan-HER, an Antibody Mixture Simultaneously Targeting EGFR, HER2, and HER3, Effectively Overcomes Tumor Heterogeneity and Plasticity. Clin Cancer Res. 2015;21:4110-22
30. Qian Y, Gong Y, Fan Z, Luo G, Huang Q, Deng S. et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol. 2020;13:130
31. Leshchiner ES, Parkhitko A, Bird GH, Luccarelli J, Bellairs JA, Escudero S. et al. Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc Natl Acad Sci U S A. 2015;112:1761-6
32. Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7:585-7
33. Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM. et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci U S A. 2019;116:2551-60
34. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR. et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021;11:142-57
35. Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, Zhang ZY. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 2017;77:5701-5
36. Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20:453-9
37. Garcia Fortanet J, Chen CH, Chen YN, Chen Z, Deng Z, Firestone B. et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J Med Chem. 2016;59:7773-82
38. Bagdanoff JT, Chen Z, Acker M, Chen YN, Chan H, Dore M. et al. Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J Med Chem. 2019;62:1781-92
39. Lu H, Liu C, Velazquez R, Wang H, Dunkl LM, Kazic-Legueux M. et al. SHP2 Inhibition Overcomes RTK-Mediated Pathway Reactivation in KRAS-Mutant Tumors Treated with MEK Inhibitors. Mol Cancer Ther. 2019;18:1323-34
40. Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D. et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018;20:1064-73
41. Ijaz M, Wang F, Shahbaz M, Jiang W, Fathy AH, Nesa EU. The Role of Grb2 in Cancer and Peptides as Grb2 Antagonists. Protein Pept Lett. 2018;24:1084-95
42. Yin J, Cai Z, Zhang L, Zhang J, He X, Du X. et al. A recombined fusion protein PTD-Grb2-SH2 inhibits the proliferation of breast cancer cells in vitro. Int J Oncol. 2013;42:1061-9
43. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548-51
44. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI. et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383:1207-17
45. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129-38
46. Ariston Gabriel AN, Wang F, Jiao Q, Yvette U, Yang X, Al-Ameri SA. et al. The involvement of exosomes in the diagnosis and treatment of pancreatic cancer. Mol Cancer. 2020;19:132
47. Kaur S, Singh SP, Elkahloun AG, Wu W, Abu-Asab MS, Roberts DD. CD47-dependent immunomodulatory and angiogenic activities of extracellular vesicles produced by T cells. Matrix Biol. 2014;37:49-59
48. Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498-503
49. Chaft JE, Litvak A, Arcila ME, Patel P, D'Angelo SP, Krug LM. et al. Phase II study of the GI-4000 KRAS vaccine after curative therapy in patients with stage I-III lung adenocarcinoma harboring a KRAS G12C, G12D, or G12V mutation. Clin Lung Cancer. 2014;15:405-10
50. Schlick K, Kiem D, Greil R. Recent Advances in Pancreatic Cancer: Novel Prognostic Biomarkers and Targeted Therapy-A Review of the Literature. Biomolecules. 2021;11:1469
51. Wang JP, Wu CY, Yeh YC, Shyr YM, Wu YY, Kuo CY. et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open-label, prospective trial. Oncotarget. 2015;6:18162-73
52. Van Cutsem E, Hidalgo M, Canon JL, Macarulla T, Bazin I, Poddubskaya E. et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int J Cancer. 2018;143:2053-64
53. Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM. et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50:2072-81
54. Degirmenci U, Wang M, Hu J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells. 2020;9:198
55. Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S. et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041-6
56. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-65
57. Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y. et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016;370:332-44
58. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401-9
59. Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S. et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell. 2015;28:384-98
60. Okaniwa M, Hirose M, Arita T, Yabuki M, Nakamura A, Takagi T. et al. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J Med Chem. 2013;56:6478-94
61. Sun Y, Alberta JA, Pilarz C, Calligaris D, Chadwick EJ, Ramkissoon SH. et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19:774-85
62. Saturno G, Lopes F, Niculescu-Duvaz I, Niculescu-Duvaz D, Zambon A, Davies L. et al. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann Oncol. 2021;32:269-78
63. Roskoski R Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol Res. 2019;142:151-68
64. Markham A, Keam SJ. Selumetinib: First Approval. Drugs. 2020;80:931-7
65. Shirley M. Encorafenib and Binimetinib: First Global Approvals. Drugs. 2018;78:1277-84
66. Signorelli J, Shah Gandhi A. Cobimetinib. Ann Pharmacother. 2017;51:146-53
67. Zeiser R. Trametinib. Recent Results Cancer Res. 2014;201:241-8
68. Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer. 2009;125:2332-41
69. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E. et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250-6
70. Dai B, Meng J, Peyton M, Girard L, Bornmann WG, Ji L. et al. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011;71:3658-68
71. Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y. et al. Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov. 2016;6:754-69
72. Kozar I, Margue C, Rothengatter S, Haan C, Kreis S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim Biophys Acta Rev Cancer. 2019;1871:313-22
73. Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B. et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther. 2018;11:7095-107
74. Arozarena I, Wellbrock C. Overcoming resistance to BRAF inhibitors. Ann Transl Med. 2017;5:387
75. Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev. 2021;92:102137
76. Nagathihalli NS, Castellanos JA, Lamichhane P, Messaggio F, Shi C, Dai X. et al. Inverse Correlation of STAT3 and MEK Signaling Mediates Resistance to RAS Pathway Inhibition in Pancreatic Cancer. Cancer Res. 2018;78:6235-46
77. Lee JW, Zhang Y, Eoh KJ, Sharma R, Sanmamed MF, Wu J. et al. The Combination of MEK Inhibitor With Immunomodulatory Antibodies Targeting Programmed Death 1 and Programmed Death Ligand 1 Results in Prolonged Survival in Kras/p53-Driven Lung Cancer. J Thorac Oncol. 2019;14:1046-60
78. Buscà R, Christen R, Lovern M, Clifford AM, Yue JX, Goss GG. et al. ERK1 and ERK2 present functional redundancy in tetrapods despite higher evolution rate of ERK1. BMC Evol Biol. 2015;15:179
79. Blake JF, Burkard M, Chan J, Chen H, Chou KJ, Diaz D. et al. Discovery of (S)-1-(1-(4-Chloro-3-fluorophenyl)-2-hydroxyethyl)-4-(2-((1-methyl-1H-pyrazol-5-yl)amino)pyrimidin-4-yl)pyridin-2(1H)-one (GDC-0994), an Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J Med Chem. 2016;59:5650-60
80. Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ. et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther. 2017;16:2351-63
81. Aronchik I, Dai Y, Labenski M, Barnes C, Jones T, Qiao L. et al. Efficacy of a Covalent ERK1/2 Inhibitor, CC-90003, in KRAS-Mutant Cancer Models Reveals Novel Mechanisms of Response and Resistance. Mol Cancer Res. 2019;17:642-54
82. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4:64
83. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P. et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997-1007
84. Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M. et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood. 2018;131:888-98
85. Flinn IW, Hillmen P, Montillo M, Nagy Z, Illés Á, Etienne G. et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 2018;132:2446-55
86. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med. 2019;380:1929-40
87. Burris HA 3rd, Flinn IW, Patel MR, Fenske TS, Deng C, Brander DM. et al. Umbralisib, a novel PI3Kδ and casein kinase-1ε inhibitor, in relapsed or refractory chronic lymphocytic leukaemia and lymphoma: an open-label, phase 1, dose-escalation, first-in-human study. Lancet Oncol. 2018;19:486-96
88. O'Neil BH, Scott AJ, Ma WW, Cohen SJ, Aisner DL, Menter AR. et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann Oncol. 2015;26:1923-9
89. Banerji U, Dean EJ, Pérez-Fidalgo JA, Batist G, Bedard PL, You B. et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and in PIK3CA-Mutated Breast and Gynecologic Cancers. Clin Cancer Res. 2018;24:2050-9
90. Aghajanian C, Bell-McGuinn KM, Burris HA 3rd, Siu LL, Stayner LA, Wheler JJ. et al. A phase I, open-label, two-stage study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of the oral AKT inhibitor GSK2141795 in patients with solid tumors. Invest New Drugs. 2018;36:1016-25
91. Chen Y, Zhou X. Research progress of mTOR inhibitors. Eur J Med Chem. 2020;208:112820
92. Xu T, Sun D, Chen Y, Ouyang L. Targeting mTOR for fighting diseases: A revisited review of mTOR inhibitors. Eur J Med Chem. 2020;199:112391
93. Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457-67
94. Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421-34
95. Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G. et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal. 2014;7:ra42
96. Jiang Z, Zhou C, Cheng L, Yan B, Chen K, Chen X. et al. Inhibiting YAP expression suppresses pancreatic cancer progression by disrupting tumor-stromal interactions. J Exp Clin Cancer Res. 2018;37:69
97. Salcedo Allende MT, Zeron-Medina J, Hernandez J, Macarulla T, Balsells J, Merino X. et al. Overexpression of Yes Associated Protein 1, an Independent Prognostic Marker in Patients With Pancreatic Ductal Adenocarcinoma, Correlated With Liver Metastasis and Poor Prognosis. Pancreas. 2017;46:913-20
98. Yoo W, Lee J, Jun E, Noh KH, Lee S, Jung D. et al. The YAP1-NMU Axis Is Associated with Pancreatic Cancer Progression and Poor Outcome: Identification of a Novel Diagnostic Biomarker and Therapeutic Target. Cancers (Basel). 2019;11:1477
99. Drexler R, Küchler M, Wagner KC, Reese T, Feyerabend B, Kleine M. et al. The clinical relevance of the Hippo pathway in pancreatic ductal adenocarcinoma. J Cancer Res Clin Oncol. 2021;147:373-91
100. Kim MH, Kim J, Hong H, Lee SH, Lee JK, Jung E. et al. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. Embo j. 2016;35:462-78
101. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH. et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780-91
102. Zhao Y, Khanal P, Savage P, She YM, Cyr TD, Yang X. YAP-induced resistance of cancer cells to antitubulin drugs is modulated by a Hippo-independent pathway. Cancer Res. 2014;74:4493-503
103. Chen M, Wang M, Xu S, Guo X, Jiang J. Upregulation of miR-181c contributes to chemoresistance in pancreatic cancer by inactivating the Hippo signaling pathway. Oncotarget. 2015;6:44466-79
104. Janse van Rensburg HJ, Azad T, Ling M, Hao Y, Snetsinger B, Khanal P. et al. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018;78:1457-70
105. Hsu PC, Miao J, Wang YC, Zhang WQ, Yang YL, Wang CW. et al. Inhibition of yes-associated protein down-regulates PD-L1 (CD274) expression in human malignant pleural mesothelioma. J Cell Mol Med. 2018;22:3139-48
106. Kim MH, Kim CG, Kim SK, Shin SJ, Choe EA, Park SH. et al. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res. 2018;6:255-66
107. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA. et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300-5
108. Zhao X, Wang X, Fang L, Lan C, Zheng X, Wang Y. et al. A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett. 2017;402:61-70
109. Donohue E, Thomas A, Maurer N, Manisali I, Zeisser-Labouebe M, Zisman N. et al. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. J Cancer. 2013;4:585-96
110. Konstantinou EK, Notomi S, Kosmidou C, Brodowska K, Al-Moujahed A, Nicolaou F. et al. Verteporfin-induced formation of protein cross-linked oligomers and high molecular weight complexes is mediated by light and leads to cell toxicity. Sci Rep. 2017;7:46581
111. Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Győrffy B. et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8:ra98
112. Lu W, Wang J, Li Y, Tao H, Xiong H, Lian F. et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur J Med Chem. 2019;184:111767
113. He L, Zhang J, Zhao J, Ma N, Kim SW, Qiao S. et al. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv Nutr. 2018;9:493-504
114. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H. et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717-29
115. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S. et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429-33
116. Li S, Song Y, Quach C, Guo H, Jang GB, Maazi H. et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun. 2019;10:1693
117. Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP. et al. Author Correction: Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2020;26:982
118. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A. et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620-7
119. Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW. et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A. 2019;116:4508-17
120. Rabanal-Ruiz Y, Otten EG, Korolchuk VI. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017;61:565-84
121. Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep. 2013;3:3230
122. Lafaro KJ, Melstrom LG. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am J Pathol. 2019;189:44-57
123. Gu D, Schlotman KE, Xie J. Deciphering the role of hedgehog signaling in pancreatic cancer. J Biomed Res. 2016;30:353-60
124. Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS. et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell. 2016;165:910-20
125. Bahra M, Kamphues C, Boas-Knoop S, Lippert S, Esendik U, Schüller U. et al. Combination of hedgehog signaling blockage and chemotherapy leads to tumor reduction in pancreatic adenocarcinomas. Pancreas. 2012;41:222-9
126. Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H. et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725-35
127. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR. et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2015;33:4284-92
128. De Jesus-Acosta A, Sugar EA, O'Dwyer PJ, Ramanathan RK, Von Hoff DD, Rasheed Z. et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer. 2020;122:498-505
129. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol. 2020;17:527-40
130. Jiang H, Xu M, Li L, Grierson P, Dodhiawala P, Highkin M. et al. Concurrent HER or PI3K Inhibition Potentiates the Antitumor Effect of the ERK Inhibitor Ulixertinib in Preclinical Pancreatic Cancer Models. Mol Cancer Ther. 2018;17:2144-55
131. Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Yamano M. et al. Synthesis and biological evaluation of pyrido[3',2':4,5]furo[3,2-d]pyrimidine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem Lett. 2007;17:2438-42
132. Freitag H, Christen F, Lewens F, Grass I, Briest F, Iwaszkiewicz S. et al. Inhibition of mTOR's Catalytic Site by PKI-587 Is a Promising Therapeutic Option for Gastroenteropancreatic Neuroendocrine Tumor Disease. Neuroendocrinology. 2017;105:90-104
133. Venkatesan AM, Chen Z, dos Santos O, Dehnhardt C, Santos ED, Ayral-Kaloustian S. et al. PKI-179: an orally efficacious dual phosphatidylinositol-3-kinase (PI3K)/mammalian target of rapamycin (mTOR) inhibitor. Bioorg Med Chem Lett. 2010;20:5869-73
134. Wang Z, Huang Y, Zhang J. Molecularly targeting the PI3K-Akt-mTOR pathway can sensitize cancer cells to radiotherapy and chemotherapy. Cell Mol Biol Lett. 2014;19:233-42
135. Chuang FC, Wang CC, Chen JH, Hwang TZ, Yeh SA, Su YC. PI3k inhibitors (BKM120 and BYL719) as radiosensitizers for head and neck squamous cell carcinoma during radiotherapy. PLoS One. 2021;16:e0245715
136. Glorieux M, Dok R, Nuyts S. The influence of PI3K inhibition on the radiotherapy response of head and neck cancer cells. Sci Rep. 2020;10:16208
137. Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F. et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med. 2015;7:735-53
138. Mehra S, Deshpande N, Nagathihalli N. Targeting PI3K Pathway in Pancreatic Ductal Adenocarcinoma: Rationale and Progress. Cancers (Basel). 2021;13:4434
139. Liu L, Wang WQ, Lou WH. [Ten directions for improving clinical therapeutic effect of pancreatic cancer in the future]. Zhonghua Wai Ke Za Zhi. 2022;60:10-6
140. Loong HH, Du N, Cheng C, Lin H, Guo J, Lin G. et al. KRAS G12C mutations in Asia: a landscape analysis of 11,951 Chinese tumor samples. Transl Lung Cancer Res. 2020;9:1759-69
141. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW. et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med. 2021;384:2382-93
142. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A. et al. EGFR Blockade Reverts Resistance to KRAS(G12C) Inhibition in Colorectal Cancer. Cancer Discov. 2020;10:1129-39
Author contact
![]() Corresponding authors: Wei Cheng, MD, Department of Hepatobiliary Surgery, Hunan Provincial People's Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, 410005 Hunan Province, China. Xiangyue Hospital Affiliated to Hunan Institute of Parasitic Diseases, National Clinical Center for Schistosomiasis Treatment, Yueyang 414000, Hunan Province, China. Translational Medicine Laboratory of Pancreas Disease of Hunan Normal University, Changsha 410005, China. Email: chengweiedu.cn. Kang Chen, Ph.D., Department of Hepatobiliary Surgery, Hunan Provincial People's Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, 410005 Hunan Province, China. Translational Medicine Laboratory of Pancreas Disease of Hunan Normal University, Changsha 410005, China. Email: chenkang1029com.
Corresponding authors: Wei Cheng, MD, Department of Hepatobiliary Surgery, Hunan Provincial People's Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, 410005 Hunan Province, China. Xiangyue Hospital Affiliated to Hunan Institute of Parasitic Diseases, National Clinical Center for Schistosomiasis Treatment, Yueyang 414000, Hunan Province, China. Translational Medicine Laboratory of Pancreas Disease of Hunan Normal University, Changsha 410005, China. Email: chengweiedu.cn. Kang Chen, Ph.D., Department of Hepatobiliary Surgery, Hunan Provincial People's Hospital, The First Affiliated Hospital of Hunan Normal University, Changsha, 410005 Hunan Province, China. Translational Medicine Laboratory of Pancreas Disease of Hunan Normal University, Changsha 410005, China. Email: chenkang1029com.