Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2024; 15(11):3272-3283. doi:10.7150/jca.94669 This issue Cite
Review
Neutrophil Extracellular Traps in Breast Cancer: Roles in Metastasis and Beyond
Xi Xu, Xinyu Wang, Zhuomeng Zheng, Yuxuan Guo, Guangchun He, Yian Wang, Shujun Fu, Chanjuan Zheng ![]() , Xiyun Deng
, Xiyun Deng ![]()
Key Laboratory of Translational Cancer Stem Cell Research, Department of Pathophysiology, Hunan Normal University School of Medicine, Changsha, Hunan, China.
Received 2024-1-25; Accepted 2024-4-12; Published 2024-4-23
Abstract

Despite advances in the treatment of breast cancer, the disease continues to exhibit high global morbidity and mortality. The importance of neutrophils in cancer development has been increasingly recognized. Neutrophil extracellular traps (NETs) are web-like structures released into the extracellular space by activated neutrophils, serving as a potential antimicrobial mechanism for capturing and eliminating microorganisms. The roles played by NETs in cancer development have been a subject of intense research in the last decade. In breast cancer, current evidence suggests that NETs are involved in various stages of cancer development, particularly during metastasis. In this review, we try to provide an updated overview of the roles played by NETs in breast cancer metastasis. These include: 1) facilitating systemic dissemination of cancer cells; 2) promoting cancer-associated inflammation; 3) facilitating cancer-associated thrombosis; 4) facilitating pre-metastatic niche formation; and 5) awakening dormant cancer cells. The translational implications of NETs in breast cancer treatment are also discussed. Understanding the relationship between NETs and breast cancer metastasis is expected to provide important insights for developing new therapeutic strategies for breast cancer patients.
Keywords: Neutrophil extracellular trap (NET), Breast cancer, Tumor immunity, Therapeutic target, Tumor microenvironment
1. Introduction
Breast cancer has become the most common malignancy worldwide, with the highest incidence and mortality rates among women globally, and metastasis being the primary cause of most breast cancer-related deaths [1]. At each stage of this complex process, malignant cells must also confront and resist attacks from the host immune system. Despite advances in targeted therapy and immunotherapy, breast cancer, particularly triple-negative breast cancer (TNBC), still exhibits high invasiveness, metastasis, and recurrence [2]. Therefore, the search for novel therapeutic strategies in breast cancer is of paramount importance.
Neutrophils are the most abundant white blood cells in the bloodstream, comprising up to 70% of circulating white blood cells in humans [3, 4]. They serve as the frontline warriors against invading microorganisms and are a major component of the body's innate immune system [5]. Tumor-associated neutrophils (TANs) have been increasingly recognized as significant contributors to tumor biology. Neutrophils paradoxically play a tumor-suppressive or tumor-promoting role in cancer immunobiology, depending on the states of neutrophils and the specific context involved [6]. The importance of neutrophils in diverse types of cancer including breast cancer is further illustrated by a recent study indicating that neutrophils exhibit diverse cancer-related functions including inflammation, angiogenesis, and antigen presentation [7].
In 2004, Brinkmann et al. observed a unique form of neutrophil degranulation, termed neutrophil extracellular trap (NET), as a novel mechanism for neutrophils to fight against microorganisms [8]. In 2012, Demers et al. first reported the role of NETs in cancer showing that hematological as well as solid tumors induce an increase in peripheral blood neutrophils that are associated with NET formation [9]. Since then, it has been widely accepted that NETs play a crucial role in the development of various types of malignancies [10]. The roles of NETs in breast cancer have been extensively studied in the last decade, which have become a subject of several nice reviews [5, 11-13]. In this review, we summarize recent advances regarding the roles of NETs in breast cancer with a special focus on metastasis-related events.
2. Pathways of NET Formation
NETs are composed of DNA, histones, and granule proteins such as neutrophil elastase (NE), myeloperoxidase (MPO), cathepsin G, and other enzymatically active proteins. Current evidence suggests that two primary pathways are involved in the formation of NETs. The first pathway, termed suicidal or lytic NETosis (Figure 1a), occurs when stimulated by triggers such as phorbol 12-myristate 13-acetate (PMA) and extracellular microbes [6, 14-16]. This pathway involves the stimulation of protein kinase C (PKC) and Raf-MEK-ERK signaling, activating NADPH oxidase (NOX) and the production of reactive oxygen species (ROS) in neutrophils [17-19]. Upon ROS stimulation, NE escapes from the granules and translocates to the nucleus, where NE cleaves histones and promotes chromatin decondensation. Subsequently, MPO binds to chromatin and collaborates with NE to induce plasma membrane rupture and NET formation [20].
Pathways of NET formation. (a) Different stimuli such as PMA, antibodies (such as autoantibodies), or cholesterol crystals can induce suicidal NETosis, which occurs a few hours after stimulation. Following the activation of NOX, ROS are generated and PAD4 is activated, followed by chromatin decondensation. Subsequently, NE and MPO are translocated from granules to the nucleus, promoting further chromatin unfolding and subsequent disruption of the nuclear membrane. In addition, NE activates GSDMD, forming pores on granules and plasma membranes, resulting in membrane rupture and cell death. (b) Vital NETosis can be induced by Staphylococcus aureus within minutes through complement receptors and TLR2 ligands. Alternatively, vital NETosis can be induced via TLR4 activation by Escherichia coli. PLTs indirectly activated through TLR4 also contribute to this process. In this pathway, NOX activity is not required. PAD4 is activated, inducing chromatin decondensation. Nuclei containing DNA are extruded without disrupting the plasma membrane, and decondensed chromatin is transported via vesicles for the expulsion of nuclear DNA. The intact cell membrane of neutrophils allows for the survival of non-nucleated neutrophils while retaining physiological functions such as phagocytosis. DAMPs that amplify ongoing immune reactions are also involved in inducing vital NETosis. PMA, phorbol 12-myristate 13-acetate; NOX, NADPH oxidase; ROS, reactive oxygen species; PAD4, peptidylarginine deiminase 4; NE, neutrophil elastase; MPO, myeloperoxidase; GSDMD, Gasdermin D; TLR2, Toll-like receptor 2; PLTs, platelets; TLR4, Toll-like receptor 4; DAMPs, damage-associated molecular patterns.
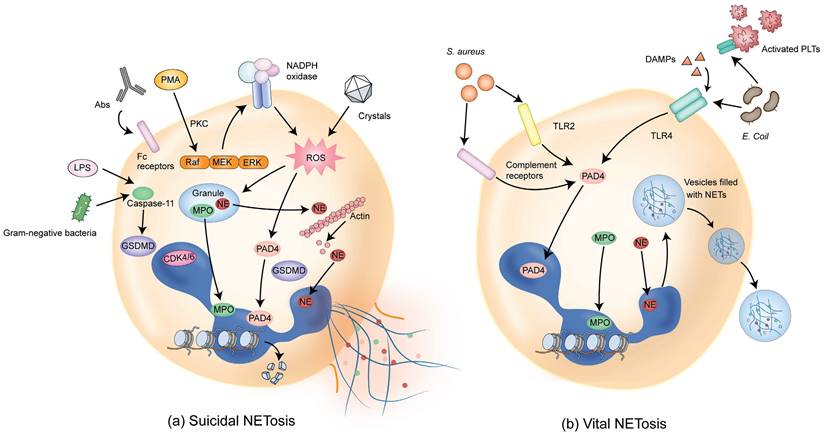
Furthermore, the activation of peptidylarginine deiminase 4 (PAD4), possibly together with the cell cycle proteins such as CDK4/6, promotes histone cleavage and chromatin decondensation through citrullination [21-23] and initiates nuclear envelope rupture and subsequently membrane rupture [24, 25]. Cytoplasmic NE also activates Gasdermin D (GSDMD), which is known to be able to form pores in the granule membrane and the plasma membrane during pyroptosis [26], promoting NE release into the cytoplasm and assisting in NET expulsion [27]. With the rupture of the cell membrane, the chromatin decorated with granule proteins is released into the extracellular space, forming NETs.
The second type of NETosis, termed vital or non-lytic NETosis (Figure 1b), involves the expulsion of nuclear chromatin accompanied by the release of granule proteins through degranulation. This type of cell death-independent NETosis occurs more rapidly, typically within minutes, and can be stimulated by triggers such as activated platelets, microorganisms including Staphylococcus aureus, and complement proteins [28-30]. In this pathway, NOX activity is not required and DNA-containing nuclear vesicles are extruded without disrupting the plasma membrane, thus preserving neutrophil functionality [6, 31].
While both pathways result in the formation of web-like structures that trap microorganisms or non-infectious pathogens, several differences exist in NET formation between suicidal and vital NETosis [32]. First, the nature of the stimulation is different between these two pathways. Suicidal NETosis is mostly induced by PMA, whereas vital NETosis is stimulated by microbial-specific molecular patterns and recognized by host pattern recognition receptors, such as toll-like receptors (TLRs). Second, different signaling pathways are involved in the two pathways. Suicidal NETosis requires activation of the Raf-MEK-ERK pathway and NOX-dependent production of ROS. In contrast, vital NETosis is ROS-independent and requires vesicular trafficking of DNA from within the nucleus to the extracellular space. Third, plasma membrane rupture is required and occurs in suicidal but not vital NETosis. Fourth, there exists difference in the time duration required for the formation of NETs. Vital NETosis executes more rapidly compared with suicidal NETosis (< 1 hour vs 2 - 4 hours). Fifth, the outcomes are different between suicidal and vital NETosis. Neutrophils undergoing suicidal NETosis could no longer be recruited and, therefore, cannot be used for sustainable host immune defense. On the contrary, vital NETosis sustains neutrophil functionality related to innate immunity in terms of detecting, capturing, and restraining target microorganisms [32].
3. Roles of NETs in Breast Cancer Metastasis
Metastasis is a multi-step process in which cancer cells spread from one site to another in the body, often leading to incurability of the disease and adverse outcomes. Levels of circulating NETs are higher in patients with metastatic disease compared with those with localized breast cancer [33]. Studies show that NETs play important roles in each step of cancer metastasis. The first connection between NETs and cancer metastasis came from animal studies showing that NETs promote cancer liver metastasis [34, 35]. NETs cause alterations in the basement membrane and the ECM and, thus, can enhance the invasive capacity of breast cancer cells [36]. In this section, we will delve into the functions and effects of NETs in the various aspects of breast cancer metastasis (Figure 2).
Roles of NETs in breast cancer metastasis. In breast cancer, NETs play different functions in various aspects of cancer metastasis. See the main text for detailed description of each of the functions played by NETs in breast cancer metastasis.
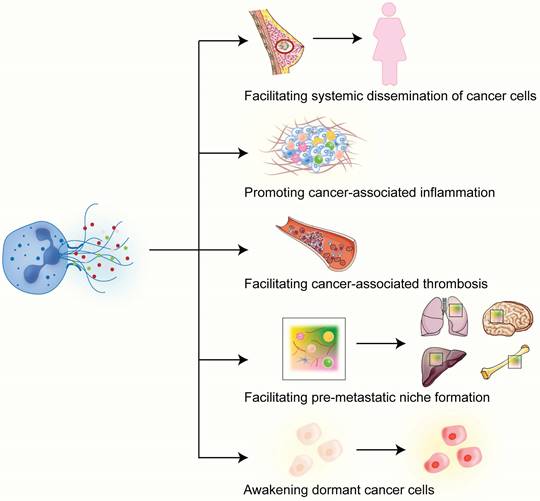
3.1 Facilitating systemic dissemination of cancer cells
Tumor cells possess the ability to spread to different parts of the body through the bloodstream or the lymphatic system, maintaining their vitality during this journey. They can survive in the circulation and later extravasate [5]. It has been suggested that NETs play a critical role in the hematogenous spread of tumors. NETs can induce morphological changes in endothelial cells, leading to increased vascular permeability, which facilitates cancer cell extravasation and promotes breast-to-lung metastasis [37]. Kolaczkowska and colleagues found that the attachment of circulating NETs to blood components increases the efficiency of cancer cell extravasation, allowing cancer cells to cross the endothelial barrier [38]. Furthermore, NETs may impact the metastatic process through interactions with circulating tumor cells (CTCs). It has been shown that CTCs may become enveloped by the DNA fibers of NETs, leading to their lodging in the liver, thereby forming micro-metastatic foci in a short period [34]. A further study has shown that during this process, NETs interact with integrin β1 on the surface of tumor cells, promoting the formation of metastatic foci and the spread of cancer cells [39]. The interaction between CTCs and TANs results in endothelial cell contraction, increased permeability, and malignant cell extravasation [40].
3.1.1 NETs promote cancer dissemination through epithelial-to-mesenchymal transition (EMT)
Epithelial-to-mesenchymal transition (EMT) is a process through which epithelial cells acquire mesenchymal properties, endowing cancer cells with invasive and metastatic potential [41]. Martins-Cardoso et al. found that NET-treated breast cancer cells have increased levels of N-cadherin and fibronectin together with a decreased level of E-cadherin in addition to the morphological changes typical of EMT [42]. Since EMT is associated with the cancer stem cell properties, they further found that in addition to promoting EMT, NETs also induce the acquisition of the CD44high/CD24low phenotype in MCF7 breast cancer cells, suggestive of enhanced stem cell properties [43].
3.1.2 NET-DNA receptor CCDC25 promotes tumor cell dissemination
The DNA component of NETs (NET-DNA) can act as a chemotactic factor, attracting cancer cells disseminated from the primary site. Yang and colleagues discovered a transmembrane protein called Coiled-Coil Domain-Containing Protein 25 (CCDC25), which serves as a receptor for NET-DNA on cancer cells. CCDC25 is overexpressed in breast cancer and can sense NET-DNA to promote breast cancer liver metastasis [44]. In addition to functioning as a receptor for NET-DNA, CCDC25 also activates the ILK-β-PARVIN pathway, attracting cancer cells and enhancing their motility. Furthermore, knockout of the CCDC25 gene in cells has been shown to eliminate NET-mediated breast cancer metastasis [44].
Cholesterol is generated through the mevalonate (MVA) pathway via a series of enzymatic steps. In this process, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) is the rate-limiting enzyme. Cholesterol is generally considered as a risk factor for breast cancer and cholesterol biosynthesis is related to stem cell characteristics in breast cancer [45, 46]. One study found that cholesterol biosynthesis is closely associated with NET formation and breast cancer metastasis [47]. Furthermore, the expression of CCDC25 in breast cancer tissues is positively correlated with levels of HMGCR and citrullinated histone H3 (H3cit), and high expression of CCDC25 and HMGCR is associated with adverse prognosis in breast cancer patients. Cholesterol biosynthesis promotes the expression of CCDC25 and the formation of NETs in a lipid raft-dependent manner [47]. Following treatment with cholesterol biosynthesis inhibitors or DNase I, a significant reduction in CCDC25 expression was observed [47]. Additionally, the tumor suppressor gene apoptosis-stimulating of p53 protein 2 (ASPP2) interferes with cholesterol biosynthesis and indirectly affects the expression of CCDC25 and NET formation, weakening the stem cell characteristics, EMT, and invasive capabilities of breast cancer cells. Downregulation of ASPP2 has been shown to promote the transformation of epithelial cells into mesenchymal cells and enhance the invasive abilities of breast cancer cells [48].
3.1.3 Cathepsin C promotes breast cancer lung metastasis through NET formation
Xiao and colleagues found that elevated expression of cathepsin C (also known as dipeptidyl peptidase 1, DDP1), a lysosomal cysteine protease essential for the catalytic activation of several serine proteases, is associated with lung metastatic capabilities in multiple breast cancer cell lines [49, 50]. Clinically, high cathepsin C expression is correlated with lung metastasis and decreased overall survival in breast cancer patients [51, 52]. The expression and secretion of cathepsin C are related to NET formation and lung metastasis in human breast tumors. Cathepsin C activates the serine protease PR3 on the surface of neutrophils, leading to the processing of IL-1β and the activation of nuclear factor NF-κB in neutrophils. These changes result in the upregulation of IL-6 and CCL3, promoting neutrophil recruitment and triggering an inflammatory cascade and NET formation [51]. Simultaneously, cancer cells support the growth of lung metastases through the NET-mediated degradation of TSP-1. Importantly, the second-generation inhibitor of cathepsin C, AZD7986, can effectively disrupt NETs and prevent lung metastasis of breast cancer [51, 53].
3.1.4 NETs play a crucial role in angiogenesis
In addition to the above-mentioned roles played by NETs in breast cancer cells, NETs also play a role in angiogenesis [54]. Under stimulation by angiopoietin 1/2, the number of NETs produced by neutrophils is approximately 2.5 times higher than in their natural state, further promoting angiogenesis [55]. NETs can damage vascular endothelial cells, activate inflammatory responses, and promote angiogenesis. By evaluating the activity of tumor-related biological pathways, it has been found that NET formation is synchronized with angiogenesis [56, 57]. NETs also enhance endothelial cell proliferation and further contribute to the formation of tubular structures [58]. It has been demonstrated that histones within NETs promote endothelial cell proliferation and new blood vessel formation. The application of histone-binding agents prevents histone-induced angiogenesis [59].
3.2 Promoting cancer-associated inflammation
While neutrophils release NETs to capture pathogens, persistent infections, and inflammatory environments can lead to excessive NET production, thereby promoting the development of various inflammatory diseases. This is primarily mediated through the activation of inflammasomes [19, 60]. Similarly, studies have found interactions between inflammasomes and NETs in cancer. It has been demonstrated that NET-associated serine proteases, such as NE, can act as alternative enzymes for processing IL-1β and IL-18 associated with inflammasomes. This subsequently leads to the inactivation of progranulin (PGRN) and the activation of MMP9 in cancer [61, 62]. Lu et al. also pointed out the relevance of NETs to the feedforward loop of inflammation, which includes the recruitment of inflammatory cells, activation of signaling pathways, and the production of inflammatory factors [63]. Inflammation within the tumor microenvironment is crucial for breast cancer and its metastatic development [64-67].
In the liver metastasis of breast cancer, neutrophils release NETs through interactions with platelets and the NF-κB signaling pathway [11, 68, 69]. NETs serve as chemoattractants to draw CTCs, thereby promoting liver metastasis. Furthermore, the release of NETs into the circulation can cause endothelial cell damage and platelet activation, stimulating other neutrophils to release NETs [70, 71]. Therefore, the occurrence of the inflammatory feedback loop in the liver, driven by NETs, promotes breast cancer liver metastasis. Lu and colleagues utilized a doxorubicin-loaded liposome-encapsulated low-molecular-weight heparin-astaxanthin nanoparticle (LA/DOX NP) to inhibit NET formation, thereby blocking the tumor's inflammatory and immune-suppressive microenvironment and, as a result, inhibiting breast cancer liver metastasis [63]. These interventions in the inflammatory feedback loop hold promise as strategies to restrain breast cancer metastasis.
3.3 Facilitating cancer-associated thrombosis
Cancer is often associated with a hypercoagulable state and increased thrombosis is one of the most common comorbidities associated with cancer. Indeed, cancer-associated thrombosis (CAT) is the second leading cause of death for cancer patients, second only to the cancer itself [72]. It has been shown that chromatin released into the bloodstream through NETs can promote coagulation and thrombus formation [9, 73]. Thrombosis leads to poor prognosis in cancer patients and cancer-induced fatalities [9]. Mounting evidence suggests that neutrophils and NETs play a significant role in thrombus formation, particularly in CAT, as neutrophils are often reshaped with an increased count, making them more prone to generating NETs in cancer [9, 73-76]. It is estimated that cancer patients face a 4-to-7 times higher risk of developing venous thrombosis compared with non-cancer patients [77, 78]. Cancer patients who experience venous thrombosis typically have a poorer prognosis [79, 80]. Cancer therapies such as surgery, chemotherapy, and angiogenesis inhibitors can further exacerbate CAT [81].
There is significant correlation between elevated NET markers and the hypercoagulable state [5, 59]. It has become well-acknowledged that cancer patients exhibit higher levels of NET biomarkers compared with non-cancer patients [82] and cancer-associated NETs promote platelet capturing and increase tissue factor (TF) activity, thereby leading to CAT [74-76, 83]. Contrarily, activated platelets can also stimulate the formation of NETs, establishing a positive feedback loop between NETs and CAT [84].
In this context, the inhibition of NETs by drugs or gene manipulation (PAD-/- mice) can reduce thrombosis formation in mice [85-87]. Using recombinant human DNase I (rhDNase I) to degrade NETs can prevent thrombosis formation in a mouse model of tumorigenesis. It should be noted that while short-term treatment prevented venous thrombosis formation, long-term treatment reduced survival rates. These results suggest the potential therapeutic efficacy of rhDNase I in treating CAT, but its long-term use needs to be carefully assessed [88]. Another study provided initial evidence that low-molecular-weight heparin blocks the adhesion of activated platelets to neutrophils via the P-selectin pathway, thereby inhibiting the generation of NETs [63]. Gomes and colleagues also demonstrated that in NET-dependent breast cancer models, inhibiting the production and secretion of IL-1β related to inflammasomes can alleviate CAT [85]. These findings suggest that targeting NETs may be a potential and promising approach to reduce thrombosis, limiting tumor progression and metastasis.
3.4 Facilitating pre-metastatic niche formation
The pre-metastatic niche (PMN) in cancer typically forms after tumor cells leave the primary site and enter the circulatory system but have not yet reached the metastatic site. This suggests that the PMN is a microenvironment with characteristics of inflammation, immune suppression, and vascular leakage that favor the initiation of metastasis before tumor cells arrive at specific distant organs [89-92]. NETs are believed to play a crucial role in the PMN, helping to prepare the "soil" for the "seeds" of metastasis, sequestering CTCs, and promoting metastasis [34, 93]. Within the PMN, fibronectin, along with glycoproteins and proteoglycans like tenascin C, osteopontin, and versican, predominantly contributes to the alterations within the ECM [94]. NETs play a role in this process, influencing the electrostatic charge and conformation of fibronectin and collagen during citrullination. This effect is mediated by the PAD4 enzyme derived from NETs during the formation of the PMN [10, 95]. Furthermore, NETs equipped with proteases are highly correlated with the growth and invasiveness of aggressive tumors. However, this high metastatic potential can be eliminated by DNase I treatment [5, 68, 96].
Recently, there have been studies highlighting the role of NETs in promoting the formation of PMN in breast cancer. Zhou et al. found that the loss of the β3 adrenergic receptor (ADRB3) gene affects the PMN of malignant tumor cells, while ADRB3-induced NETs can persist for an extended period and contribute to the generation of PMN, thereby protecting disseminated tumor cells (DTC) from rejection by cytotoxic T lymphocytes [97]. In the pre-metastatic stage, the rate-limiting enzyme 2-hydroxyacid oxidase 1 (HAO1) involved in oxalate synthesis is upregulated in the lung alveolar epithelial cells, leading to the accumulation of oxalate in lung tissues. This accumulation activates NOX, inducing neutrophils to produce NETs, thereby promoting the formation of the PMN [98]. Furthermore, it has also been shown that resident mesenchymal stem cells (MSCs) play a crucial role in the formation of the PMN in the lungs when breast cancer cells migrate to this organ. MSCs have the ability to recruit neutrophils to the lungs and, with their assistance, generate complement C3 to transform these neutrophils into NETs. A further study has revealed that the upregulation of C3 in the lung's PMN microenvironment during tumor development is driven by Th2 cytokines via the STAT6 signaling pathway. Therefore, there is potential to reduce the likelihood of breast cancer metastasizing to the lungs by intervening in the Th2-STAT6-C3-NET cascade [99].
Another study related to breast cancer metastasis focuses on a subset of neutrophils known as tumor-associated aged neutrophils (Naged, CXCR4+ CD62Llow), which play a critical role in the formation of the pre-metastatic microenvironment. The study used flow cytometry and immunohistochemistry in 206 patients and multiple mouse models to investigate the distribution of Naged. Naged promotes the generation of NETs by activating the histone deacetylase SIRT1 through the tumor-secreted nicotinamide phosphoribosyltransferase (NAMPT), thereby driving tumor metastasis. By intervening in aged neutrophils and their formation pathways, it is possible to effectively reduce breast cancer lung metastasis [100]. More recently, using a mouse model of 4T1 breast cancer metastasis, Pan et al. demonstrated that chronic stress induces a pre-metastatic niche in the lungs to capture and arrest metastasizing breast cancer cells through the production of acetylcholine (Ach) by pulmonary epithelial cells. Called the CXCL2-Ach-NETosis pathway, this pathway mediates the formation of the lung pre-metastatic niche involves the chemokine CXCL2 that recruits neutrophils into the lungs followed by the production of Ach from pulmonary epithelial cells that enhances NETosis [101]. These findings highlight the significant role of NETs in breast cancer metastasis and provide a new direction for the development of targeted therapeutic strategies for this process in the future.
3.5 Awakening dormant cancer cells
Cancer cells that have spread from the primary tumor site to other tissues frequently exist in a dormant state and remain relatively silent in those tissues for extended periods. This phenomenon of tumor cell dormancy is common in most solid tumors including breast cancer [102]. These dormant cancer cells are often undetectable in clinical settings until they are awakened. Once awakened, these cancer cells can form metastasis and lead to tumor recurrence. It has been demonstrated that NETs have the potential to awaken dormant breast cancer cells, contributing to tumor recurrence and metastatic spread [103]. For example, long-term exposure to tobacco smoke or intranasal injection of lipopolysaccharide (LPS), leading to sustained inflammatory conditions, can result in the reinitiation of proliferation and the transformation of dormant breast cancer cells into invasive lung metastases. Even after remaining dormant in the lungs for a month, tumor cells can be awakened by LPS [103].
NETs induce the awakening of dormant cancer cells predominantly via remodeling the extracellular matrix (ECM). It has been demonstrated that proteases within NETs, such as NE and matrix metalloproteinase 9 (MMP9), can cleave and reshape laminin, exposing a binding site that acts as an activation site for integrin α3β1, leading to the activation of FAK/ERK/MLCK/YAP signaling in cancer cells and their reawakening, promoting proliferation. In in vitro experiments, laminins-111, -211, -411, and -511 were identified as critical ECM proteins required for NET-induced awakening of dormant breast cancer cells [103, 104]. In addition, thrombospondin-1 (TSP-1), a large glycoprotein present in the basement membrane surrounding mature blood vessels, has also been shown to regulate cancer cell dormancy and metastasis [104-106]. Degradation of TSP-1 by NE and MMP9 within NETs [103] helps eliminate the inhibitory effect of TSP-1 on the reawakening of cancer cells, thus assisting in the reawakening of cancer cells. Therefore, both the remodeling of laminins and the degradation of TSP-1 are essential for awakening dormant cancer cells [5, 103].
The cells in proximity to the remodeled laminin typically exhibit a proliferative state, while those near intact laminin remain in a dormant state. The use of specific antibodies to inhibit the new laminin epitopes can impede the reawakening of cancer cells both in vitro and in vivo [103]. Additionally, inhibiting the formation of NETs, such as through the use of PAD4 inhibitors or DNase therapy, can prevent the activation and formation of dormant cancer cells. Moreover, in vitro experiments have demonstrated that blocking NE and MMP9 can prevent cancer cells from re-entering the cell cycle and inhibit the progression of LPS-induced cancer in vivo [103]. The reawakening of dormant cancer cells is associated with the contribution of NETs to creating a favorable local microenvironment (“soil”) for the colonization of breast tumor cells (“seeds”) [107].
4. Translational Implications of NETs in Breast Cancer
4.1 NET-related gene signatures as biomarkers for prognosis prediction
Given the important roles of NETs in breast cancer development and progression particularly in the metastatic process, predicting therapeutic responses and patient outcomes using an NET-related gene signature might aid in identification of patients at risk and selection of effective therapies. Zhao et al. have explored this [108]. Through analyzing the bulk and single-cell RNA sequencing data from public databases, Huang et al. constructed a risk index with the NET-related pivotal genes to stratify triple-negative breast cancer patients into high- and low-risk groups. They found that patients of the low-risk group are enriched in Wnt signaling and were more sensitive to Wnt signaling pathway inhibitors. Furthermore, they found that BT549 cells that have a low risk index exhibit higher sensitivity to XVA-939 (a typical Wnt signaling inhibitor) [109]. These findings open up new avenue to patient stratification and personalized therapy based on the NET-related gene signature.
4.2 NET components as therapeutic targets
It has been demonstrated that NET formation induced by chemotherapy reduces therapy response in mouse models of breast cancer lung metastasis [110]. Therefore, targeting NETs to alleviate therapy resistance through various approaches has become an emerging field of great interest and is expected to be a promising strategy in breast cancer therapy (Figure 3).
Strategies for targeting the NETs. Currently evaluated or potentially available strategies for targeting the NETs to intervene breast cancer metastasis include: 1) inhibition of TLRs; 2) inhibition of CXCR1/2; 3) scavenging of ROS; 4) inhibition of PAD4; 5) blocking of GSDMD; 6) DNase therapy; 7) inhibition of HIF-1α; and 8) inhibition of MPO/NE. TLR, Toll-like receptor; CXCR, CXC chemokine receptor; ROS, reactive oxygen species; PAD4, peptidylarginine deiminase 4; GSDMD, Gasdermin D.
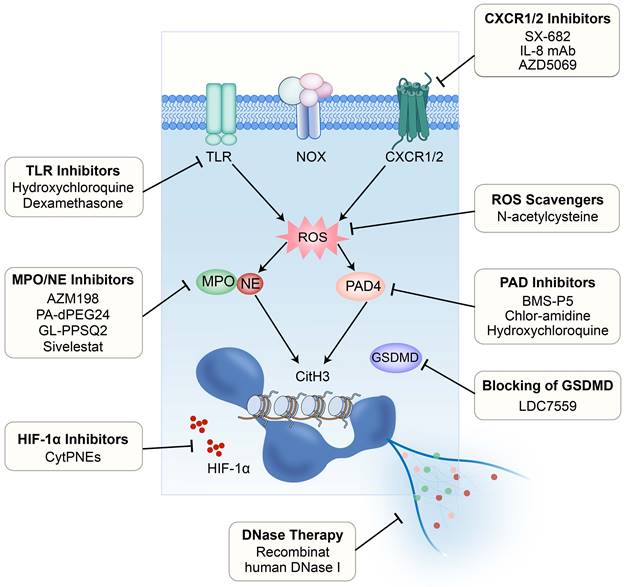
4.2.1 Targeting the DNA component or its membrane receptor
DNase has the capability to disrupt the structure of the NET by digesting its DNA component, making NETs a potential target for DNase therapy [111]. Based on this, DNase has been tested in experimental cancer models. For example, DNase treatment has been shown to disrupt CTC-NET interactions [39] and alleviate the disease burden [95] in experimental models of breast cancer. Park and colleagues further found that using nanoparticles encapsulating DNase I can inhibit the formation of NETs or degrade NETs, significantly reducing lung metastasis in a mouse model of breast cancer [68]. It should be noted that excessive inhibition of NETs may weaken the natural defense mechanisms of the immune system and increase the risk of bacterial infections [112]. Therefore, Chen and his team have designed a nanoplatform that can release DNase I via photoactivation in a controllable way. This can aid in NET degradation while retaining the interaction between tumor cells and other immune cells. Moreover, the application of this nanoplatform in the liver can prevent the capture of CTCs by NETs, thereby inhibiting metastasis [113].
Another potential intervention strategy may be based on targeting the transmembrane NET-DNA receptor CCDC25, thus reducing cancer invasiveness [44]. However, it is important to note that CCDC25 has low tissue specificity, and systemic inhibition of this receptor may have potential side effects, necessitating careful preclinical research. A potential strategy using oncolytic bacterium-mediated delivery of shCCDC25 in vivo may help reduce side effects while sustaining long-term anti-metastasis efficacy, which has shown promising results in a mouse orthotopic model of breast cancer lung metastasis [114].
4.2.2 Targeting the chemokine-receptor interaction
Neutralization of IL-8, a prominent factor triggering the formation of NETs, through monoclonal antibodies or inhibition of its receptors CXCR1/2 using pharmacological inhibitors such as SX-682, has been demonstrated to suppress the release of inflammatory mediators and the formation of NETs [115-117]. The use of AZD5069, a small molecule antagonist of CXCR2, has been shown to inhibit the infiltration of neutrophils into brain metastatic foci of breast cancer and may reduce the potential of neutrophils to promote the generation of tumor-associated NETs [118].
4.2.3 Targeting the hypoxic microenvironment
It is well known that tumor development and metastasis are associated with hypoxia, with a concomitant increase in expression and activity of hypoxia-inducible factor-1α (HIF-1α) [119]. Simultaneously, ROS generated during NET formation can inhibit the degradation of HIF-1α in tumor cells, leading to HIF-1α accumulation [6, 120]. Indeed, accumulation of HIF-1α has been observed in the CTCs of breast cancer patients [121]. Researchers have proposed a new strategy to deliver BAY 87-2243, an HIF-1α inhibitor, to cells through the chemotactic action of neutrophils on CTC-NET clusters. By incorporation into a nanoparticle known as cyto-pharmaceuticals (CytPNEs), BAY 87-2243 can be accurately delivered to circulating 4T1 tumor cells, thus suppressing the formation of metastatic foci and extending the survival of mice bearing breast cancer lung metastasis [122].
4.2.4 Targeting the pyroptosis-related pathway
Targeting the pore-forming protein GSDMD, which promotes translocation of NE to the nucleus during suicidal NETosis, has been examined. Through chemical screening, LDC7559 was discovered to specifically bind to GSDMD, reducing the activation of pathological inflammasomes and NETosis [26]. Targeting receptors such as the G-CSF receptor (G-CSFR) on tumor cells can also affect the formation of NETs. Wang et al. found that anti-G-CSFR monoclonal antibodies possess the ability to inhibit NET release without compromising immune responses against pathogens [123].
5. Concluding Remarks and Perspectives
An increasing body of evidence suggests that NETs play a crucial role in breast cancer development, particularly in metastasis. NETs not only facilitate the migration and invasion of breast tumor cells but also promote their survival and growth in the bloodstream as well as in the microenvironment. Understanding the role of NETs in breast cancer development is of great clinical importance. Various treatment strategies targeting NETs are being actively explored, aimed at enhancing immune cell responses and effectively inhibiting the growth and metastasis of breast cancer. However, further research is needed to gain a deeper understanding of the specific mechanisms of NETs in breast cancer and to evaluate the effectiveness and potential side effects of these intervention strategies.
NETs should be regarded as a double-edged sword in the development of cancer of cancer. Although the majority of current studies point to a pro-metastatic role of NETs in breast cancer, we should bear in mind that the NET, a web-like structure as implied by its name, can also trap cancer cells and restrain their spread to distant organs. Therefore, we should be cautious in designing anti-NET therapies against cancer and in explaining the results from these investigations. In addition, we need to bear in mind that most of the NET components are the factors important for normal functioning of neutrophils. Therefore, targeting the NET components may also lead to neutrophil suppression and cause side effects such as neutropenia and increased risk of life-threatening infections. Nevertheless, targeting the NETs provides a promising therapeutic strategy with the potential of restraining the metastatic spread of breast cancer. Further understanding the pathophysiology behind NETs and their obligations in breast cancer metastasis will be instrumental in designing NET-targeting approaches for effective breast cancer therapy.
Acknowledgements
Funding
This work was supported by the Natural Science Foundation of China (82173374, 82103342, 82303126), Key Project of Developmental Biology and Breeding of Hunan Province (2022XKQ0205), Natural Science Foundation of Hunan (2024JJ5281, 2024JJ6325), The Research Team for Reproduction Health and Translational Medicine of Hunan Normal University (2023JC101), and Hunan Normal University School of Medicine Open Project Fund (KF2023004, KF2023006).
Author contributions
XX and XW: Reviewing the literature and writing the original manuscript. ZZ, YG, GH, YW, and SF: Revising and editing the manuscript. CZ and XD: Revising and reviewing the manuscript. All authors have read and approved the final submitted manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Jiang T, Wang Y, Chen X, Xia W, Xue S, Gu L. et al. Neutrophil extracellular traps (NETs)-related lncRNAs signature for predicting prognosis and the immune microenvironment in breast cancer. Front Cell Dev Biol. 2023;11:1117637-52
3. Rosales C. Neutrophils at the crossroads of innate and adaptive immunity. J Leukoc Biol. 2020;108:377-96
4. Lecot P, Sarabi M, Pereira Abrantes M, Mussard J, Koenderman L, Caux C. et al. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front Immunol. 2019;10:2155-73
5. Demkow U. Neutrophil Extracellular Traps (NETs) in Cancer Invasion, Evasion and Metastasis. Cancers (Basel). 2021;13:4495-511
6. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279-87
7. Wu Y, Ma J, Yang X, Nan F, Zhang T, Ji S. et al. Neutrophil profiling illuminates anti-tumor antigen-presenting potency. Cell. 2024;187:1422-39.e24
8. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS. et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-5
9. Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA. et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A. 2012;109:13076-81
10. Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front Immunol. 2020;11:1749-55
11. Snoderly HT, Boone BA, Bennewitz MF. Neutrophil extracellular traps in breast cancer and beyond: current perspectives on NET stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res. 2019;21:145-57
12. De Meo ML, Spicer JD. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin Immunol. 2021;57:101595-604
13. Zheng C, Xu X, Wu M, Xue L, Zhu J, Xia H. et al. Neutrophils in triple-negative breast cancer: an underestimated player with increasingly recognized importance. Breast Cancer Res. 2023;25:88-99
14. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. 1996;59:229-40
15. de Andrea CE, Ochoa MC, Villalba-Esparza M, Teijeira Á, Schalper KA, Abengozar-Muela M. et al. Heterogenous presence of neutrophil extracellular traps in human solid tumours is partially dependent on IL-8. J Pathol. 2021;255:190-201
16. Burgener SS, Schroder K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb Perspect Biol. 2020;12:a037028-43
17. Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A. et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75-7
18. Huang J, Hong W, Wan M, Zheng L. Molecular mechanisms and therapeutic target of NETosis in diseases. MedComm (2020). 2022;3:e162-80
19. Li X, Xiao S, Filipczak N, Yalamarty SSK, Shang H, Zhang J. et al. Role and Therapeutic Targeting Strategies of Neutrophil Extracellular Traps in Inflammation. Int J Nanomedicine. 2023;18:5265-87
20. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677-91
21. Zhu D, Zhang Y, Wang S. Histone citrullination: a new target for tumors. Mol Cancer. 2021;20:90-106
22. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853-62
23. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205-13
24. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE. et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117:7326-37
25. Amulic B, Knackstedt SL, Abu Abed U, Deigendesch N, Harbort CJ, Caffrey BE. et al. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev Cell. 2017;43:449-62.e5
26. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S. et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3:eaar6689-701
27. Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 2019;60:721-7
28. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD. et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413-25
29. Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122:2784-94
30. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386-93
31. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L. et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. 2012;3:307-17
32. Masuda S, Nakazawa D, Shida H, Miyoshi A, Kusunoki Y, Tomaru U. et al. NETosis markers: Quest for specific, objective, and quantitative markers. Clin Chim Acta. 2016;459:89-93
33. Martinez-Cannon BA, Garcia-Ronquillo K, Rivera-Franco MM, Leon-Rodriguez E. Do circulating neutrophil extracellular traps predict recurrence in early breast cancer? Front Oncol. 2022;12:1044611-7
34. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123:3446-58
35. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K. et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016;76:1367-80
36. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120-38
37. McDowell SAC, Luo RBE, Arabzadeh A, Doré S, Bennett NC, Breton V. et al. Neutrophil oxidative stress mediates obesity-associated vascular dysfunction and metastatic transmigration. Nat Cancer. 2021;2:545-62
38. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ. et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673-85
39. Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F. et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int J Cancer. 2017;140:2321-30
40. Reymond N, d'Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858-70
41. Manfioletti G, Fedele M. Epithelial-Mesenchymal Transition (EMT). Int J Mol Sci. 2023;24:11386-91
42. Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, König S. et al. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers (Basel). 2020;12:1542-58
43. Martins-Cardoso K, Maçao A, Souza JL, Silva AG, König S, Martins-Gonçalves R. et al. TF/PAR2 Signaling Axis Supports the Protumor Effect of Neutrophil Extracellular Traps (NETs) on Human Breast Cancer Cells. Cancers (Basel). 2023;16:5-22
44. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J. et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583:133-8
45. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK. et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094-8
46. Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL. et al. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019;27:3927-38.e6
47. Tang Q, Liang B, Zhang L, Li X, Li H, Jing W. et al. Enhanced CHOLESTEROL biosynthesis promotes breast cancer metastasis via modulating CCDC25 expression and neutrophil extracellular traps formation. Sci Rep. 2022;12:17350-62
48. Wang Y, Bu F, Royer C, Serres S, Larkin JR, Soto MS. et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat Cell Biol. 2014;16:1092-104
49. Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363-71
50. Korkmaz B, Caughey GH, Chapple I, Gauthier F, Hirschfeld J, Jenne DE. et al. Therapeutic targeting of cathepsin C: from pathophysiology to treatment. Pharmacol Ther. 2018;190:202-36
51. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P. et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39:423-37.e7
52. Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C. et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20:543-56
53. Palmér R, Mäenpää J, Jauhiainen A, Larsson B, Mo J, Russell M. et al. Dipeptidyl Peptidase 1 Inhibitor AZD7986 Induces a Sustained, Exposure-Dependent Reduction in Neutrophil Elastase Activity in Healthy Subjects. Clin Pharmacol Ther. 2018;104:1155-64
54. Zhang Y, Guo L, Dai Q, Shang B, Xiao T, Di X. et al. A signature for pan-cancer prognosis based on neutrophil extracellular traps. J Immunother Cancer. 2022;10:e004210-20
55. Lavoie SS, Dumas E, Vulesevic B, Neagoe PE, White M, Sirois MG. Synthesis of Human Neutrophil Extracellular Traps Contributes to Angiopoietin-Mediated In Vitro Proinflammatory and Proangiogenic Activities. J Immunol. 2018;200:3801-13
56. Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493-8
57. Bausch D, Pausch T, Krauss T, Hopt UT, Fernandez-del-Castillo C, Warshaw AL. et al. Neutrophil granulocyte derived MMP-9 is a VEGF independent functional component of the angiogenic switch in pancreatic ductal adenocarcinoma. Angiogenesis. 2011;14:235-43
58. Li B, Liu Y, Hu T, Zhang Y, Zhang C, Li T. et al. Neutrophil extracellular traps enhance procoagulant activity in patients with oral squamous cell carcinoma. J Cancer Res Clin Oncol. 2019;145:1695-707
59. Jung HS, Gu J, Kim JE, Nam Y, Song JW, Kim HK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS One. 2019;14:e0216055-70
60. Lachowicz-Scroggins ME, Dunican EM, Charbit AR, Raymond W, Looney MR, Peters MC. et al. Extracellular DNA, Neutrophil Extracellular Traps, and Inflammasome Activation in Severe Asthma. Am J Respir Crit Care Med. 2019;199:1076-85
61. Meyer-Hoffert U, Wiedow O. Neutrophil serine proteases: mediators of innate immune responses. Curr Opin Hematol. 2011;18:19-24
62. Shao BZ, Yao Y, Li JP, Chai NL, Linghu EQ. The Role of Neutrophil Extracellular Traps in Cancer. Front Oncol. 2021;11:714357-69
63. Lu Z, Long Y, Li J, Li J, Ren K, Zhao W. et al. Simultaneous inhibition of breast cancer and its liver and lung metastasis by blocking inflammatory feed-forward loops. J Control Release. 2021;338:662-79
64. Singh R, Mishra MK, Aggarwal H. Inflammation, Immunity, and Cancer. Mediators Inflamm. 2017;2017:6027305
65. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-44
66. Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle. 2009;8:3267-73
67. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015;12:584-96
68. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J. et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8:361ra138-58
69. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. 2018;15:206-21
70. Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc Natl Acad Sci U S A. 2017;114:E9618-e25
71. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL. et al. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952-61
72. Sharma BK, Flick MJ, Palumbo JS. Cancer-Associated Thrombosis: A Two-Way Street. Semin Thromb Hemost. 2019;45:559-68
73. Cao W, Zhu MY, Lee SH, Lee SB, Kim HJ, Park BO. et al. Modulation of Cellular NAD(+) Attenuates Cancer-Associated Hypercoagulability and Thrombosis via the Inhibition of Tissue Factor and Formation of Neutrophil Extracellular Traps. Int J Mol Sci. 2021;22:12085-102
74. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr. et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880-5
75. Demers M, Wagner DD. Neutrophil extracellular traps: A new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology. 2013;2:e22946-e8
76. Demers M, Wagner DD. NETosis: a new factor in tumor progression and cancer-associated thrombosis. Semin Thromb Hemost. 2014;40:277-83
77. Stein PD, Beemath A, Meyers FA, Skaf E, Sanchez J, Olson RE. Incidence of venous thromboembolism in patients hospitalized with cancer. Am J Med. 2006;119:60-8
78. Ay C, Pabinger I, Cohen AT. Cancer-associated venous thromboembolism: Burden, mechanisms, and management. Thromb Haemost. 2017;117:219-30
79. Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med. 2000;343:1846-50
80. Khan UT, Walker AJ, Baig S, Card TR, Kirwan CC, Grainge MJ. Venous thromboembolism and mortality in breast cancer: cohort study with systematic review and meta-analysis. BMC Cancer. 2017;17:747-59
81. Khorana AA, Connolly GC. Assessing risk of venous thromboembolism in the patient with cancer. J Clin Oncol. 2009;27:4839-47
82. Thålin C, Hisada Y, Lundström S, Mackman N, Wallén H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler Thromb Vasc Biol. 2019;39:1724-38
83. Abdol Razak N, Elaskalani O, Metharom P. Pancreatic Cancer-Induced Neutrophil Extracellular Traps: A Potential Contributor to Cancer-Associated Thrombosis. Int J Mol Sci. 2017;18:487-505
84. Andrews RK, Arthur JF, Gardiner EE. Neutrophil extracellular traps (NETs) and the role of platelets in infection. Thromb Haemost. 2014;112:659-65
85. Gomes T, Várady CBS, Lourenço AL, Mizurini DM, Rondon AMR, Leal AC. et al. IL-1β Blockade Attenuates Thrombosis in a Neutrophil Extracellular Trap-Dependent Breast Cancer Model. Front Immunol. 2019;10:2088-98
86. Hisada Y, Grover SP, Maqsood A, Houston R, Ay C, Noubouossie DF. et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica. 2020;105:218-25
87. Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M. et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110:8674-9
88. Várady CBS, Oliveira AC, Monteiro RQ, Gomes T. Recombinant human DNase I for the treatment of cancer-associated thrombosis: A pre-clinical study. Thromb Res. 2021;203:131-7
89. Kaplan RN, Rafii S, Lyden D. Preparing the "soil": the premetastatic niche. Cancer Res. 2006;66:11089-93
90. Ursini-Siegel J, Siegel PM. The influence of the pre-metastatic niche on breast cancer metastasis. Cancer Lett. 2016;380:281-8
91. Medeiros B, Allan AL. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int J Mol Sci. 2019;20:2272-88
92. Wang Y, Ding Y, Guo N, Wang S. MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front Immunol. 2019;10:172-87
93. Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216:176-94
94. Paolillo M, Schinelli S. Extracellular Matrix Alterations in Metastatic Processes. Int J Mol Sci. 2019;20:4947-64
95. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A. et al. Neutrophil Extracellular Traps Accumulate in Peripheral Blood Vessels and Compromise Organ Function in Tumor-Bearing Animals. Cancer Res. 2015;75:2653-62
96. Lee J, Lee D, Lawler S, Kim Y. Role of neutrophil extracellular traps in regulation of lung cancer invasion and metastasis: Structural insights from a computational model. PLoS Comput Biol. 2021;17:e1008257-85
97. Zhou Z, Zhan J, Luo Q, Hou X, Wang S, Xiao D. et al. ADRB3 induces mobilization and inhibits differentiation of both breast cancer cells and myeloid-derived suppressor cells. Cell Death Dis. 2022;13:141-50
98. Zeng Z, Xu S, Wang F, Peng X, Zhang W, Zhan Y. et al. HAO1-mediated oxalate metabolism promotes lung pre-metastatic niche formation by inducing neutrophil extracellular traps. Oncogene. 2022;41:3719-31
99. Zheng Z, Li YN, Jia S, Zhu M, Cao L, Tao M. et al. Lung mesenchymal stromal cells influenced by Th2 cytokines mobilize neutrophils and facilitate metastasis by producing complement C3. Nat Commun. 2021;12:6202-16
100. Yang C, Wang Z, Li L, Zhang Z, Jin X, Wu P. et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J Immunother Cancer. 2021;9:e002875-92
101. Pan J, Zhang L, Wang X, Li L, Yang C, Wang Z. et al. Chronic stress induces pulmonary epithelial cells to produce acetylcholine that remodels lung pre-metastatic niche of breast cancer by enhancing NETosis. J Exp Clin Cancer Res. 2023;42:255
102. Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer. 2020;20:398-411
103. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME. et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:eaao4227-40
104. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H. et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15:807-17
105. El Rayes T, Catena R, Lee S, Stawowczyk M, Joshi N, Fischbach C. et al. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. Proc Natl Acad Sci U S A. 2015;112:16000-5
106. Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene. 2002;21:2513-24
107. Sanz-Moreno V, Balkwill FR. Mets and NETs: The Awakening Force. Immunity. 2018;49:798-800
108. Zhao J, Xie X. Prediction of prognosis and immunotherapy response in breast cancer based on neutrophil extracellular traps-related classification. Front Mol Biosci. 2023;10:1165776
109. Huang Z, Wang J, Sun B, Qi M, Gao S, Liu H. Neutrophil extracellular trap-associated risk index for predicting outcomes and response to Wnt signaling inhibitors in triple-negative breast cancer. Sci Rep. 2024;14:4232
110. Mousset A, Lecorgne E, Bourget I, Lopez P, Jenovai K, Cherfils-Vicini J. et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell. 2023;41:757-75.e10
111. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V. et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107:9813-8
112. Sadtler K, Estrellas K, Allen BW, Wolf MT, Fan H, Tam AJ. et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science. 2016;352:366-70
113. Chen J, Hou S, Liang Q, He W, Li R, Wang H. et al. Localized Degradation of Neutrophil Extracellular Traps by Photoregulated Enzyme Delivery for Cancer Immunotherapy and Metastasis Suppression. ACS Nano. 2022;16:2585-97
114. Liu LN, Chen C, Xin WJ, Li Q, Han C, Hua ZC. The oncolytic bacteria-mediated delivery system of CCDC25 nucleic acid drug inhibits neutrophil extracellular traps induced tumor metastasis. J Nanobiotechnology. 2024;22:69
115. Que H, Fu Q, Lan T, Tian X, Wei X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim Biophys Acta Rev Cancer. 2022;1877:188762-79
116. Bilusic M, Heery CR, Collins JM, Donahue RN, Palena C, Madan RA. et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. 2019;7:240-7
117. Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H. et al. A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res. 2020;22:4-12
118. Safarulla S, Madan A, Xing F, Chandrasekaran A. CXCR2 Mediates Distinct Neutrophil Behavior in Brain Metastatic Breast Tumor. Cancers (Basel). 2022;14:515-35
119. Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer. 2016;2:758-70
120. Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D. et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781-94
121. Kallergi G, Markomanolaki H, Giannoukaraki V, Papadaki MA, Strati A, Lianidou ES. et al. Hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in circulating tumor cells of breast cancer patients. Breast Cancer Res. 2009;11:R84-95
122. Zhang Y, Wang C, Li W, Tian W, Tang C, Xue L. et al. Neutrophil Cyto-Pharmaceuticals Suppressing Tumor Metastasis via Inhibiting Hypoxia-Inducible Factor-1α in Circulating Breast Cancer Cells. Adv Healthc Mater. 2022;11:e2101761
123. Wang H, Aloe C, Wilson N, Bozinovski S. G-CSFR antagonism reduces neutrophilic inflammation during pneumococcal and influenza respiratory infections without compromising clearance. Sci Rep. 2019;9:17732-43
Author contact
![]() Corresponding authors: Chanjuan Zheng, zhengchanjuanedu.cn; Xiyun Deng, dengxiyunmededu.cn.
Corresponding authors: Chanjuan Zheng, zhengchanjuanedu.cn; Xiyun Deng, dengxiyunmededu.cn.