Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2024; 15(11):3406-3417. doi:10.7150/jca.95437 This issue Cite
Research Paper
Exploring the Common Genetic Underpinnings of Chronic Pulmonary Disease and Esophageal Carcinoma Susceptibility
Dengfeng Zhang1,†, Yu zhou2,4,†, Tianxing Lu1,†, Jing Li1,†, Longyu Zhu3, Shujun Li1, ![]() , Yishuai Li2,4,
, Yishuai Li2,4, ![]() , Xiaoliang Duan2,4,
, Xiaoliang Duan2,4, ![]()
1. Department of Thoracic Surgery, The Second Hospital of Hebei Medical University, Shijiazhuang, China.
2. Department of Thoracic Surgery, Hebei Chest Hospital, Shijiazhuang, China.
3. Department of Radiotherapy, The Fourth Hospital of Hebei Medical University, Shijiazhuang, China.
4. Hebei Provincial Key Laboratory of Pulmonary Diseases, Shijiazhuang, China.
† Equal contribution.
Received 2024-2-18; Accepted 2024-4-18; Published 2024-4-29
Abstract

Background: Pulmonary diseases and esophageal cancer are highly prevalent conditions with rising incidence worldwide. Prior evidence supports shared environmental and behavioral factors, but less is known regarding potential genetic links underlying this comorbidity. This study aimed to elucidate the complex genetic relationship between chronic lung diseases and esophageal cancer risk.
Methods: Linkage disequilibrium score regression assessed the genetic correlation between esophageal cancer and asthma, COPD, and idiopathic pulmonary fibrosis leveraging extensive GWAS datasets. Pleiotropic analysis, gene-set enrichment, eQTL mapping, and mendelian randomization causality analyses were then conducted to identify specific shared genetic variants, enriched pathways, causal relationships and gene regulatory mechanisms connecting lung disease and cancer susceptibility.
Results: Significant genetic correlations were observed between esophageal cancer and both COPD and asthma, but not idiopathic pulmonary fibrosis. Further analyses identified 13 pleiotropic loci and 6 shared genes including CHRNA4, ERBB3, and SMAD3, as well as pathways related to immune function. eQTL integration highlighted 53 genes like SOCS1, FGF2, and CHRNA5 with tissue-specific regulatory effects on disease risk. Bidirectional relationships were noted, whereby genetic predisposition to asthma and COPD increased esophageal cancer risk, while cancer liability reciprocally raised pulmonary fibrosis risk.
Conclusions: These genomic analyses provide initial evidence that shared genetic factors may underpin the comorbidity between lung conditions and esophageal malignancy. The genes and pathways identified offer insights into biological mechanisms linking both diseases, aiding future screening, prevention and therapeutic efforts to mitigate this growing comorbidity burden.
Keywords: Esophageal cancer, Pulmonary disease, Genome-wide association study, Genetic correlation, Mendelian randomization analysis
Introduction
Esophageal cancer and chronic pulmonary diseases such as chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF) and asthma are highly prevalent conditions associated with substantial morbidity and mortality burdens worldwide [1-4]. Prior observational studies have noted epidemiologic links and clinical comorbidity between these diseases, suggesting potential common environmental or behavioral factors [5-7]. For instance, smoking and inflammation influence pathogenesis of both malignancy and lung disorders [7-11]. However, despite recognized shared risk determinants, the contributions of genetic factors underlying this medical comorbidity have remained less explored.
Recent advances in genomics research have enabled large-scale analyses assessing genetic contributions through methodologies including genome-wide association studies (GWAS), linkage disequilibrium score regression, Mendelian randomization, and various bioinformatic annotation approaches [12-16]. By integrating multiple genetic correlation and causality assessment techniques with functional genomic annotation, these cutting-edge analytic frameworks now allow comprehensive interrogation of potential biological mechanisms driving comorbidity from sequence variation through to gene regulatory effects [17-19].
Leveraging extensive GWAS datasets, this study therefore aimed to elucidate the complex genetic relationship between chronic pulmonary diseases and esophageal carcinoma risk. We specifically sought to determine genetic correlation, identify pleiotropic loci, assess causality, and define regulatory mechanisms that may reciprocally associate lung conditions with esophageal cancer susceptibility. Delineating these intricate genomic links facilitating comorbidity may aid future screening, prevention, and therapeutic efforts to mitigate this growing global health burden.
Methods
Lung disease data
Pulmonary disease data were derived from a collaborative network of 23 biobank repositories across 4 continents as part of the Global Biobank Meta-analysis Initiative, comprising over 2.2 million individuals. Specifically, data on idiopathic pulmonary fibrosis were obtained from a large-scale meta-analysis [20] including 11,160 cases and 1,364,410 controls, with inverse-variance weighted fixed effects meta-analyses conducted for all ancestries, each ancestry, and sex-stratified ancestries across all biobanks. Similarly, the asthma GWAS was conducted as part of the Global Biobank Meta-analysis Initiative, performing a large-scale GWAS via meta-analysis of 22 biobanks across multiple ancestries, identifying 179 asthma-associated loci, 49 of which were novel. COPD data were directly accessed from the Global Biobank Meta-analysis Initiative, comprising 58,559 European ancestry cases and 937,358 controls. Additionally, for the asthma and IPF data, we only selected individuals of European ancestry for subsequent genetic analyses (Table S1).
Esophageal cancer data
Esophageal cancer data were derived from a meta-analysis of genome-wide association studies (GWAS) across four independent studies in Europeans, North Americans, and Australians [21]. All cases were of European ancestry with histopathologically confirmed disease. The meta-analysis was conducted using fixed-effects inverse variance-weighted methods, comprising 4,112 esophageal adenocarcinoma cases and 17,159 representative controls from European, North American, and Australian GWAS (Table S1). Stringent SNP quality control was implemented: (i) non-biallelic SNPs and those with ambiguous alleles were excluded; (ii) SNPs without rs-identifiers were removed; (iii) duplicate SNPs or those not present in 1000 Genomes or with allele mismatches were deleted; (iv) due to complex LD structure, SNPs within the major histocompatibility complex region (chr6: 28.5-33.5Mb) were excluded for LDSC analyses; (v) SNPs with minor allele frequency (MAF) < 0.01 were retained.
Genetic correlation analysis
Linkage disequilibrium score regression (LDSC) [22] and high-density linkage disequilibrium (HDL) [23] methods were utilized to assess shared polygenic architecture between traits, with LD scores calculated from European ancestry samples in the 1,000 Genomes Project phase 3 as the reference panel [24]; the HDL reference set comprised 1,029,876 quality-controlled HapMap3 SNPs.
We further utilized stratified LD score regression with data from different immune cell types to examine SNP heritability enrichment for esophageal cancer and pulmonary diseases, assessing whether specific cell types in these tissues demonstrated significant genetic enrichment. Data for 292 immune cell types (including B cells, gamma delta T cells, alpha beta T cells, innate lymphocytes, myeloid cells, stromal cells, and stem cells) were obtained from the ImmGen Consortium [25]. After adjusting for the baseline model and all gene sets, we used the p-value of the z-score of the regression coefficients to evaluate significance of the SNP heritability enrichment estimates for each tissue and cell type.
Pleiotropic analysis under composite null hypothesis (PLACO)
PLACO is a novel SNP-level approach that can investigate pleiotropic loci between complex traits using only summary-level genotype-phenotype association statistics [26]. We computed the squared Z-scores for each variant and removed SNPs with extremely high Z2 (>80). Additionally, considering potential correlations between pulmonary diseases and esophageal cancer, we estimated the correlation matrix of Z. The omnibus test of pleiotropy was then performed using the level-α horizontal intersection-union test (IUT) method. The final p-value for the IUT test was the maximum of the p-values for testing H0 versus H1.
Based on the PLACO results, we further mapped the identified loci to nearby genes to explore potential shared biological mechanisms for these pleiotropic variants. We performed MAGMA Generalized Gene-Set Analysis of GWAS Data on genes overlapping or in proximity to pleiotropic loci identified by PLACO [27], to identify candidate pathways enriched for pleiotropic effects, as well as tissue enrichment of pleiotropic genes. Functional mapping and annotation by FUMA genome-wide association study (GWAS) [28] were utilized to determine biological roles of pleiotropic loci. A series of pathway enrichment analyses mapping annotated genes were conducted based on the Molecular Signatures Database (MSigDB) to elucidate functionality [29]. eQTL analyses incorporated SNP-gene association data including esophageal tissue, lung tissue, and whole blood.
Mendelian randomization analysis
We used the clumping program in PLINK software (6) to select all loci independently associated with the disease at genome-wide significance (P<5×10^-8) as instrumental variables (IVs), with an r^2 threshold of 0.001 within a 10,000kb window. To ensure the strength of the IVs, we calculated the r^2 and F-statistic for each IV [30]. The F-statistic was computed as: where r^2 represents the proportion of variance explained by the IV, n is the sample size, and k is the number of SNPs. The primary method adopted for Mendelian randomization (MR) was inverse variance weighted (IVW), which requires the IVs to satisfy 3 assumptions: (1) the IV should be associated with exposure; (2) the IV should not be associated with confounders of the exposure-outcome association; (3) the IV affects the outcome only through the exposure. We conducted several sensitivity analyses. First, the IVW and MR-Egger's Q test can detect potential violations of assumptions through heterogeneity between the IV estimates [31]. Second, we applied MR-Egger to estimate directional pleiotropy according to its intercept, ensuring genetic variants are independently associated with exposure and outcome [32]. We increased the stability and robustness of results through additional analyses using MR methods with different modeling assumptions and strengths (DIVW, MR-RAPS, weighted median and weighted mode). All statistical analyses were performed in R 3.5.3, with MR analyses utilizing the MendelianRandomization package [33].
Results
A flow diagram outlining the full analysis process is provided in Figure 1.
Flow diagram outlining the full analysis process.
Genetic correlations
LDSC genetic correlation analyses revealed significant genetic correlations between pulmonary diseases (COPD and asthma) and esophageal cancer: COPD (rg = 0.217, P = 4.22E-5), asthma (rg = 0.158, P = 2.00E-4); however, the genetic correlation between idiopathic pulmonary fibrosis and esophageal cancer was not significant (rg = 0.071, P = 0.5761). HDL analysis also corroborated relationships between esophageal cancer and COPD (rg = 0.197, P = 4.27E-04) and asthma (rg = 0.143, P = 0.005) (Table S2). The LDSC intercepts excluded the possibility of sample overlap.
Identification of pleiotropy loci and genes
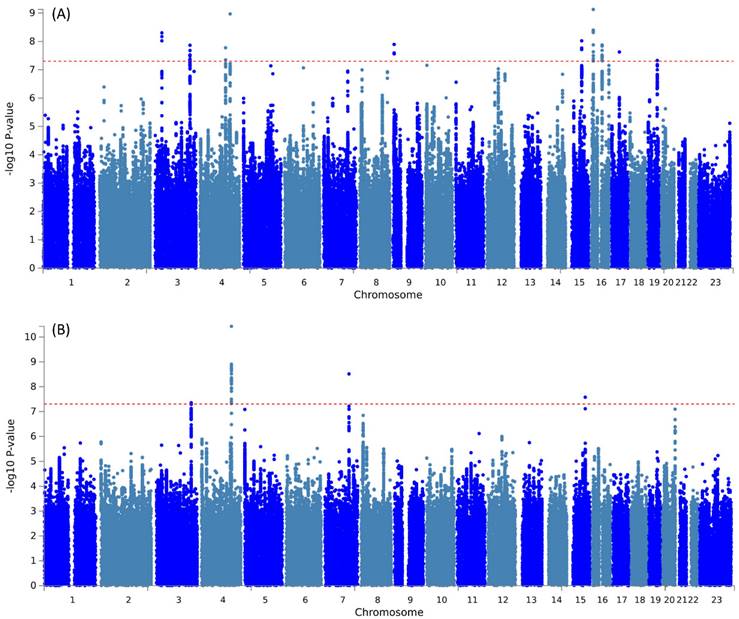
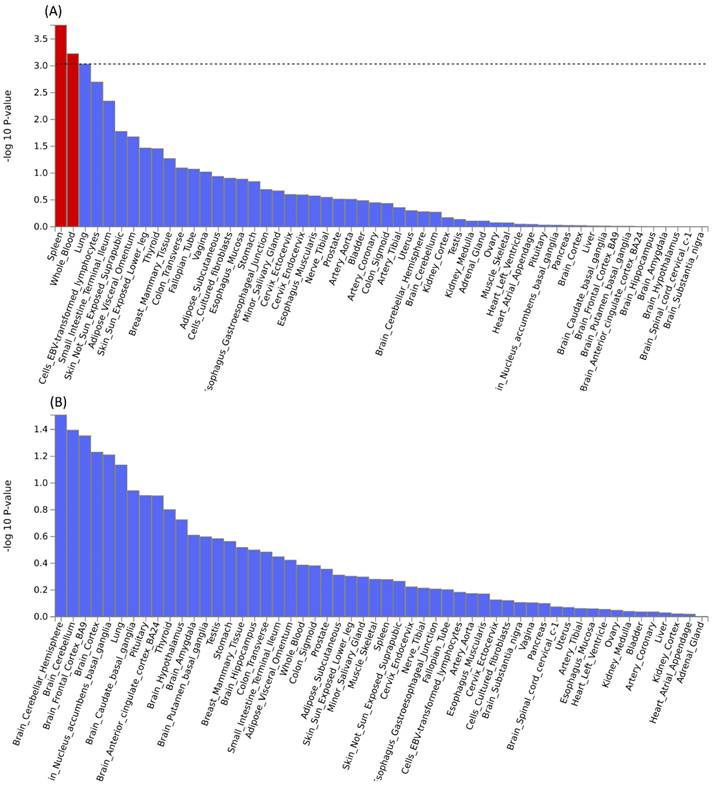
Further pleiotropic analysis (PLACO) was undertaken between the two diseases, and the Manhattan plots are shown in Figure 2. As summarized in Table 1, we identified 13 pleiotropic loci (9 for asthma, 4 for COPD), and 4q31.21 was shared between the two phenotypes (Table S3). Notably, the QQ plots showed no genomic inflation (Figure S1). In addition, basic information for each genomic risk locus can be found in Figure S2. We identified that among the 9 genomic loci associated with asthma, the locus at 15:67395918-68218648 harbors the highest number of SNPs. Similarly, within the 4 genomic loci associated with COPD, the locus at 15:78712119-79080798 has the highest number of SNPs. Regarding the functional impacts of pleiotropic SNPs on genes, please refer to Figure S3. We observed that the functional impact of pleiotropic SNPs on genes is predominantly concentrated in intronic and intergenic regions. Specifically, the pleiotropic SNPs between asthma and esophageal cancer primarily affect gene function in intronic regions, whereas those between COPD and esophageal cancer exert their main influence on gene function in intergenic regions. Finally, regional plots for each risk locus are presented in Figures S4 to S16. Gene-set enrichment analysis using MAGMA on the pleiotropic results revealed the top 10 significantly enriched gene sets as shown in Table 2, involving pathways such as alpha beta T cell differentiation, alpha beta T cell activation, positive regulation of RNA metabolic process, chromatin, Foxp3 in COVID19 and positive regulation of transcription by RNA polymerase II. Moreover, tissue-specific MAGMA analysis indicated that the most significant enrichment evidence in both diseases appeared in spleen and whole blood tissues, which are closely related to immune processes (Figure 3). Importantly, this section of MAGMA gene-set and tissue-specific analyses was based on the complete SNP p-value distribution.
Manhattan plot illustrating the pleiotropic loci between pulmonary diseases and esophageal cancer.
Information on the thirteen identified pleiotropic loci.
| Trait pairs | GenomicLocus | Chromosome: start-end | rsID | P | Mapped Genes |
|---|---|---|---|---|---|
| Asthma&EAC | 3p22.3 | 3:32923640-33185407 | rs6764245 | 4.97E-09 | CCR4, GLB1 |
| Asthma&EAC | 3q26.2 | 3:168585686-168976021 | rs879394 | 1.36E-08 | RP11-368I23.4, RP11-152C17.1 |
| Asthma&EAC | 4q27 | 4:122884909-123727295 | rs72687029 | 1.67E-08 | KIAA1109 |
| Asthma&EAC | 4q31.21 | 4:145227600-146177041 | rs1032296 | 1.07E-09 | RP11-361D14.2, KRT18P51 |
| Asthma&EAC | 9p24.1 | 9:5652270-6585872 | rs62556407 | 2.76E-08 | KIAA2026 |
| Asthma&EAC | 15q22.33 | 15:67395918-68218648 | rs2289791 | 9.50E-09 | SMAD3 |
| Asthma&EAC | 16p13.13 | 16:11004363-11467218 | rs12923849 | 7.46E-10 | CLEC16A |
| Asthma&EAC | 16q12.2 | 16:53579222-53848561 | rs1558902 | 1.33E-08 | FTO |
| Asthma&EAC | 19q13.32 | 19:46150182-46470434 | rs7256524 | 4.70E-08 | FBXO46 |
| COPD&EAC | 3q26.2 | 3:168587408-168976021 | rs13078090 | 4.44E-08 | RP11-152C17.1, MECOM |
| COPD&EAC | 4q31.21 | 4:145227600-146177041 | rs1032296 | 3.72E-11 | RP11-361D14.2, KRT18P51 |
| COPD&EAC | 7q31.2 | 7:116910447-117449242 | rs10278953 | 3.06E-09 | ASZ1 |
| COPD&EAC | 15q25.1 | 15:78712119-79080798 | rs564585 | 2.66E-08 | CHRNA5 |
Results of gene set analysis (top 10), with significant results (Pbon < 0.05 after multiple corrections) highlighted in red.
| Trait pairs | Gene Sets | N genes | Beta | SE | P | Pbon |
|---|---|---|---|---|---|---|
| COPD&EAC | REACTOME_HIGHLY_CALCIUM_PERMEABLE_NICOTINIC_ACETYLCHOLINE_ RECEPTORS | 9 | 1.515 | 0.336 | 3.38E-06 | 0.058 |
| COPD&EAC | GOBP_BEHAVIORAL_RESPONSE_TO_NICOTINE | 8 | 1.337 | 0.316 | 1.20E-05 | 0.204 |
| COPD&EAC | REACTOME_HIGHLY_CALCIUM_PERMEABLE_POSTSYNAPTIC_NICOTINIC_ ACETYLCHOLINE_RECEPTORS | 11 | 1.273 | 0.304 | 1.45E-05 | 0.246 |
| COPD&EAC | REACTOME_VESICLE_MEDIATED_TRANSPORT | 657 | 0.132 | 0.032 | 1.74E-05 | 0.297 |
| COPD&EAC | GOBP_RESPONSE_TO_NICOTINE | 44 | 0.574 | 0.140 | 1.98E-05 | 0.337 |
| COPD&EAC | REACTOME_MEMBRANE_TRAFFICKING | 618 | 0.127 | 0.033 | 5.44E-05 | 0.925 |
| COPD&EAC | BROWNE_HCMV_INFECTION_4HR_DN | 241 | 0.182 | 0.053 | 2.69E-04 | 1.000 |
| COPD&EAC | DAZARD_UV_RESPONSE_CLUSTER_G2 | 25 | 0.588 | 0.173 | 3.50E-04 | 1.000 |
| COPD&EAC | SA_PTEN_PATHWAY | 17 | 0.690 | 0.206 | 4.13E-04 | 1.000 |
| COPD&EAC | MULLIGHAN_NPM1_SIGNATURE_3_DN | 156 | 0.223 | 0.067 | 4.24E-04 | 1.000 |
| Asthma&EAC | GOBP_ALPHA_BETA_T_CELL_DIFFERENTIATION | 116 | 0.461 | 0.088 | 7.24E-08 | 1.23E-03 |
| Asthma&EAC | GOBP_ALPHA_BETA_T_CELL_ACTIVATION | 165 | 0.358 | 0.073 | 5.27E-07 | 0.009 |
| Asthma&EAC | GOBP_POSITIVE_REGULATION_OF_RNA_METABOLIC_PROCESS | 1756 | 0.105 | 0.022 | 9.20E-07 | 0.016 |
| Asthma&EAC | GOCC_CHROMATIN | 1249 | 0.123 | 0.026 | 1.19E-06 | 0.020 |
| Asthma&EAC | WP_FOXP3_IN_COVID19 | 15 | 1.251 | 0.266 | 1.30E-06 | 0.022 |
| Asthma&EAC | GOBP_POSITIVE_REGULATION_OF_TRANSCRIPTION_BY_RNA_POLYMERASE_II | 1187 | 0.123 | 0.027 | 1.65E-06 | 0.028 |
| Asthma&EAC | ZWANG_CLASS_3_TRANSIENTLY_INDUCED_BY_EGF | 215 | 0.296 | 0.065 | 2.94E-06 | 0.050 |
| Asthma&EAC | GOBP_POSITIVE_REGULATION_OF_MACROMOLECULE_BIOSYNTHETIC_PROCESS | 1836 | 0.097 | 0.022 | 3.01E-06 | 0.051 |
| Asthma&EAC | GOMF_PURINE_NUCLEOTIDE_BINDING | 1914 | 0.091 | 0.021 | 5.04E-06 | 0.086 |
| Asthma&EAC | GOMF_TRANSCRIPTION_REGULATOR_ACTIVITY | 1643 | 0.103 | 0.023 | 5.07E-06 | 0.086 |
Tissue-specific analysis based on genome-wide pleiotropy using MAGMA.
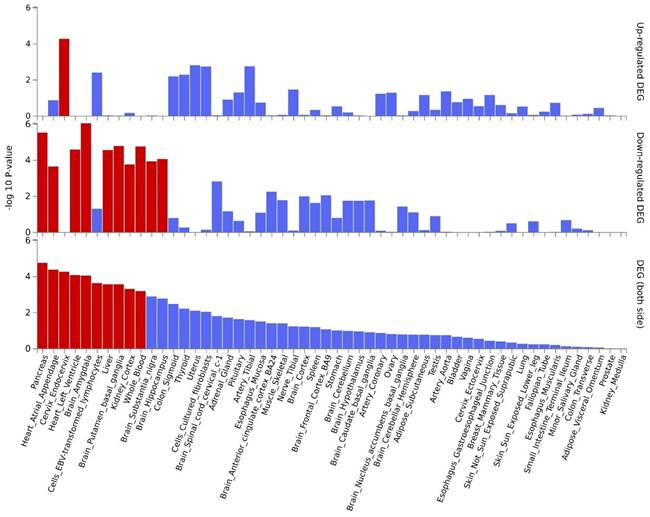
By leveraging the location information of lead SNPs, we successfully mapped nearby genes associated with these pleiotropic risk loci (Table 1). Subsequently, we conducted further MAGMA gene test and identified 6 significantly pleiotropic genes (Figure S17 and Table S4): RIN3, SMAD3, AAGAB, ERBB3, LPP, and CHRNA4. Details on the nearby and MAGMA genes are provided in Table S5. The QQ plot (Figure S18) did not show genomic inflation, indicating the results are credible. The expression patterns of pleiotropic genes across different tissues are shown in Figure S19, demonstrating differential expression of these genes in some tissues including EBV transformed lymphocytes cells, whole blood, spleen, esophagus, etc. Tissue-specific enrichments are detailed in Figure S20. Pathway analysis results can be found in Figure S21, while enrichments for cell types are shown in Figure S22. The key pathways involved include: the receptor complex, regulation of chemotaxis, the nuclear speckle, and regulation of growth.
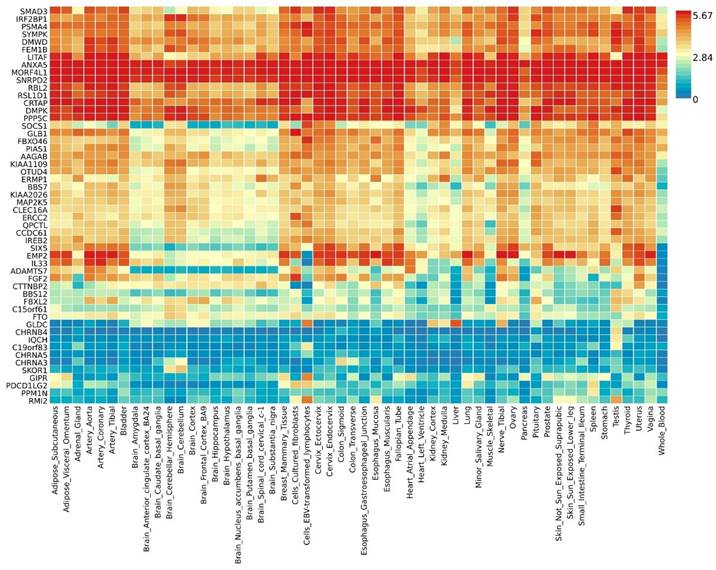
By further leveraging eQTL information (including esophageal tissue, lung tissue, spleen, and whole blood data), we successfully identified eQTL genes associated with these pleiotropic risk loci (Table S6). A total of 53 relevant genes were identified, including several widely-studied molecules such as SMAD3, SOCS1, ERCC2, IL33, FGF2, FTO, CHRNA5, and PPM1N, with 44 genes related to asthma and 9 genes related to chronic obstructive pulmonary disease (COPD). Expression patterns of pleiotropic eQTL genes across tissues can be found in Figure 4. In various tissues, notable expression is observed for genes such as LITAF, ANXA5, MORF4L1, and SNRPD2. Conversely, genes including CHRNB4, IQCH, C19orf83, and CHRNA5 exhibit significantly reduced expression across multiple tissues. Enrichments of these genes in different tissues are detailed in Figure 5, showing significant enrichments in whole blood, esophagus colon, lung, brain, skeletal muscle, etc. Pathway enrichment analysis results can be found in Figure S23 and Figure S24. Enrichment analysis implicated various pathways including behavioral response to nicotine, fat cell differentiation, the nuclear speckle, response to fatty acids, myeloid cell differentiation, and others.
Expression profiles of pleiotropic eQTL genes across different tissues.
Tissue-specific enrichment analysis based on pleiotropic eQTL genes.
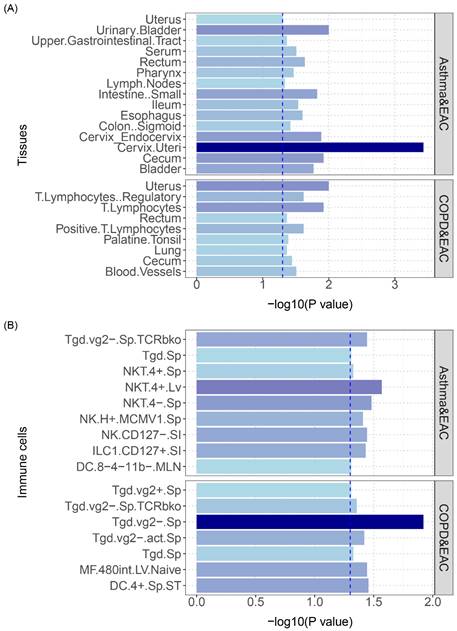
Tissue and cell-type specific enrichment of SNP heritability
We subsequently utilized stratified linkage disequilibrium score regression (S-LDSC) to analyze genetics data specific to particular tissue or cell types. This approach estimates the genetic contribution of different cell types or tissues, which aids in pinpointing the source of genetic associations with traits or diseases. It enables interpretation of genome-wide polygenic signals (GWAS) in the context of a particular tissue or cell type by elucidating in which tissue or cell type genetic variation has a functional effect, thereby furthering understanding of the biological mechanisms underlying complex traits and diseases.
The analysis results are shown in Figure 6: asthma and COPD had overlapping polygenicity for EAC in the uterus and lymphocyte tissues, with the cervical tissue having the highest significance; additionally, asthma and COPD showed overlapping polygenicity for EAC in NK and Tgd cells, with Tgd cells having the highest significance; asthma and EAC exhibited the most enrichment in NK cells, indicating that NK cells play a vital role in the developmental processes of both diseases; COPD and EAC displayed the most enrichment in Tgd cells, signifying that this cell type is critical in the disease courses of both (specific results are shown in Table S7 and Table S8).
(A) S-LDSC enrichment analysis identifies tissues with significant heritability enrichment between pulmonary diseases and esophageal cancer. (B) S-LDSC enrichment analysis identifies immune cells with significant heritability enrichment between pulmonary diseases and esophageal cancer. The blue dashed line represents the significance threshold of 0.05.
Causal associations
Finally, we performed causal inference on the relationship between lung diseases and esophageal cancer using two-sample MR methods, which supported the significant association between the two, with genetic instruments used listed in Table S9. Sensitivity analyses are shown in Table 3, and full sensitivity analysis results are in Table S10.
Outline of results of the Mendelian Randomization (MR) analysis.
| Exposures | Outcomes | Methods | Estimate | P | Heterogeneity test | |
|---|---|---|---|---|---|---|
| Estimate | P | |||||
| Asthma | EAC | IVW | 1.194 (1.048, 1.361) | 0.008 | 131.336 | 0.189 |
| DIVW | 1.198 (1.041, 1.378) | 0.012 | ||||
| MR-RAPS | 1.201 (1.039, 1.388) | 0.013 | ||||
| Weighted mode | 1.242 (0.892, 1.727) | 0.199 | ||||
| Weighted median | 1.225 (0.996, 1.508) | 0.055 | ||||
| MR-Egger (slope) | 1.182 (0.824, 1.696) | 0.360 | ||||
| MR-Egger (intercept) | 0 (-0.016, 0.017) | 0.953 | ||||
| EAC | Asthma | IVW | 1.065 (1.022, 1.11) | 0.003 | 1.088 | 0.297 |
| DIVW | 1.067 (1.02, 1.117) | 0.005 | ||||
| MR-RAPS | 1.066 (1.018, 1.116) | 0.007 | ||||
| EAC | IPF | IVW | 1.203 (1.019, 1.42) | 0.029 | 0.046 | 0.831 |
| DIVW | 1.21 (1.013, 1.445) | 0.036 | ||||
| MR-RAPS | 1.203 (1.008, 1.435) | 0.040 | ||||
Causal effect analysis of asthma on esophageal cancer showed consistent results across three MR methods (IVW, DIVW, MR-RAPS) that asthma has a causal effect on esophageal cancer. The heterogeneity test P was 0.189, indicating no heterogeneity existed. The MR-Egger intercept P>0.05, suggesting no influence from horizontal pleiotropy. Reverse causal effect analysis of esophageal cancer on asthma also showed significant causal effects between the two, with consistent results from three MR methods (IVW, DIVW, MR-RAPS). The heterogeneity test P was 0.297, indicating no heterogeneity. The bidirectional causal relationships further validate potential pleiotropy between the two diseases. Additionally, causal effect analysis of esophageal cancer on IPF showed consistent results from three MR methods (IVW, DIVW, MR-RAPS) that esophageal cancer has a causal effect on IPF. The heterogeneity test P was 0.831, indicating no heterogeneity existed.
Discussion
In this large-scale analysis, we identified significant genetic correlations and pleiotropic loci between esophageal cancer and pulmonary diseases including COPD and asthma by leveraging GWAS summary statistics. Enrichment analysis implicated various pathways including behavioral response to nicotine, fat cell differentiation, the nuclear speckle, response to fatty acids, myeloid cell differentiation, and others, which may be involved in the pleiotropic mechanisms between diseases. Causal inferences supported bidirectional relationships, where genetic liability for asthma and COPD increased esophageal cancer risk, while liability for esophageal cancer conversely increased asthma and IPF susceptibility. Our integrated approach combining multiple state-of-the-art methodologies provides robust evidence for potential shared genetic components across these diseases.
Prior observational studies have noted epidemiologic connections between esophageal carcinoma and chronic respiratory illnesses including COPD, IPF, and asthma, suggesting potential shared environmental or behavioral factors [34-38]. For example, several large cohort studies found a significantly heightened risk of esophageal cancer among COPD patients compared to controls without airflow obstruction [5, 39]. Likewise, population-based analyses have revealed a nearly 3-fold higher prevalence of asthma among esophageal cancer cases versus matched controls. Proposed common contributors to pathogenesis of both malignancy and lung disease include tobacco use, which damages the esophageal mucosa while promoting airway inflammation and remodeling [40-43]. Additionally, systemic inflammatory states may facilitate esophageal carcinogenesis while also exacerbating pulmonary disease severity [44, 45]. These epidemiologic observations provide clinical evidence complementing the genetic correlations identified here, further supporting that shared etiologic factors likely underpin the co-occurrence of these conditions. Elucidating the intricate biological mechanisms linking esophageal carcinoma and chronic lung diseases may contribute to improved prevention, screening and management for patients at high risk of this comorbidity.
Regarding the pleiotropic loci we identified, some of these loci have previously been reported in association with esophageal cancer or pulmonary diseases. For instance, the pleiotropic locus in the 15q25 region has been found to be associated with lung cancer and COPD [46-49]. The pleiotropic locus at 4q31.21 has also been reported to influence susceptibility to asthma [50]. In addition, some of the newly discovered pleiotropic loci, such as those located at 3p21.31 and 10q22.3, have not been previously reported in association with these two types of diseases. This may represent potential common genetic factors between the newly discovered esophageal cancer and pulmonary diseases in our study. Overall, these results provide further support for the shared genetic architecture between esophageal cancer and pulmonary diseases, contributing to our understanding of the relationship between these two conditions. Subsequent functional studies are needed to validate the impact of these newly discovered pleiotropic loci on diseases and to elucidate their potential mechanisms in the onset of diseases.
Our research findings suggest the potential existence of shared genetic mechanisms between esophageal cancer and pulmonary diseases. Enrichment analysis indicates that these pleiotropic loci involve various biological pathways, such as behavioral responses to nicotine [51-54], adipocyte differentiation [55, 56], nuclear speckles [57, 58], responses to fatty acids [59, 60], and granulocyte differentiation [61], among others. These pathways are associated with processes like smoking, inflammation, and immune function, contributing to an explanation of the interconnections between diseases. Additionally, tissue and cell-type analyses reveal that the genetic correlations between esophageal cancer and pulmonary diseases predominantly originate from cervical tissue, lymphocytes, NK cells, and Tgd cells, among others. This implies a potential pivotal role of these tissues and immune cells in the onset of diseases.
Our study had several key strengths. The utilization of immense GWAS sample sizes across diseases enhanced statistical power to uncover subtle shared genetic effects that may have been difficult to detect in smaller-scaled efforts, overcoming barriers like residual confounding that can distort observational studies. Additionally, the random assortment of genetic variants avoids reverse causation, a key advantage of MR designs. However, some limitations should be considered when interpreting the results. It is important to note that the predominantly European ancestry of the study participants may limit the generalizability of the findings to other populations. The available GWAS data were predominately of European ancestry, restricting generalizability, with further diversity needed to determine if findings extend to other populations. The use of summary statistics also precluded stratified analyses to pinpoint specific at-risk subgroups. While offering statistical power, the high genomic stringency of methods like PLACO may miss small-effect pleiotropic loci that contribute meaningfully to comorbidity overall. Even for significant hits, deciphering the precise biological functionality through which variants influence both traits remains challenging. By demonstrating shared genetic underpinnings across multiple independent large-scale GWAS, our work provides initial evidence towards biological mechanisms that may reciprocally link esophageal cancer and chronic lung conditions. Future research should explore whether lifestyle or pharmaceutical interventions modifying genetic risk could mitigate progression across both diseases. With further confirmation in diverse cohorts and functional validation, clinically translating findings could enable earlier targeted screening and prevention efforts for those most vulnerable to this comorbidity.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to acknowledge the reviewers for their helpful comments on this paper.
Funding
This study was supported by Science and Technology Plan Project of Hebei Province (No. 223777106D), Youth Project of the National Health Commission (No. 20230108), Hebei Provincial Government sponsored the Project Of Training Excellent Talents in Clinical Medicine (No. 303-2022-27-37) and Hebei Provincial 2024 government-funded Clinical Medicine Excellent Talents Training Projects (No. ZF2024177).
Data availability statement
The original contributions presented in the study are publicly available. Lung disease data sources: Full joint meta-analysis summary statistics and GBMI meta-analysis results are available for download at https://www.globalbiobankmeta.org/resources and can be browsed at the PheWeb Browser http://results.globalbiobankmeta.org. Esophageal cancer data sources: https://www.ebi.ac.uk/gwas/studies/GCST003739. Further inquiries can be directed to the corresponding authors.
Author contributions
Xiaoliang Duan and Shujun Li designed the study. Dengfeng Zhang and Yu Zhou downloaded and analyzed the data. Dengfeng Zhang and Yishuai Li wrote the manuscript. Jing Li, Longyu Zhu and Tianxing Lu revised the manuscript. The final manuscript has been approved by all authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: a Cancer Journal For Clinicians. 2021;71:209-49
2. Venkatesan P. GOLD COPD report: 2024 update. Lancet Respir Med. 2024;12:15-6
3. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA. et al. Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients With Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA. 2018;319:2299-307
4. Stempel DA, Szefler SJ. Is the assessment of asthma treatment efficacy sufficiently comprehensive? J Allergy Clin Immunol. 2024;153:629-36
5. Zhou J, Fang P, Liang Z, Li X, Luan S, Xiao X. et al. Causal relationship between lung diseases and risk of esophageal cancer: insights from Mendelian randomization. J Cancer Res Clin Oncol. 2023;149:15679-86
6. Cameron ME, Ayzengart AL, Oduntan O, Judge SM, Judge AR, Awad ZT. Low Muscle Mass and Radiodensity Associate with Impaired Pulmonary Function and Respiratory Complications in Patients with Esophageal Cancer. J Am Coll Surg. 2023;236:677-84
7. Rong Y, Hao Y, Xue J, Li X, Li Q, Wang L. et al. Comparison of complications and long-term survival after minimally invasive esophagectomy versus open esophagectomy in patients with esophageal cancer and chronic obstructive pulmonary disease. Frontiers In Oncology. 2022;12:934950
8. Li N, Sohal D. Current state of the art: immunotherapy in esophageal cancer and gastroesophageal junction cancer. Cancer Immunol Immunother. 2023;72:3939-52
9. Garcia-Ryde M, van der Burg NMD, Larsson CE, Larsson-Callerfelt A-K, Westergren-Thorsson G, Bjermer L. et al. Lung Fibroblasts from Chronic Obstructive Pulmonary Disease Subjects Have a Deficient Gene Expression Response to Cigarette Smoke Extract Compared to Healthy. Int J Chron Obstruct Pulmon Dis. 2023;18:2999-3014
10. Pacheco Da Silva E, Varraso R, Lenzotti A-M, Fezeu LK, Sit G, Galan P. et al. Household Use of Green Cleaning Products, Disinfecting Wipes, and Asthma Control Among Adults. J Allergy Clin Immunol Pract. 2024 12
11. Kato M, Mochizuki H, Kama Y, Kusuda S, Okada K, Yoshihara S. et al. Palivizumab prophylaxis in preterm infants and subsequent wheezing/asthma: 10-year follow-up study. Pediatr Pulmonol. 2024;59:743-9
12. Mosquera-Rendón J, Moreno-Herrera CX, Robledo J, Hurtado-Páez U. Genome-Wide Association Studies (GWAS) Approaches for the Detection of Genetic Variants Associated with Antibiotic Resistance: A Systematic Review. Microorganisms. 2023 11
13. Xu Z, Wu X, Xiao C, Zhang W, Yan P, Yang C. et al. Observational and genetic analyses of the bidirectional relationship between depression and hypertension. J Affect Disord. 2024;348:62-9
14. Louck LE, Cara KC, Klatt K, Wallace TC, Chung M. The Relationship of Circulating Choline and Choline-Related Metabolite Levels with Health Outcomes: A Scoping Review of Genome-Wide Association Studies and Mendelian Randomization Studies. Adv Nutr. 2024;15:100164
15. Gomes B, Singh A, O'Sullivan JW, Schnurr TM, Goddard PC, Loong S. et al. Genetic architecture of cardiac dynamic flow volumes. Nat Genet. 2024;56:245-57
16. Wang A, Shen J, Rodriguez AA, Saunders EJ, Chen F, Janivara R. et al. Characterizing prostate cancer risk through multi-ancestry genome-wide discovery of 187 novel risk variants. Nat Genet. 2023;55:2065-74
17. Cui G, Li S, Ye H, Yang Y, Jia X, Lin M. et al. Gut microbiome and frailty: insight from genetic correlation and mendelian randomization. Gut Microbes. 2023;15:2282795
18. Speed D, Balding DJ. SumHer better estimates the SNP heritability of complex traits from summary statistics. Nat Genet. 2019;51:277-84
19. Stauffer E-M, Bethlehem RAI, Dorfschmidt L, Won H, Warrier V, Bullmore ET. The genetic relationships between brain structure and schizophrenia. Nat Commun. 2023;14:7820
20. Partanen JJ, Häppölä P, Zhou W, Lehisto AA, Ainola M, Sutinen E. et al. Leveraging global multi-ancestry meta-analysis in the study of idiopathic pulmonary fibrosis genetics. Cell Genom. 2022;2:100181
21. Gharahkhani P, Fitzgerald RC, Vaughan TL, Palles C, Gockel I, Tomlinson I. et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett's oesophagus: a large-scale meta-analysis. Lancet Oncol. 2016;17:1363-73
22. Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R. et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236-41
23. Ning Z, Pawitan Y, Shen X. High-definition likelihood inference of genetic correlations across human complex traits. Nat Genet. 2020;52:859-64
24. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO. et al. A global reference for human genetic variation. Nature. 2015;526:68-74
25. Open-source ImmGen. mononuclear phagocytes. Nat Immunol. 2016;17:741
26. Ray D, Chatterjee N. A powerful method for pleiotropic analysis under composite null hypothesis identifies novel shared loci between Type 2 Diabetes and Prostate Cancer. PLoS Genetics. 2020;16:e1009218
27. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219
28. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826
29. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-50
30. Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40:755-64
31. Thompson SG, Sharp SJ. Explaining heterogeneity in meta-analysis: a comparison of methods. Stat Med. 1999;18:2693-708
32. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32:377-89
33. Patel A, Ye T, Xue H, Lin Z, Xu S, Woolf B. et al. MendelianRandomization v0.9.0: updates to an R package for performing Mendelian randomization analyses using summarized data. Wellcome Open Res. 2023;8:449
34. Sharma JR, Dubey A, Yadav UCS. Cigarette smoke-induced galectin-3 as a diagnostic biomarker and therapeutic target in lung tissue remodeling. Life Sci. 2024;339:122433
35. Wang T, Duan W, Jia X, Huang X, Liu Y, Meng F. et al. Associations of combined phenotypic ageing and genetic risk with incidence of chronic respiratory diseases in the UK Biobank: a prospective cohort study. Eur Respir J. 2024 63
36. Rezaeeyan H, Arabfard M, Rasouli HR, Shahriary A, Gh BFNM. Evaluation of common protein biomarkers involved in the pathogenesis of respiratory diseases with proteomic methods: A systematic review. Immun Inflamm Dis. 2023;11:e1090
37. Chen C-H, Wang C-Y, Chen C-Y, Wang Y-H, Chen K-H, Lai C-C. et al. The influence of prior use of inhaled corticosteroids on COVID-19 outcomes: A systematic review and meta-analysis. PloS One. 2024;19:e0295366
38. Hirons B, Rhatigan K, Kesavan H, Turner RD, Birring SS, Cho PSP. Cough in chronic lung disease: a state of the art review. J Thorac Dis. 2023;15:5823-43
39. Wang X, Wright Z, Wang J, Roy S, Fass R, Song G. Elucidating the Link: Chronic Obstructive Pulmonary Disease and the Complex Interplay of Gastroesophageal Reflux Disease and Reflux-Related Complications. Medicina (Kaunas). 2023 59
40. Gémes N, Balog JÁ, Neuperger P, Schlegl E, Barta I, Fillinger J. et al. Single-cell immunophenotyping revealed the association of CD4+ central and CD4+ effector memory T cells linking exacerbating chronic obstructive pulmonary disease and NSCLC. Front Immunol. 2023;14:1297577
41. Khoj L, Zagà V, Amram DL, Hosein K, Pistone G, Bisconti M. et al. Effects of cannabis smoking on the respiratory system: A state-of-the-art review. Respir Med. 2024;221:107494
42. Niu J, Guo W, Lu A, Han G, Wang G, Peng B. et al. Comparison with gastric cancer-associated genes reveals the role of ferroptosis-related genes in eosinophils of asthma patients: A bioinformatic study. Medicine. 2023;102:e35002
43. Zhang X, Gao L, Meng H, Zhang A, Liang Y, Lu J. Obesity alters immunopathology in cancers and inflammatory diseases. Obes Rev. 2023;24:e13638
44. Wang H-K, Wei Q, Yang Y-L, Lu T-Y, Yan Y, Wang F. Clinical usefulness of the lymphocyte-to-monocyte ratio and aggregate index of systemic inflammation in patients with esophageal cancer: a retrospective cohort study. Cancer Cell International. 2023;23:13
45. Liu J, Gao D, Li J, Hu G, Liu J, Liu D. The Predictive Value of Systemic Inflammatory Factors in Advanced, Metastatic Esophageal Squamous Cell Carcinoma Patients Treated with Camrelizumab. OncoTargets and Therapy. 2022;15:1161-70
46. Ji X, Bossé Y, Landi MT, Gui J, Xiao X, Qian D. et al. Identification of susceptibility pathways for the role of chromosome 15q25.1 in modifying lung cancer risk. Nat Commun. 2018;9:3221
47. Kaur-Knudsen D, Bojesen SE, Tybjærg-Hansen A, Nordestgaard BG. Nicotinic acetylcholine receptor polymorphism, smoking behavior, and tobacco-related cancer and lung and cardiovascular diseases: a cohort study. J Clin Oncol. 2011;29:2875-82
48. Young RP, Hopkins RJ, Hay BA, Epton MJ, Black PN, Gamble GD. Lung cancer gene associated with COPD: triple whammy or possible confounding effect? Eur Respir J. 2008;32:1158-64
49. Kim W, Prokopenko D, Sakornsakolpat P, Hobbs BD, Lutz SM, Hokanson JE. et al. Genome-Wide Gene-by-Smoking Interaction Study of Chronic Obstructive Pulmonary Disease. Am J Epidemiol. 2021;190:875-85
50. Hirota T, Takahashi A, Kubo M, Tsunoda T, Tomita K, Doi S. et al. Genome-wide association study identifies three new susceptibility loci for adult asthma in the Japanese population. Nat Genet. 2011;43:893-6
51. Wang M, Li Y, Xiao Y, Yang M, Chen J, Jian Y. et al. Nicotine-mediated OTUD3 downregulation inhibits VEGF-C mRNA decay to promote lymphatic metastasis of human esophageal cancer. Nat Commun. 2021;12:7006
52. Li Y, Wang M, Yang M, Xiao Y, Jian Y, Shi D. et al. Nicotine-Induced ILF2 Facilitates Nuclear mRNA Export of Pluripotency Factors to Promote Stemness and Chemoresistance in Human Esophageal Cancer. Cancer Research. 2021;81:3525-38
53. Truong LN, Wilson Santos E, Zheng Y-M, Wang Y-X. Rieske Iron-Sulfur Protein Mediates Pulmonary Hypertension Following Nicotine/Hypoxia Coexposure. Am J Respir Cell Mol Biol. 2024;70:193-202
54. Błach J, Siedliński M, Sydor W. Immunology in COPD and the use of combustible cigarettes and heated tobacco products. Eur J Med Res. 2023;28:397
55. Broekman W, Roelofs H, Zarcone MC, Taube C, Stolk J, Hiemstra PS. Functional characterisation of bone marrow-derived mesenchymal stromal cells from COPD patients. ERJ Open Res. 2016 2
56. Katoh M, Katoh M. AP1- and NF-kappaB-binding sites conserved among mammalian WNT10B orthologs elucidate the TNFalpha-WNT10B signaling loop implicated in carcinogenesis and adipogenesis. Int J Mol Med. 2007;19:699-703
57. Ware SA, Kliment CR, Giordano L, Redding KM, Rumsey WL, Bates S. et al. Cell-free DNA levels associate with COPD exacerbations and mortality. Respir Res. 2024;25:42
58. Yan L, Cui Y, Feng J. Biology of Pellino1: a potential therapeutic target for inflammation in diseases and cancers. Front Immunol. 2023;14:1292022
59. Lan Y, Ma Q, Luo G, Yang H, Li Y, Zhang Q. Epicardial adipose tissue in patients with chronic obstructive pulmonary disease: systematic review with meta-analysis and trial sequential analysis. BMC Pulm Med. 2023;23:241
60. Capitão C, Coutinho D, Neves PM, Capelas ML, Pimenta NM, Santos T. et al. Protein intake and muscle mass maintenance in patients with cancer types with high prevalence of sarcopenia: a systematic review. Support Care Cancer. 2022;30:3007-15
61. Sanchez-Azofra A, Gu W, Masso-Silva JA, Sanz-Rubio D, Marin-Oto M, Cubero P. et al. Inflammation biomarkers in OSA, chronic obstructive pulmonary disease, and chronic obstructive pulmonary disease/OSA overlap syndrome. J Clin Sleep Med. 2023;19:1447-56
Author contact
![]() Corresponding authors: Shujun Li, Yishuai Li, Xiaoliang Duan. Email: lishujun2333com, liyishuai66com, dxlhbchestcom.
Corresponding authors: Shujun Li, Yishuai Li, Xiaoliang Duan. Email: lishujun2333com, liyishuai66com, dxlhbchestcom.