Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2025; 16(4):1040-1050. doi:10.7150/jca.103297 This issue Cite
Research Paper
Characteristics and function of the gut microbiota in patients with rectal neuroendocrine tumors
Yue Gao*, Hongxia Zheng*, Mujie Ye*, Guozhi Zhou, Jinhao Chen, Xinyun Qiang, Jianan Bai, Xintong Lu, Qiyun Tang ![]()
Department of Geriatric Gastroenterology, Neuroendocrine Tumor Center, The First Affiliated Hospital of Nanjing Medical University, No. 300 Guangzhou Road, Nanjing 210029, Jiangsu Province, China.
* These authors contributed equally.
Received 2024-8-1; Accepted 2024-12-1; Published 2025-1-1
Abstract

The gut microbiota plays a significant role in the initiation and progression of tumors, but its role in rectal neuroendocrine tumors (rNETs) remains unclear. Fecal samples were collected from 19 healthy individuals and 21 rNET patients,with the rNET cohort further divided into metastatic (rNETs-M) and non-metastatic (rNETs-nM) groups. Using metagenomic high-throughput sequencing, we analyzed the diversity, species composition, and functional characteristics of the gut microbiota. We applied a random forest model to identify potential microbial biomarkers for predicting rNET and specifically distinguishing rNETs-M cases. Alpha diversity analysis revealed that species diversity was lower in the rNETs group than in the control group. In contrast, the rNETs-M group exhibited higher species diversity than the rNETs-nM group. Beta diversity analysis demonstrated significant differences in community structure between the rNETs and control groups between the rNET-M and rNETs-nM groups. Notably, in the rNETs group, the abundance of potential pathogens such as Escherichia coli and Shigella was elevated.Furthermore, the rNETs-M group exhibited an increased abundance of potential pathogens such as Alistipes. KEGG enrichment analysis indicated that these distinct microbiota play a significant role in environmental information processing, genetic information processing, and metabolic pathways. Random forest analysis and ROC curve results identified Lachnospira pectinoschiza (AUC=0.885), Parasutterella muris (AUC=0.862), Sodaliphilus pleomorphus(AUC=0.956), Methylobacterium iners (AUC=0.971) as biomarkers with significant discriminatory value.
Keywords: rectal neuroendocrine tumors, gut microbiome, liver metastasis, metagenomic sequencing
Introduction
Neuroendocrine neoplasms (NENs) are highly heterogeneous tumors originating from neuroendocrine cells and peptidergic neurons that produce polypeptide hormones and bioactive amines [1]. NENs can occur in any part of the body, with gastroenteropancreatic neuroendocrine tumors (GEP-NENs) being the most common, accounting for 55%-70% of all NENs [2]. In recent years, the incidence of GEP-NENs has continued to rise, increasing from 1.09 per 100,000 in 1973 to 6.98 per 100,000 in 2012, making it the second most common digestive system cancer [3]. The rectum is a frequent site for NENs, and because patients with rectal neuroendocrine tumors (rNETs) often lack obvious clinical symptoms, timely diagnosis and treatment can be challenging [4]. Although the prognosis of rNETs is generally better compared to most tumors, over 20% of patients typically present with distant metastasis at diagnosis, with liver metastasis being the most common site [5]. Studies have shown that patients with liver metastasis have a significantly lower five-year survival rate compared to those without liver metastasis [6].
In recent years, increasing research has shown that microbes play an important role in human health and disease, with the gut microbiota producing many essential metabolites to maintain host and gut homeostasis [7]. With the advancement of metagenomic sequencing technology, studies have found that the gut microbiota can promote tumor initiation and progression through DNA mutations, activation of oncogenic pathways, promotion of chronic inflammation, supplementation of systemic pathways, and initiation of metastasis [8]-[10]. Research on gut microbiota in NENs is increasing, with metagenomic sequencing of fecal samples from patients with midgut NENs and healthy controls showing differences in gut microbiota composition and function, indicating a potential link between the gut microbiota and midgut NENs [11]. A study found that rNETs patients exhibited abnormal depletion of microbial species and weakened connectivity, as well as abnormal accumulation of lipids and lipid-like molecules, suggesting that the occurrence of rNETs may be related to a weakened gut microbiota activity in energy metabolism, vitamin biosynthesis, and transport [12].
In this study, we used metagenomic sequencing to analyze the species-level composition of gut microbiota and their potential functions and metabolic characteristics in patients with rNETs, as well as in those with liver metastasis. The results may help identify biomarkers and potential therapeutic targets for rNETs with liver metastasis.
Materials and methods
Research subjects
Stool samples were obtained from 21 patients at the Neuroendocrine Tumor Center, the First Affiliated Hospital of Nanjing Medical University, and 19 healthy individuals at the physical examination center.
The inclusion criteria for the control group were as follows: (1) age >18 years old and <80 years old; (2) negative results on OC-qFIT ultra-sensitive quantitative fecal occult blood tests. The inclusion criteria for the rNETs group were as follows: (1) Pathologically diagnosed with rNETs; (2) Imaging diagnosis of liver space-occupying lesions.
Exclusion criteria are as follows: (1) Patients who had taken antibiotics, corticosteroids, or probiotics within 3 months before sample collection; (2) Patients who have undergone abdominal surgery or other invasive treatments within 3 months before sample collection; (3) Patients who had taken cathartic agents or underwent enema treatment before sample collection; (4) Personal history of cancer, family history or personal history of inflammatory bowel disease; (5) lactation or pregnancy; (6) preoperative radiotherapy or neoadjuvant chemotherapy; (7) Patients who have undergone fecal bacteria transplantation in the past; (8) Individuals with mental illness; (9) incomplete information or lack of informed consent; (10)Established or suspected history of alcohol or drug abuse.
Fecal sample collection
In accordance with the predefined inclusion and exclusion criteria, a total of 40 fecal samples meeting the specified standards were collected and promptly preserved in a -80 ℃ refrigerator for future analysis. Before sample collection, all participating patients provided informed consent. This study has received approval from the Ethics Review Committee of the First Affiliated Hospital of Nanjing Medical University.
Experimental methods
DNA extraction
Genomic DNA from the sample was extracted using the HiPure Bacterial DNA Kit (Guangzhou, China) following the manufacturer's protocol. The quality of the DNA was assessed using Qubi (Thermo Fisher Scientific, Waltham, MA) and Nanodrop (Thermo Fisher Scientific, Waltham, MA).
Illumina sequencing
The qualified genomic DNA was initially fragmented to 350 bp through acoustic shearing, followed by terminal repair, A-tailing, and ligation of Illumina sequencing adapters using the NEBNext® Ultra™DNA Library Prep Kit (NEB, USA). Subsequently, 300-400bp DNA fragments were amplified and enriched via PCR. The resulting PCR products were purified using the AMPure XP system (Beckman Coulter, Brea CA., USA). Sequencing libraries were evaluated with an Agilent 2100 bioanalyzer (Agilent Santa Clara CA.) and quantified using real-time PCR before sequencing on an Illumina Novaseq 6000 sequencer employing a PE 150 sequencing strategy.
Statistical method
Quality assessment of intestinal flora sequencing
The raw data obtained after sequencing were filtered based on the following criteria: (1) removal of reads containing adapters; (2) elimination of reads with an N ratio exceeding 10%; (3) exclusion of low-quality reads, defined as those where the number of bases with a quality score Q≤20 accounted for more than 50% of the entire read. All samples' Q20 and Q30 values exceeded 95%, indicating high data quality suitable for subsequent sequencing.
Analysis of species composition
In the R language, the VennDiagram package is utilized to construct Venn diagrams illustrating common endemic species among species in different groups and to distinguish between species unique to each group, disregarding abundance and focusing solely on presence or absence. Visualizations were created for two species groups at both phylum and genus levels, while circus plots were employed to demonstrate abundance correlations between samples and species.
Alpha diversity analysis
Chao1, ACE, Shannon, Simpson index were calculated using Python scikit-bio package [13](version 0.5.6). Alpha index comparison between groups was calculated by Welch's t-test and Wilcoxon rank test in R project Vegan package[13]. Alpha index comparison among groups was computed by Tukey's HSD test and Kruskal-Wallis H test in R project Vegan package[13].
Beta diversity analysis
Bray-curtis distance matrix based on gene/taxon/function abundance was generated by R Vegan package [13]. Multivariate statistical techniques including PCA (principal component analysis), PCoA (principal coordinates analysis) and NMDS (non-metric multidimensional scaling) of Bray- curtis distances were calculated using R vegan package [13] and plotted using R ggplot2 package [14]. Adonis (also called Permanova) and Anosim test was calculated using R project Vegan package [13]. Heatmap graph were plotted using R Pheatmap package[15].
Difference analysis
Basic analysis
Between groups Venn analysis was performed in R project VennDiagram package [16] and upset plot was performed in R project UpSetR package [17]to identify unique and common species or functions. Species/functions comparison between groups was calculated by welch's t-test and wilcoxon rank test in R project Vegan package[13]. Species/functions comparison among groups was computed by ANOVA (analysis of variance) in R project Vegan package[13].
Personalized analysis
Species comparison between groups was calculated by Metastats (version 20090414) [18]. Differentially enriched KEGG pathways were identified according to their reporter score from the Zscores of individual Kos (KEGG Orthologs). An absolute value of reporter score = 1.96 or higher (95% confidence according to a normal distribution) was used as a detection threshold for pathways that differed significantly in abundance [19]. Biomarker features of species and functions in each group were screened by LEfSe software (version 1.0) [20]. Ternary plot of species was plotted using R ggtern package [21] based on tukey HSD test using R Vegan package[13].
Results
Library sequencing data and gene prediction
The sequencing yielded a total of 3,743,818,196 original reads. Following pre-processing, the dataset retained 3,700,381,650 clean reads available for analysis, averaging 92,509,541 reads per sample (Figure S1A). The average number of bases per sample before quality control was 14,039,318,235 bp; after quality control, it was reduced to an average of 13,829739561 bp. 345743489 high-quality reads were obtained following host sequence screening (Figure S1B). Subsequently, the giant hit software was utilized for assembling the effective readbeing 2724.575, with an average continuous length of each sample being 2724.575 and 2724.575 bp without significant inter-group differences (Figure S1C).
Diversity of the intestinal microbiota
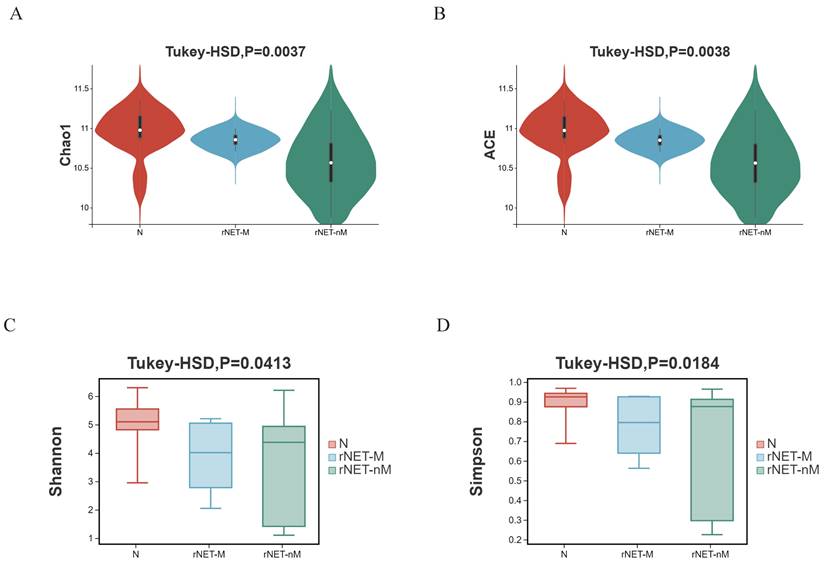
Alpha diversity illustrates the box chart and P-values for comparing indicators: (A) Chao1 index, (B) ACE index, (C) Shannon index, and (D) Simpson index. The Chao1 and ACE indices exclusively assess species richness, with higher values indicating greater diversity. In contrast, The Shannon index and Simpson index provide a comprehensive reflection of species richness and evenness. A higher value indicates a greater sample balance. Comparisons were conducted between the rNETs group and the control group, the rNETs-M group and the rNETs-nM group, and among the rNET group, the rNETs-M group, and the control group. Statistically significant differences in the four indexes were observed between the rNETs and control groups (Fig. S2). When comparing the stool samples of patients in the rNETs-M group and rNETs-nM group, a statistically significant difference was observed in the ACE index and Chao1 index. However, no significant difference was found in the Shannon index and Simpson index (Fig. S3). Statistically significant differences were observed among the three groups for all four indexes (Fig. 1).
(A-D) Illustrates the alpha diversity indexes, including the Chao1 index (A), ACE index (B), Shannon index (C), and Simpson index (D) of the rNETs group, rNETs-M group, and control group.
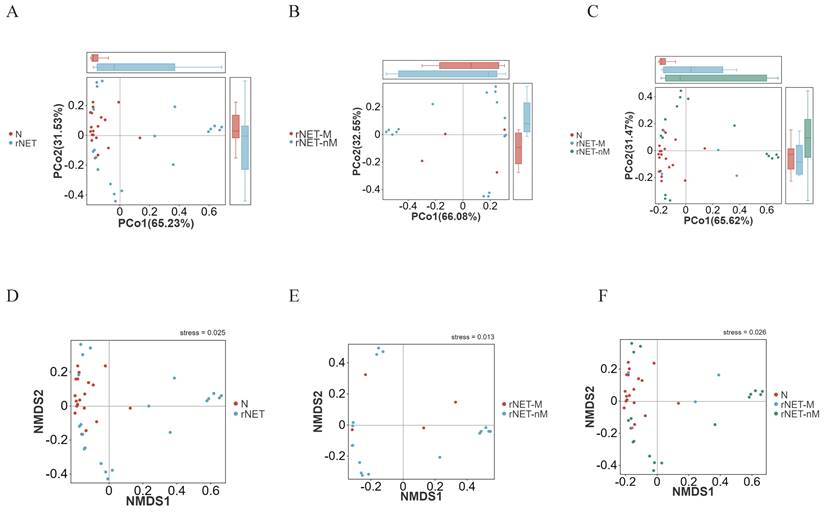
The structural similarity was explored using PCoA analysis of beta diversity. Factor 1 explained 65.23% of the total variance in both the rNETs group and control group (Fig. 2A). In comparison, Factor 2 accounted for 31.53% of the total variance. In contrast, Factor 1 explained 32.55% of the total variance in the rNETs-M group and rNETs-nM (Fig. 2B), with Factor 2 accounting for 66.08% of the total variation. The rNETs, rNETs-M, and control groups (Fig. 2C) exhibited two primary axes (factors). Specifically, Factor 1 contributed 65.62% of the total variation, whereas Factor 2 accounted for 31.47%. Subsequently, NMDS analysis was conducted based on Bray-Curtis matrix obtained from PCoA analysis. The stress values for all three NMDS analysis groups, rNETs group and control group (Fig. 2D), rNETs-M group and rNETs-nM (Fig. 2E), as well as rNETs group, rNETs-M group and control group (Fig. 2F) were all below <0.2 indicating reliable results from variance analysis.
(A-C) Principal coordinates analysis (PCoA) plot depicting (A) N vs rNETs, (B) rNETs-M vs rNETs-nM, and (C) N vs rNETs-M vs rNETs-nM. (D-E) Presents the Nonmetric Multidimensional Scaling (NMDS) plot, with (D) N vs. rNETs, (E) rNETs-M vs. rNETs-nM, and (F) N vs. rNETs-M vs. rNETs-nM.
Characteristics of the composition of intestinal microbial species
Welch's t-test was employed to compare the differences between the rNETs group and the control group. At the phylum level (Fig. 3A), Pseudomonadota exhibited enrichment in the tumor group. At the same time, Bacteroidota was more prevalent in the control group. At the genus level (Fig. 3B), bacteria enriched in the control group included Eubacterium, Dorea, Phocaeicola, Alistipes, and Lachnospira. Conversely, Escherichia and Shigella were present in higher abundance in the tumor group. Finally, at the species level (Fig. 3C), Alistipes putredinis, Bacteroides uniformis, and Dorea longicatena were found to be enriched in the control group. In contrast, Escherichia coli, Shigella sonnei, and Eggerthella lenta were identified as predominant strains within the tumor group.
(A-E) Displays the Welch t-test results for microbial species in the intestinal microbiota of the groups N vs rNETs at the phylum levels (A), N vs rNETs at the genus levels(B), N vs rNETs at the species levels(C), rNETs-nM vs rNETs-M at the genus level (D), rNETs-nM vs rNETs-Mat the species level (E).
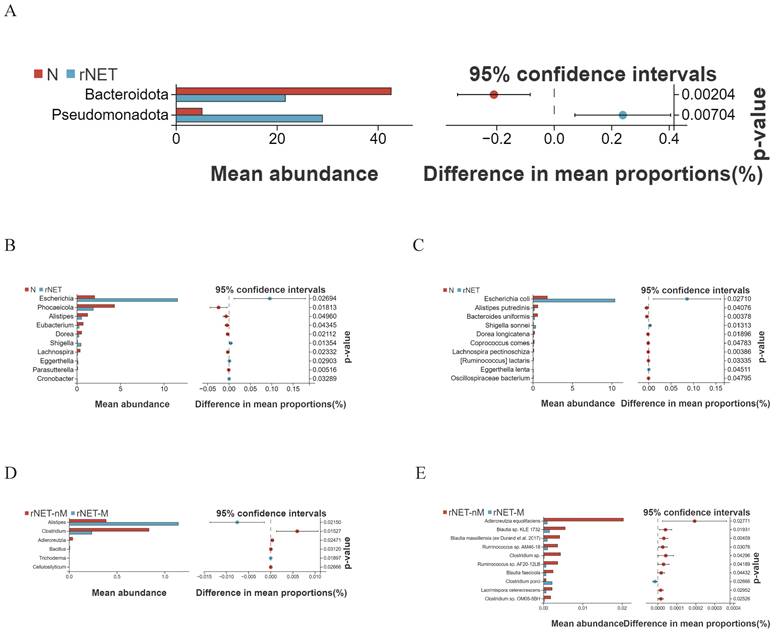
Welch's t-test revealed distinct differences in flora between the rNETs-M and rNETs-nM groups. No statistically significant species were identified at the phylum level. At the genus level (Fig. 3D), Clostridium was found to be abundant in the rNETs-nM group, while Alistipes exhibited higher abundance in the rNETs-M group. At the species level (Fig. 3E), bacteria enriched in the rNETs-nM group included Adlercreutzia equolifaciens, Blautia faecicola, and Clostridium porci, whereas these were more prevalent in the rNETs-M group.
In the case of multiple groups, to elucidate species differences among different groups, we constructed the Wayne diagram (Fig. S4) based on species abundance information from the samples, depicting taxonomic distribution at phylum, genus, and species levels to illustrate shared endemic information across samples. At the phylum level (Fig. S4A), 62 phyla were common to all three groups, with 13 unique to the healthy group and 4 bacteria genera unique to the primary group; no specific bacteria were found in the liver metastasis group. At the genus level (Fig. S4B), 714 bacteria genera were common across all three groups, with 102 unique to the healthy group and 73 unique to the primary group. Five bacteria genera were endemic in the liver metastasis group. At the species level (Fig. S4C), there were 2773 common bacterial species; among these, 472 were endemic in the healthy group and 392 in primary tumors, while only 33 bacterial genera showed endemism in liver metastases. Trend analysis revealed no gradual increase at the phylum level but demonstrated a gradual increase at the genus level (Table 1) and species level (Table 2); these differences were statistically significant.
Abundance and corresponding p-values of bacteria genera across incremental levels
| Genus | N | rNETs-nM | rNETs-M | p-value | q-value |
|---|---|---|---|---|---|
| Renibacterium | 0 | 1.00E-06 | 6.00E-06 | 0.036225282 | 0.360552309 |
| Gordonia | 1.20E-05 | 3.70E-05 | 7.00E-05 | 0.010743804 | 0.235198286 |
| Oenococcus | 1.10E-05 | 0.000105 | 0.000477 | 0.044365664 | 0.406185644 |
| Agathobacter | 0.0964947 | 0.287 | 0.4364 | 0.022394941 | 0.288912813 |
| Eisenbergiella | 0.003993 | 0.015599 | 0.04345 | 0.038245884 | 0.36733829 |
| Paraprevotella | 0.027287 | 0.05427 | 0.234523 | 0.004934687 | 0.235198286 |
| Parabacteroides | 0.577711 | 0.700713 | 1.615925 | 0.02339924 | 0.289841212 |
Abundance and corresponding p-values of bacteria species across incremental levels.
| Species | N | rNETs-nM | rNETs-M | p-value | q-value |
|---|---|---|---|---|---|
| Renibacterium salmoninarum | 0 | 1.00E-06 | 6.00E-06 | 0.036225282 | 0.359924545 |
| Gordonia amicalis | 1.10E-05 | 3.20E-05 | 5.20E-05 | 0.019375022 | 0.283663272 |
| Collinsella sp. AF25-2LB | 3.80E-05 | 0.000288 | 0.00106 | 0.026768661 | 0.317168087 |
| Eggerthella sp. 1_3_56FAA | 8.30E-05 | 0.00018 | 0.00024 | 0.011361349 | 0.205207055 |
| Enterococcus aquimarinus | 0 | 4.00E-06 | 2.10E-05 | 0.039325427 | 0.377869186 |
| Clostridium sp. Marseille-P3244 | 0.000349 | 0.000534 | 0.001267 | 0.037507518 | 0.369175123 |
| Agathobacter rectalis | 0.0964326 | 0.286765 | 0.4363 | 0.022386961 | 0.300935721 |
| Eisenbergiella tayi | 0.001754 | 0.004429 | 0.031125 | 0.027771232 | 0.324154178 |
| Lachnoclostridium sp. An118 | 0.000117 | 0.000296 | 0.000776 | 0.010485772 | 0.202037667 |
| Bacteroides fragilis | 0.407747 | 0.840575 | 5.681125 | 0.005329296 | 0.202037667 |
| Bacteroides thetaiotaomicron | 0.0386905 | 0.208833 | 0.534475 | 0.015736526 | 0.258623855 |
| Bacteroides uniformis | 0.0640721 | 0.20402 | 0.242825 | 0.003417602 | 0.202037667 |
| Phocaeicola coprocola | 0.0178226 | 0.089142 | 1.172925 | 0.007671662 | 0.202037667 |
| Alistipes putredinis | 0.0668617 | 0.152854 | 0.364675 | 0.012966307 | 0.223445597 |
| Fusobacterium varium | 0.000929 | 0.006405 | 0.02401 | 0.007214647 | 0.202037667 |
Differential functional analysis of the gut microbiota
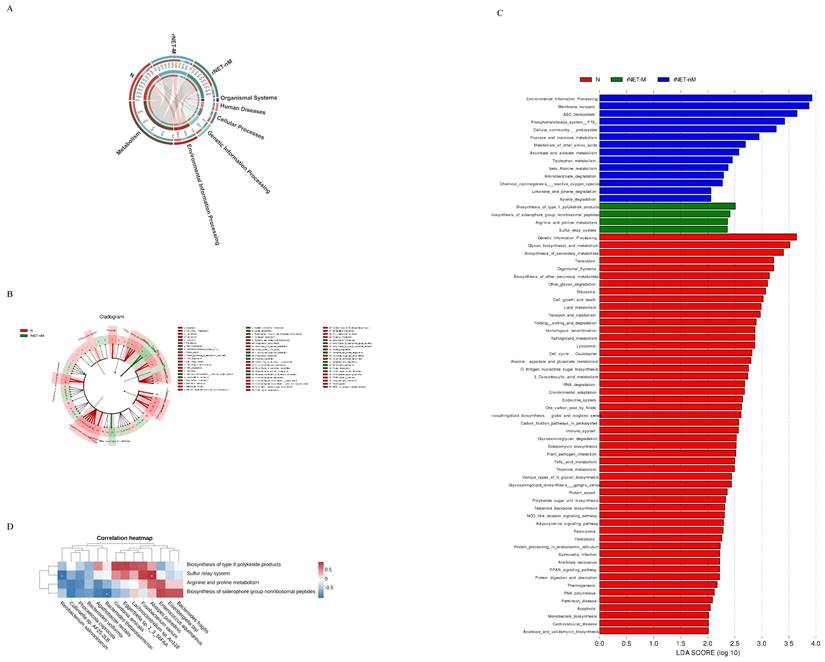
In terms of exploring the functional differences of the flora, we annotated the total number of genes in all the statistical samples to the pathway database. Our analysis revealed that most of the genes were associated with metabolic functions (Fig. 4A). was depicted as a branching tree graph (Fig. 4B). LEfSe was employed to assess and compare functional differences across each group. Differential function analysis using KEGG database identified significant pathways related to Environmental Information Processing, Membrane Transport, and ABC Transporters (Fig. 4C). Furthermore, within the metastasis group, key pathways included Biosynthesis_of_type_II_polyketide_products, Biosynthesis_of_siderophore_group_nonribosomal_peptides, Arginine_and_proline_metabolism, and Sulfur_relay_system. Spearman correlation analysis (Fig. 4D) was conducted to examine associations between bacterial flora at increasing species levels and rNETs-M group bacterial flora. The results indicated a positive correlation between Alistipes putredinis and the Sulfur_relay_system pathway while Renibacterium salmoninarum showed a negative correlation with this pathway. Additionally, Bacteroides thetaiotaomicron negatively correlated with the Biosynthesis_of_siderophore_group_nonribosomal_peptides pathway.
(A) Cladogram analysis of functional differences among three groups, (B) Cladogram analysis of functions among three groups, and (C) LDA score analysis of functions within the three groups. Additionally, (D) Heatmap showing the correlation between gut microbiota and metabolic functions.
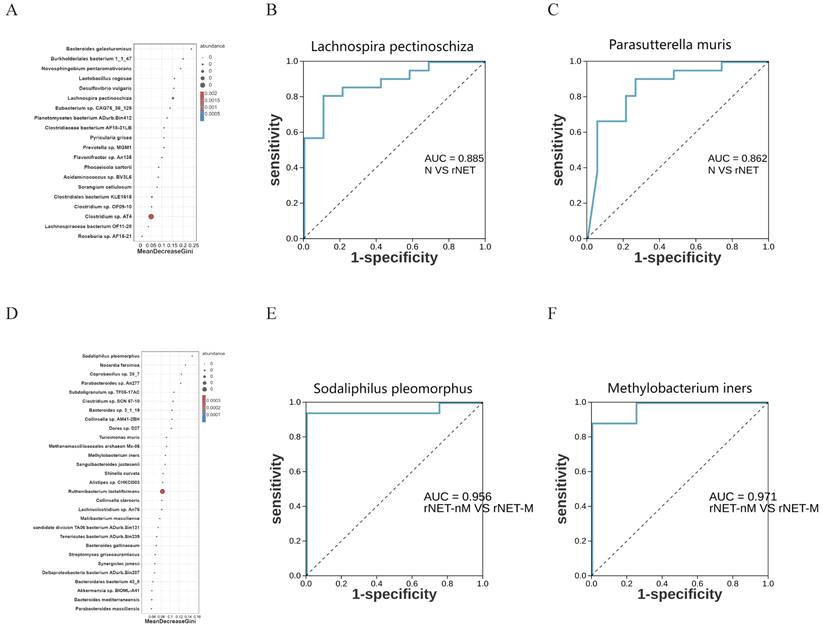
Random forest model for vegetation and receiver operating characteristic (ROC) analysis
Random forest model analysis was employed to investigate the potential of microbial flora as a predictive factor for rNETs and liver metastases. To further assess the utility of intestinal microbes as biomarkers, the area under the curve (AUC) was utilized for analysis. At the species level, random forest model analysis revealed that Lachnospira pectinoschiza was pivotal in identifying rNETs (Fig. 5A). Receiver Operating Characteristic (ROC) curve analysis was conducted for different species at the species level to evaluate the sensitivity of the differential flora in diagnosing rNETs, as indicated by the area under the curve. Lachnospira pectinoschiza exhibited an AUC of 0.885 [95%CI (0.781-0.988)] (Fig. 5B).Parasutterella muris demonstrated an AUC of 0.862 [95% CI (0.744-0.981)](Fig. 5C), indicating its potential as a significant biomarker for distinguishing the rNETs group from the control group. Furthermore, the random forest analysis highlighted Sodaliphilus pleomorphus as the most influential in differentiating rNETs-M (Fig. 5D). Receiver Operating Characteristic (ROC) curve analysis was conducted at the species level, revealing that Sodaliphilus pleomorphus achieved an AUC of 0.956 [95% CI (0.865-1)] (Fig. 5E), and Methylobacterium iners attained an AUC of 0.971 [95% CI (0.901-1)] (Fig. 5F). These findings underscore their significance as crucial biomarkers for identifying the rNETs-nM group.
Illustrates the random forest model and ROC analysis of flora. Panel A shows the random forest model of the control, rNETs group, and groups, while panel B presents the ROC analysis of Lachnospira pectinoschiza. Additionally, panel C displays the Parasutterella ROC analysis of muris, and panel D depicts the random forest model of the rNETs-nM group and rNETs-M group. Furthermore, panels E and F showcase the ROC analyses of Sodaliphilus pleomorphic and Methylobacterium liners, respectively.
Discussion
As increasing evidence suggests a link between gut microbiota dysbiosis and tumors, NENs have also garnered widespread attention. Compared to previous studies based on 16S rRNA gene sequencing, our research is the first to use metagenomic sequencing analysis to describe the gut microbiota composition at the species level in rNETs patients with liver metastasis, revealing the gene functions and potential metabolic pathways of gut microbes.
Compared to prior research [12], our study found significant differences in both α-diversity and β-diversity between the rNETs group and the control group, suggesting notable shifts in microbial community structure. Compared to healthy individuals, the richness and diversity of gut microbiota in tumor patients were significantly reduced. The rNETs-M group showed higher Chao1 and ACE indices than the rNETs-nM group, indicating increased gut microbiota diversity in patients with liver metastasis, which may be related to tumor metastasis to the liver. Song W et al. [22] found that, compared to primary liver cancer, the relative abundance of Fusobacterium nucleatum was significantly higher in liver metastases from colorectal cancer, and that Fusobacterium nucleatum and other symbiotic bacteria such as Bacteroides and Prevotella species colonize liver metastases as cancer cells spread. This suggests that an increase in Fusobacterium nucleatum and symbiotic bacteria may promote colorectal cancer growth and liver metastasis.
There is a dynamic balance between the microbiota and host gastrointestinal system health and disease. When microbial homeostasis is disrupted, it can lead to disease onset[23]. At the phylum level, the dominant bacterial phyla in the gut microbiota of the rNETs group and the control group are Bacteroidota and Pseudomonadota. In the rNETs group, Pseudomonadota is upregulated, while Bacteroidota is downregulated. At the genus level, gut microbiota in the rNETs group, including Escherichia and Shigella, are upregulated. At the species level, Escherichia coli, Shigella sonnei, and Eggerthella lenta are upregulated, which may be associated with various diseases. Previous research data indicate an association between inflammatory bowel disease (IBD) and intestinal NENs, with tumor risk increasing sevenfold compared to the general population [24]. Studies have reported that the onset of IBD is related to the depletion of multiple beneficial bacteria, particularly the reduction of certain symbiotic bacteria, such as an imbalance in Clostridia and Firmicutes proportions and decreased Bacteroidota abundance [25]. Our results suggest that Bacteroidota is downregulated in the rNETs group compared to the control group. Therefore, whether supplementing beneficial Bacteroidota can reduce the incidence of rNETs requires further investigation. Previous studies have found that Eggerthella lenta is more abundant in high-grade gastroenteropancreatic neuroendocrine tumors (GEP-NETs) than in low-grade GEP-NETs[26], leading us to speculate that Eggerthella lenta may be closely associated with tumor development. Clinically, rNETs patients may experience non-specific symptoms such as abdominal pain and diarrhea[27], which may be related to Shigella sonnei invading host cells, mediating multiple signaling pathways, recruiting inflammatory factors and immune cells, colonizing, and damaging colonic epithelial cells[28][29]. The abundance of Alistipes is elevated in the liver metastasis group; Lu Y et al. found that Alistipes could upregulate downstream target genes of β-catenin, activate the Wnt signaling pathway, and promote colorectal cancer metastasis[30]. Whether it can lead to liver metastasis of rNETs still needs further investigation.
To describe functional changes in the gut microbiome, we performed metagenomic functional annotation of KEGG pathways, revealing that metabolic function is the primary pathway in all three groups. LEfSe analysis showed that the 2_Oxocarboxylic_acid_metabolism pathway is upregulated in healthy individuals. Clostridium butyricum can promote the production of short-chain fatty acids (SCFAs) via the 2_Oxocarboxylic_acid_metabolism pathway, thereby repairing the gut barrier in mice with ulcerative colitis[31]. The ABC_transporters pathway is upregulated in the primary tumor group, with studies indicating that multidrug resistance (MDR) in tumor cells to chemotherapy drugs is associated with overexpression of ABC_transporters[32]. This overexpression can promote the efflux of chemotherapeutic drugs and reduce their accumulation within tumor cells[33]. Additionally, ABC_transporters can regulate the tumor immune microenvironment (TIME) by transporting various cytokines, thereby controlling antitumor immunity and sensitivity to anticancer drugs[34]. The Arginine_and_proline_metabolism pathway is significantly upregulated in the liver metastasis group, and changes in gut microbiota structure can induce intracellular toxin accumulation, leading to amino acid dysbiosis and gut barrier dysfunction by interfering with the Arginine_and_proline_metabolism pathway, potentially causing gut diseases[35]. Whether this promotes liver metastasis requires further validation.
Several consensus guidelines suggest that measuring serum CgA concentration is a reliable biomarker for identifying GEP-NENs[36][37]. However, elevated CgA levels have also been observed in some non-GEP-NENs, including lung cancer, breast cancer, and prostate cancer[38]. Previous studies have confirmed the heterogeneity of microbiome characteristics in aggressive disease groups, and an integrated analysis of microbiome markers from multiple cohorts has shown improved accuracy in early screening and diagnosis[39]. We developed a random forest model to confirm the potential of the gut microbiota in predicting rNETs. Lachnospira pectinoschiza and Parasutterella muris can serve as microbiome markers indicative of rNETs, while Sodaliphilus pleomorphus and Methylobacterium iners can be markers indicative of rNETs-M. From a clinical perspective, individuals exhibiting a "high-risk" microbiome profile may require additional colonoscopy to confirm the diagnosis.
This study has some limitations. First, all rNETs patients were from hospitals in a specific region, resulting in a limited sample size. Therefore, further validation through large-scale clinical studies across multiple regions is needed to confirm these preliminary findings. Second, our observations are primarily data-driven and only involve changes in the gut microbiota, so further in vivo and in vitro downstream validation is necessary.
Conclusion
Metagenomic sequencing technology in this study revealed significant disparitiesin intestinal microbiota composition between patients with rNETs and the general population. Certain specific alterations in the flora maybe closely associatedwith the onset and metastasis of rNETs, offering potential insights for the diagnosis and treatment of such tumors.
Supplementary Material
Supplementary figures.
Acknowledgements
Funding
This work was supported by National Natural Science Foundation of China (no.82373180 and 82303453), Science Foundation Project of Ili & Jiangsu Joint Institute of Health (Grant No. yl2020lhms05 and yl2023ms10), Jiangsu Province Hospital (the Frist Affiliated Hospital with Nanjing Medical University) ClinicalCapacity Enhancement Project (Grant No. JSPH-MA-2021-4).
Ethics approval and consent to participate
The feces was obtained with patients inform and was approved by The First Affiliated Hospital of Nanjing Medical University (Jiangsu Province Hospital).
Data availability statement
All of the data of this study are available from the corresponding author.
Author contributions
TQ and YM: conceptualization, methodology, software; GY, ZH, LX and YM: datacuration, writing-original draft preparation; GY, ZH, ZG, CJ, QX, BJ, LX: visualization, investigation; TQ and YM: writing-reviewing and editing; GY, ZH and ZG: software, validation; TQ and YM: supervision. All authors read and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cives M, Strosberg JR. Gastroenteropancreatic Neuroendocrine Tumors. CA Cancer J Clin. 2018;68:471-87
2. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y. et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017;3:1335-42
3. Das S, Dasari A. Epidemiology, Incidence, and Prevalence of Neuroendocrine Neoplasms: Are There Global Differences? Curr Oncol Rep. 2021;23:43
4. Garcia-Carbonero R, Capdevila J, Crespo-Herrero G, Díaz-Pérez JA, Martínez Del Prado MP, Alonso Orduña V. et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): results from the National Cancer Registry of Spain (RGETNE). Ann Oncol. 2010;21:1794-803
5. Riihimäki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. The epidemiology of metastases in neuroendocrine tumors. Int J Cancer. 2016;139:2679-86
6. Frilling A, Modlin IM, Kidd M, Russell C, Breitenstein S, Salem R. et al. Recommendations for management of patients with neuroendocrine liver metastases. Lancet Oncol. 2014;15:e8-21
7. Xue C, Chu Q, Zheng Q, Yuan X, Su Y, Bao Z. et al. Current understanding of the intratumoral microbiome in various tumors. Cell Rep Med. 2023;4:100884
8. Ansari S, Yamaoka Y. Role of vacuolating cytotoxin A in Helicobacter pylori infection and its impact on gastric pathogenesis. Expert Rev Anti Infect Ther. 2020;18:987-96
9. Heng WS, Pore M, Meijer C, Hiltermann TJN, Cheah SC, Gosens R. et al. A unique small cell lung carcinoma disease progression model shows progressive accumulation of cancer stem cell properties and CD44 as a potential diagnostic marker. Lung Cancer. 2021;154:13-22
10. Fong W, Li Q, Yu J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene. 2020;39:4925-43
11. Mulders MCF, Audhoe AS, Van Koetsveld PM, Feelders RA, Hofland LJ, de Herder WW. et al. Midgut neuroendocrine tumor patients have a depleted gut microbiome with a discriminative signature. Eur J Cancer. 2024;197:113472
12. Hu W, Chen ZM, Li XX, Lu L, Yang GH, Lei ZX. et al. Faecal microbiome and metabolic signatures in rectal neuroendocrine tumors. Theranostics. 2022;12:2015-27
13. Oksanen J, Blanchet FG, Kindt R, Legendre P, Stevens MHH. Vegan: Community Ecology Package. R package, version 1.17-4. 2010
14. Wickham H. ggplot2. Wiley Interdisciplinary Reviews: Computational Statistics. 2011;3:180-5
15. Kolde R. pheatmap: Pretty Heatmaps. 2015;1(7).
16. Chen H, Boutros PC. Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12: 35. BMC Bioinformatics. 2011;12:35
17. Conway Jake, R Lex, Alexander Gehlenborg. et al. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33(18):2938-2940
18. White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352
19. Patil KR, Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc Natl Acad Sci U S A. 2005;102:2685-9
20. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60
21. Hamilton NE, Ferry M. ggtern: Ternary Diagrams Using ggplot2. Journal of statistical software. 2018;87:1-17
22. Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O. et al. Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis. Adv Mater. 2018;30:e1805007
23. Perdijk O, Azzoni R, Marsland BJ. The microbiome: an integral player in immune homeostasis and inflammation in the respiratory tract. Physiol Rev. 2024;104:835-79
24. Bojesen RD, Riis LB, Høgdall E, Nielsen OH, Jess T. Inflammatory Bowel Disease and Small Bowel Cancer Risk, Clinical Characteristics, and Histopathology: A Population-Based Study. Clin Gastroenterol Hepatol. 2017;15:1900-7.e2
25. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14:573-84
26. Mohamed A, Asa SL, McCormick T, Al-Shakhshir H, Dasari A, Mauricio R. et al. The Role of the Microbiome in Gastroentero-Pancreatic Neuroendocrine Neoplasms (GEP-NENs). Curr Issues Mol Biol. 2022;44:2015-28
27. Vietti Violi N, Duran R, Demartines N, Sempoux C, Guiu B, Bize PE. et al. Local recurrence rate in patients with colorectal cancer liver metastasis after wedge resection or percutaneous radiofrequency ablation. Int J Hyperthermia. 2018;34:1020-8
28. Boero E, Vezzani G, Micoli F, Pizza M, Rossi O. Functional assays to evaluate antibody-mediated responses against Shigella: a review. Front Cell Infect Microbiol. 2023;13:1171213
29. Kotloff KL, Riddle MS, Platts-Mills JA, Pavlinac P, Zaidi AKM. Shigellosis. Lancet. 2018;391:801-12
30. Lu Y, Cui A, Zhang X. Commensal microbiota-derived metabolite agmatine triggers inflammation to promote colorectal tumorigenesis. Gut Microbes. 2024;16:2348441
31. Zhao M, Xie X, Xu B, Chen Y, Cai Y, Chen K. et al. Paeonol alleviates ulcerative colitis in mice by increasing short-chain fatty acids derived from Clostridium butyricum. Phytomedicine. 2023;120:155056
32. Fan Y, Tao T, Guo Z, Wah To KK, Chen D, Wu S. et al. Lazertinib improves the efficacy of chemotherapeutic drugs in ABCB1 or ABCG2 overexpression cancer cells in vitro, in vivo, and ex vivo. Mol Ther Oncolytics. 2022;24:636-49
33. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. 2017;36:293-303
34. Erin N, Grahovac J, Brozovic A, Efferth T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist Updat. 2020;53:100715
35. Song G, Gan Q, Qi W, Wang Y, Xu M, Li Y. Fructose Stimulated Colonic Arginine and Proline Metabolism Dysbiosis, Altered Microbiota and Aggravated Intestinal Barrier Dysfunction in DSS-Induced Colitis Rats. Nutrients. 2023;15:782
36. Pavel M, Öberg K, Falconi M, Krenning EP, Sundin A, Perren A. et al. Gastroenteropancreatic neuroendocrine neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31:844-60
37. de Mestier L, Lepage C, Baudin E, Coriat R, Courbon F, Couvelard A. et al. Digestive Neuroendocrine Neoplasms (NEN): French Intergroup clinical practice guidelines for diagnosis, treatment and follow-up (SNFGE, GTE, RENATEN, TENPATH, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, SFR). Dig Liver Dis. 2020;52:473-92
38. Uhlig R, Dum D, Gorbokon N, Menz A, Büscheck F, Luebke AM. et al. Synaptophysin and chromogranin A expression analysis in human tumors. Mol Cell Endocrinol. 2022;555:111726
39. Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M. et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25:667-78
Author contact
![]() Corresponding author: Qiyun Tang, Department of Geriatric Gastroenterology, Neuroendocrine Tumor Center, The First Affiliated Hospital of Nanjing Medical University, Institute of Neuroendocrine Tumor, Nanjing Medical University, NO.300 Guangzhou Road, Nanjing, China. Email: qytangedu.cn.
Corresponding author: Qiyun Tang, Department of Geriatric Gastroenterology, Neuroendocrine Tumor Center, The First Affiliated Hospital of Nanjing Medical University, Institute of Neuroendocrine Tumor, Nanjing Medical University, NO.300 Guangzhou Road, Nanjing, China. Email: qytangedu.cn.